Abstract
Despite surgery, chemotherapy, and radiotherapy treatments, the children, adolescents, and young adults who are diagnosed with metastasized Ewing sarcoma face a dismal prognosis. Amyloid precursor-like protein 2 (APLP2) has recently been implicated in the survival of cancer cells and in our current study, APLP2’s contribution to the survival of Ewing sarcoma cells was examined. APLP2 was readily detected in all Ewing sarcoma cell lines analyzed by western blotting, with the TC71 Ewing sarcoma cells expressing the lowest level of APLP2 among the lines. While irradiation induces apoptosis in TC71 Ewing sarcoma cells (as we determined by quantifying the proportion of cells in the sub-G1 population), transfection of additional APLP2 into TC71 decreased irradiation-induced apoptosis. Consistent with these findings, in parallel studies, we noted that isolates of the TC71 cell line that survived co-culture with lymphokine-activated killer (LAK) cells (which kill by inducing apoptosis in target cells) displayed increased expression of APLP2, in addition to smaller sub-G1 cell populations after irradiation. Together, these findings suggest that APLP2 lowers the sensitivity of Ewing sarcoma cells to radiotherapy-induced apoptosis and that APLP2 expression is increased in Ewing sarcoma cells able to survive exposure to cytotoxic immune cells.
Introduction
Ewing sarcoma is a pediatric cancer in the small round blue cell family of tumors. Ewing sarcoma patients with regional lymph node involvement tend to have a particularly low duration of survival after diagnosis, and the patients who relapse with Ewing sarcoma have a very unfavorable prognosis.Citation1,Citation2 The nature of the progenitor cell(s) for Ewing sarcoma is unclear, but there is evidence that the disease may arise from mesenchymal stem cells.Citation3,Citation4 Ewing sarcomas possess characteristic gene fusions as a result of chromosomal translocations between EWS and ETS family member genes. The products of these gene fusions are thought to act as abnormal transcription factors that induce expression of some specific genes and repress the expression of others, contributing to a tumorigenic phenotype.Citation5-Citation7
Standard multi-modal therapy for Ewing sarcoma includes radiation, surgery, and chemotherapy, treatments that can be accompanied by severe side effects.Citation8,Citation9 Even with the best of available treatments, Ewing sarcoma is often fatal, demonstrating that the tumor cells are capable of resisting current treatments. Although Ewing sarcoma tumors were initially identified as radiosensitive,Citation10 high-dose radiation therapy is recommended for Ewing sarcoma patients.Citation11 Higher doses of radiation come at a cost, carrying an increased risk for secondary malignancies, such as various types of bone tumor or leukemia.Citation12 To date, expression of certain molecules involved in the regulation of apoptosis or in the pathogenesis of Ewing sarcoma (including the most commonly observed Ewing sarcoma gene fusion product, EWS-FLI1) have been shown to influence the radiosensitivity of Ewing sarcoma cells.Citation7,Citation13-Citation15 Radiotherapy can be more effective when combined with agents that increase sensitivity of cells to radiation effects (a.k.a. chemical radiosensitizers).Citation16 Thus, more effective therapies with reduced toxicity and improved patient survival can be achieved through a greater understanding of the molecular basis of Ewing sarcoma cell survival.
Ewing sarcoma cells have been demonstrated to be highly susceptible to lysis by LAK cells, which are generated by exposure of leukocytes to high-dose interleukin-2, and include cells with natural killer cell markers and ones with T cell markers.Citation17-Citation19 In addition to the susceptibility of Ewing sarcoma cells to LAK cells, there is evidence for LAK cell effectiveness against other pediatric cancers (neuroblastoma and rhabdomyosarcoma), as well as against lymphoma, hepatocellular carcinoma, pancreatic cancer, glioblastoma, and colon cancer.Citation20-Citation27 However, as with all anti-tumor therapies, there is some selection for tumor resistance and evasion that occurs with the use of immunotherapies, including immunotherapies based on cytotoxic effector cells such as LAK cells. Understanding the nature of resistance to immunotherapies, as well as to conventional therapies such as chemotherapy and radiotherapy, is necessary in order for new approaches to overcome tumors to be successfully developed.
An ideal molecular target increases the susceptibility of tumor cells to apoptotic death mediated by more than one source (e.g., radiation therapy and cytotoxic cells). Amyloid precursor-like protein 2 (APLP2) has been implicated as a pro-survival mediator in Chinese hamster ovary cells transfected with APLP2 and in normal mouse sperm.Citation28 Our laboratory recently demonstrated that APLP2 is a pro-survival factor in pancreatic cancer cells and also showed that APLP2 reduces the surface expression of the major histocompatibility complex (MHC) class I molecule (which is vital for T cell recognition and lysis of tumor cells) by an endocytic mechanism.Citation29-Citation33 Findings from our laboratory and others show that APLP2 is elevated in a variety of cancer cell lines (e.g., pancreatic and prostate cancer cell lines) and in human pancreatic and colorectal cancer tissues.Citation29,Citation32-Citation36 Therefore, we hypothesized that APLP2 might be a pro-survival factor in Ewing sarcoma cells.
In these studies, we have identified APLP2 as a protein that is amply expressed in several Ewing sarcoma cell lines. Overexpression of APLP2 in Ewing sarcoma cells reduced the percentage of cells in the sub-G1 population (i.e., the apoptotic population) following irradiation. Furthermore, although most Ewing sarcoma cells were found to be susceptible to lysis by LAK cells, some Ewing sarcoma cells were able to survive incubation with LAK cells, indicating resistance. We have demonstrated that cell lines derived from the resistant tumor cell population have substantially increased expression of APLP2. We also noted that a relatively low proportion of the LAK-resistant Ewing sarcoma cells exposed to gamma radiation had sub-G1 DNA content. These findings suggest that the radiation-exposed LAK-resistant Ewing sarcoma cells are relatively resistant to acquiring signs of apoptosis. Collectively, our findings suggest that APLP2 has a regulatory role in the survival of Ewing sarcoma cells.
Results
APLP2 suppresses the induction of irradiation-induced apoptosis
On the basis of previous findings on APLP2 expression in cancer cells and its role in cell survival,Citation29,Citation34-Citation36 we investigated APLP2 expression in several Ewing sarcoma cell lines and detected substantial levels of APLP2 by immunoblotting (). In addition to the cell lines for which data are shown in (A4573, RD-ES, and TC-71), we also found substantial expression of APLP2 in the Ewing sarcoma cell lines Hs822.T, Hs863.T, A673, and RD-ES (data not shown). We next sought to determine the influence of APLP2 on the apoptosis of irradiated TC71 Ewing sarcoma cells. To begin, we examined the effect of increasing doses of radiation on the apoptosis of untransfected TC71 cells expressing endogenous levels of APLP2. Following irradiation, a dose-dependent accumulation of the population in sub-G1 (a hallmark of apoptotic cellsCitation37) was found in the irradiated cells, with the full effect being observed at 20–25 Gy (). We then assessed various transfection approaches to upregulate the expression of APLP2 in the TC71 cells (which, as shown in , endogenously expresses a lower level of APLP2 than some other Ewing sarcoma cell lines). Among the transfection methods that we tested, the optimal results for transient transfection of a full-length APLP2 cDNA were obtained with Lipofectamine, which produced a clear increase in APLP2 expression (). To test the effect of APLP2 on irradiation-induced apoptosis, TC71 cells were transfected with APLP2 and subsequently exposed to radiation. As indicated in , transfection with APLP2 (compared with transfection with vector alone) caused a substantial reduction in the percentage of irradiated cells with sub-G1 DNA content (i.e., apoptotic cells). These data suggest that APLP2 has an anti-apoptotic effect in irradiated Ewing sarcoma cells.
Figure 1. Substantial levels of APLP2 are present in Ewing sarcoma cell lines. Lysates of the Ewing sarcoma cell lines A4573, RD-ES, and TC-71 were immunoblotted with a polyclonal antiserum specific for APLP2 or with an antiserum for actin (as a loading control). MDA-MB435s (melanoma cell line) and 721.221 (B lymphoblastoid cell line) were included as controls expressing known high or low (respectively) levels of APLP2. The white line indicates the position of an intervening lane that was removed from the figure. The data shown are representative of results from 2 experiments that included these 3 Ewing sarcoma cell lines analyzed together and are representative of over 10 experiments that included various other groups of Ewing sarcoma cell lines.
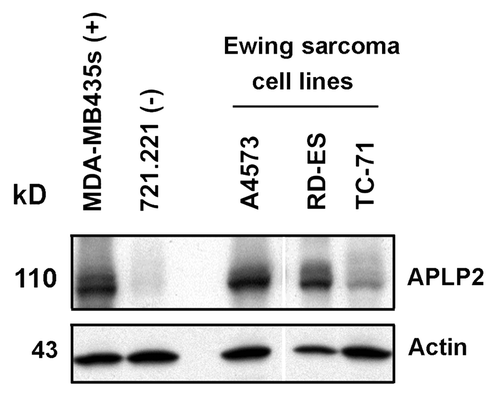
Figure 2. Irradiated TC71 Ewing sarcoma cells undergo apoptosis with a maximum sub-G1 population at 20–25 Gy. TC71 cells were seeded in 60 mm2 dishes at least 24 h prior to irradiation at the indicated dose, and the cells were in the log phase of growth at the time of irradiation. Twenty-four hours post-irradiation, the cells were fixed and stained with propidium iodide, and their DNA content was determined by flow cytometry. Cell cycle analysis was performed using ModFit software. (A) A dose-dependent increase in apoptotic cells was observed up to 20–25 Gy. Error bars denote the standard deviation; n = 2. (B) Representative, bright-field images of cells in culture 24 h post-irradiation at the indicated radiation dosage. The data shown are representative of results from 3 experiments.
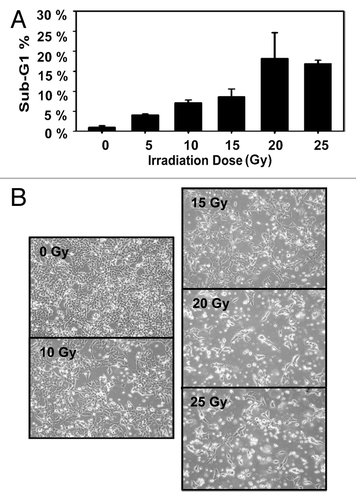
Figure 3. APLP2 reduces the sensitivity of TC71 Ewing sarcoma cells to irradiation-mediated apoptosis. (A) TC71 cells were seeded at 5 × 106 cells per 100 mm dish, and upon reaching 40–50% confluence at about 24 h they were transfected with the pCMV-Tag4A vector alone or with pCMV-Tag4A-APLP2. The transfected cells were collected at 48 h and lysed for use in western blots for APLP2 (and for actin, as a control). (B) TC71 cells at 40–50% confluence were transfected with the pCMV-Tag4A empty vector or with pCMV-Tag4A-APLP2 and incubated for 48 h. The cells were then irradiated (0 Gy or 20 Gy), incubated for an additional 24 h, harvested, fixed, stained with propidium iodide, and analyzed for DNA content by flow cytometry. The results in the graph depict the percentage of cells with sub-G1 DNA content in APLP2-overexpressing cells vs. vector only-transfected cells. Duplicate samples were used and error bars denote the percent confidence interval. The results shown are representative of results from 3 separate experiments.
Ewing sarcoma cell lines escaping LAK cell lysis have increased APLP2 expression
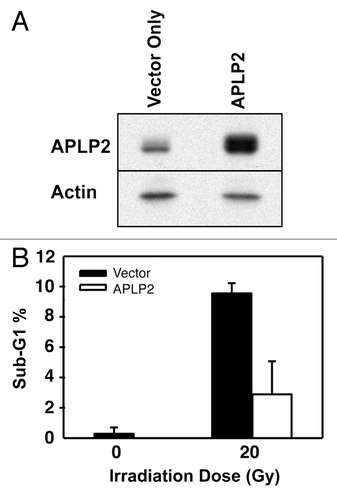
In addition to challenging Ewing sarcoma cells with radiation, we extended our studies by challenging the survival of Ewing sarcoma cells with cytotoxic immune cells. As tools for our experiments, we isolated Ewing sarcoma cells that failed to be cleared by cytotoxic immune cells, since they represent an immune-evasive population. To generate cytotoxic cell populations for our use, we cultured human peripheral blood leukocytes, with or without high-dose IL-2, for 3 d to generate LAK cells or control effector cells, respectively. Similar to observations made before in many laboratories, both populations contained cells with surface markers characteristic of cytotoxic T lymphocytes, NK, and NK-T cells (), with elevated CD56 expression on the NK cells in the IL-2-activated cell population (), consistent with previous reports.Citation39-Citation41
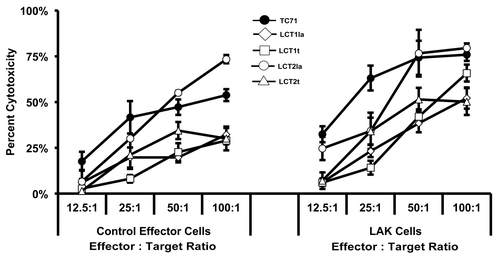
Figure 4. Ewing sarcoma cell lines are lysed by LAK cells. (A) Effector cell populations (control effector cells and LAK cells) were characterized for their composition of cytotoxic cells by cell surface markers: cytotoxic T lymphocytes (CTLs) (CD3+CD8+CD56-); natural killer (NK) cells (CD3-CD56+); and natural killer T (NKT) cells (CD3+CD8+CD56+). Error bars denote the standard error of the mean, with n = 5. (B) NK cells within the LAK cell population had enhanced CD56 surface expression, compared with NK cells within the control effector cell population (P < 0.001). The mean fluorescence units (MFU) from staining of the NK cells in the population with phycoerythrin (PE)-conjugated, anti-CD56 antibody is shown. Error bars denote the standard error of the mean, with n = 5. For (A and B), the flow cytometric analysis for assessment of T cell and NK cell markers on the effector cells was performed once, with multiple samples per analytical point as indicated. (C) Peripheral blood leukocytes were cultured for 3 d in the presence (LAK cells) or absence of high-dose IL-2 (control effector cells) and then incubated with 51Cr-labeled Ewing sarcoma TC71 (circle) and RD-ES (triangle) cell lines. Labeled K562 cells served as a positive control. 51Cr release from lysed cells was measured and the percent cytotoxicity was calculated for each set of triplicate wells (as described in the Materials and Methods section). The error bars represent the standard error of the mean; n = 6. The data shown are representative of results from 3 similar experiments.
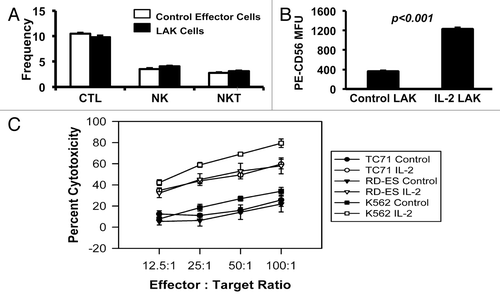
At the maximum level of exposure to cytotoxic effectors (4 h at a 100:1 effector:target ratio), 79% or 34% of the K562 cells were lysed by the cytotoxic cells generated in the presence of IL-2 (LAK cells) or absence of IL-2 (control effector cells), respectively (). Furthermore, both TC71 and RD-ES Ewing sarcoma cell lines reached approximately 60% or 20% lysis following the maximum exposure to the cytotoxic cells generated in the presence or absence of IL-2, respectively (). These results support previous observations that Ewing sarcoma cells are susceptible to lysis by NK cells and LAK cells.Citation17,Citation18,Citation42-Citation44 Notably, while the majority of TC71 Ewing sarcoma cells exposed to LAK cells were killed when a high effector:target ratio was used, some of the TC71 cells survived exposure to the immune cells and represent a refractory population.
To generate lines from LAK-resistant cells, a separate co-culture (at a ratio of 10 LAK cells per 1 unlabeled TC71 cell) was set up, and then the effector LAK cells were removed after 5 d. From the remaining LAK-evasive TC71 cells, we established four lines (LCT1la, LCT1t, LCT2la, and LCT2t). In a separate cytotoxicity assay, we found that three of the four cell lines (LCT1la, LCT1t, and LCT2t) were resistant both to LAK effector cells and to control effector cells when re-challenged (). As demonstrated in , the LCT2la line was more resistant than TC71 at 2 of the 4 effector:target ratios for both the control effector cells and the LAK cells, and analysis of variance (ANOVA) showed a significant difference (P = 0.0451) between TC71 and LCT2la as LAK cell targets. By ANOVA, the control effector cells also showed a trend to difference (P = 0.0671) for killing LCT2la vs. TC71, although statistical significance at a threshold of p < 0.05 was not reached. Thus LCT2la was not as resistant as the other LCT lines, but still showed some increase in resistance relative to TC71. Furthermore, the observation that the test results with lower effector:target ratios showed a greater distinction between TC71 and the LCT cell lines is consistent with the low effector:target ratio (10:1) used to originally select the LCT cell lines.
Figure 5. Ewing sarcoma cell lines escaping LAK cell-mediated destruction were established. The LCT2la cell line did not have impaired resistance to LAK cytotoxicity; however, the remaining three cell lines were more resistant to LAK cytotoxicity, compared with TC71 cells. Peripheral blood mononuclear cells (PBMC) were obtained from healthy donor apheresis and used to generate LAK cells following three days in culture with 1000 U/mL recombinant human interleukin (IL)-2. Control effector cells were PBMC cultured in the absence of IL-2. The results obtained using the TC71 cells as targets are shown by the lines with dark circles. The results obtained using the LCT lines as targets are shown by lines with open symbols (as indicated on the figure for each of the 4 LCT lines). Sensitivity of LCT cell lines to LAK-mediated cytotoxicity was determined by the chromium-51 release assay. Line graphs represent the mean percent cytotoxicity for the indicated cell line under the given condition. Significance was determined using two-way ANOVA for independent samples. Error bars denote the standard error of the mean, n = 2,3. The experiment for which results are shown in this figure was performed once, and within the experiment for which results are shown in this figure, similar results were obtained with multiple sample points and over a range of 4 E:T ratios with 2 separate cytotoxic effector populations (i.e., with LAK cells and with control effector cells).
LAK-resistant Ewing sarcoma cells express elevated levels of APLP2 and are more resistant to irradiation-induced apoptosis
The TC71 cell lines evading LAK-cell cytotoxicity were tested for expression of APLP2 by western blotting. We found that APLP2 was strongly elevated in all 4 LAK-resistant cell lines (by a 2- to 7-fold increase) compared with the parental TC71 cells ( and data not shown). We also examined whether an increase in APLP2 homologs (APP and APLP1) had occurred in the LAK-resistant cells and found no detectable APP and no increase in APLP1 (data not shown). These findings demonstrate that among the Ewing sarcoma cell lines derived from LAK-escaping cells, there was an increase in total and surface APLP2 expression, but not in APP or APLP1 expression.
Figure 6. Ewing sarcoma cells that survived co-culture with LAK cells express higher levels of APLP2 and have more resistance to irradiation-induced apoptosis. (A) Lysates of the TC71 cell line and LAK-escaping LCT1la, LCT2la, and LCT2t cell lines were immunoblotted for APLP2 or actin. The results shown are representative of the findings from two separate experiments. (B) All four LCT cell lines have reduced sensitivity to irradiation-mediated apoptosis, compared with TC71 cells. Log-phase cells were exposed to 25 Gy gamma radiation, then cultured for 24 h prior to propidium iodide staining and DNA content analysis. Error bars denote the standard error of the mean; n = 3. Statistical significance was determined for all LAK-escaping cell lines compared with the parental TC71 cell line using the Student t test. The data shown were acquired in 2 independent experiments.
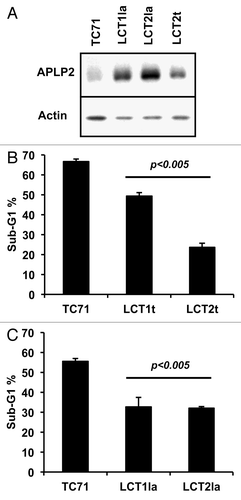
As mentioned above, when APLP2 was overexpressed in the TC71 cell line, the cells became more resistant to irradiation-mediated apoptosis (). Therefore, we examined if our LAK-escaping cell lines, which had elevated APLP2 expression, were also less susceptible to apoptosis subsequent to irradiation. Indeed, at 24 h post-irradiation, all four LAK-escaping cell lines demonstrated reduced sub-G1 DNA content (), compared with the original TC71 cell line. Thus, these data demonstrate that the Ewing sarcoma cell lines that evaded cytotoxic destruction have elevated APLP2 expression and are also less susceptible to irradiation-induced apoptosis.
Discussion
In our study, we investigated the ability of APLP2 expressed in Ewing sarcoma cell lines () to serve an anti-apoptotic function within Ewing sarcoma cells, before or following a laboratory model mimicking radiotherapy. Overexpression of APLP2 in Ewing sarcoma cells, as a result of APLP2 transfection, reduced the sub-G1 population following radiation ( and ). Therefore, APLP2 is capable of regulating the induction of apoptosis in Ewing sarcoma cells after irradiation. The precise mechanism whereby APLP2 suppresses radiation-mediated apoptosis in Ewing sarcoma cells has not been determined, but might involve interactions with Fe65 proteins. Fe65 associates with a sequence derived from the APLP2 C-terminus, and some studies have implicated this complex in gene transcription.Citation38 Thus, in our studies, increased APLP2 expression may have upregulated transcription of unidentified survival-related genes by a mechanism involving association with Fe65. Fe65 is also known to be necessary for Tip-60-mediated histone H4 acetylation at DNA strand breaks, and interaction between Fe65 and APP is necessary for this function of Fe65 in DNA repair.Citation45 Therefore, an alternative possibility is that APLP2, as an APP homolog, has a role equivalent to APP in this process, in which case our results could be due to increased APLP2/Fe65 interactions that upregulate DNA repair.
The LAK-escaping Ewing sarcoma cell lines, isolated from the TC71 cell line, that we generated displayed elevated expression of APLP2 ( and data not shown). In agreement with our results using Ewing sarcoma cells transfected with APLP2, these LAK-evasive cell lines (three of which maintained resistance to cytotoxic cells post-incubation, ) also demonstrated reduced irradiation-mediated apoptosis (relative to control TC71 cells) (). Thus, through immune selection, we were able to isolate an endogenous population of Ewing sarcoma cells that displayed elevated expression of APLP2 and had increased resistance to the pro-apoptotic effects of ionizing radiation. These data complement our findings with transient APLP2 transfections indicating that APLP2 decreases the apoptotic sensitivity of irradiated Ewing sarcoma cells ().
As mentioned above in the Introduction, our laboratory has also shown that APLP2 can facilitate the endocytosis of MHC class I molecules, which are antigen-presenting molecules that trigger lysis of tumor cells by T cells.Citation29-Citation33 In contrast to their activation of T cells, MHC class I molecule interaction with inhibitory receptors on NK cells downregulates NK cell cytotoxic function.Citation46,Citation47 Because LAK cell populations include both T cells and NK cells (e.g., see ), selection of tumor cells by co-culture with LAK cells could potentially provide pressure both for increase and decrease of cell-surface MHC class I expression. In our future studies, using Ewing sarcoma cells cultured with NK cells or with cytotoxic T cells (each as an isolated effector population), we will examine whether there is a positive or negative correlation of the evasion of either (or both) of these effector populations with APLP2 expression and with the surface expression of MHC class I molecules. In these studies, we will also examine whether NK cell selection pressure on Ewing sarcoma cells affects APLP2 expression in correlation with changes in the surface expression of other NK inhibitory receptors (besides MHC class I molecules) and with changes in the surface expression of NK activating receptors.Citation46,Citation47 The increased expression of APLP2 in LAK-evasive Ewing sarcoma cells supports the possibility that APLP2 might be capable of reducing the susceptibility of Ewing sarcoma cells to destruction mediated by cytotoxic immune cells as a result of APLP2’s anti-apoptotic effects, since cytotoxic cells kill by an apoptotic mechanism.Citation48-Citation50 However, further experiments will be required to solidify a role for APLP2 in the immune-evasive properties of Ewing sarcomas and to obtain a total picture of how APLP2’s combined anti-apoptotic and pro-endocytic functions may figure into its advancement of Ewing sarcoma immune evasion.
In conclusion, our findings suggest that APLP2 should be considered as a factor potentially able to regulate Ewing sarcoma apoptosis, whether mediated by irradiation or cytotoxic immune effector cells. Thus, exploring the impact of therapies targeting APLP2 may present a new, unexplored field in Ewing sarcoma treatment. Strategies targeting APLP2 may have value as adjuvants that could increase Ewing sarcoma susceptibility to a variety of pro-apoptotic therapies.
Materials and Methods
Cell culture
The Ewing sarcoma cell lines A673, Hs822.T, Hs863.T, and RD-ES were purchased from ATCC (CRL-1598, CRL-7556, CRL-7598, HTB-166, respectively) and the A4573 and TC71 cell lines were gifts from Dr Eugenie Kleinerman at the University of Texas MD Anderson Cancer Center. The MDA-MB435s melanoma cell line was obtained from Dr Vinod Labhasetwar (Cleveland Clinic) and the K562 cell line from Dr Ted Hansen (Washington University School of Medicine). The cell lines A673, Hs822.T, Hs863.T, and A-4573 were cultured in DMEM supplemented with 10% v/v fetal bovine serum, 1 mM sodium pyruvate (Invitrogen, 11360), 2 mM l-glutamine (Invitrogen, 25030), 100 units/mL penicillin and 100 μg/mL streptomycin (Invitrogen, 15140). The K562, TC71, and RD-ES cell lines were cultured in RPMI 1640 (Fisher Scientific 10-04-CV) and supplemented with 10–15% v/v fetal bovine serum, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin.
To generate LAK cells, human peripheral blood leukocytes were incubated for three days at 37 °C, 5% CO2 in RPMI 1640 with 10% fetal bovine serum, 1 μL/mL β-mercaptoethanol (Sigma-Aldrich, M-7522) and 1000 units/mL IL-2 and then cells in suspension were collected. Control effector cells were generated simultaneously, by the same process as the LAK cells, except that they were cultured in the absence of IL-2. To select for LAK-escaping Ewing sarcoma cells, LAK cells and TC71 cells were co-cultured at an effector:target ratio of 10:1 in RPMI 1640 containing 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM l-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin, plus 1 μL/ml β-mercaptoethanol and 1000 units/mL IL-2. Following 5 d of culture, the effector cells (in suspension) were removed, and adherent cells were collected and cultured for 16 d in RPMI 1640 containing the additives listed above (including 1 μL/ml β-mercaptoethanol, but not IL-2). Four separate lines, designated as LCT1la, LCT1t, LCT2la, and LCT2t, were established from the cells.
Antibodies
The anti-APLP2 rabbit polyclonal antibody was purchased from Calbiochem/EMD Chemicals (171617). The anti-actin monoclonal antibody was obtained from Novus Biologicals (NBP1-41294). Secondary monoclonal antibodies used for immunoblot analysis were peroxidase-conjugated AffiniPure goat anti-mouse IgG light chain or peroxidase-conjugated IgG fraction mouse anti-rabbit IgG light chain (Jackson ImmunoResearch Laboratories, Inc., 115-035-174 and 211-032-171, respectively).
For flow cytometric characterization of cytotoxic effector cell types in control effector cell and LAK cell populations, the following monoclonal antibodies were purchased from AbD Serotec: AlexaFluor 647-conjugated mouse IgG1 anti-human CD3 (MCA463A647T), FITC-conjugated mouse IgG1 anti-human CD8 (MCA2511F), R-phycoerythrin (PE)-conjugated mouse IgG2b anti-human CD56 (1437PE), and AlexaFluor 647-conjugated mouse IgG1, isotype control (MCA928A647). Additional isotype controls used include FITC-conjugated mouse IgG1 (BD Biosciences, 551954) and PE-conjugated mouse IgG2b (eBioscience, 12-4732).
Transfections
Cells were transiently transfected with a previously described full-length APLP2 cDNA,Citation30 in the pCMV-Tag4A vector. For transfecting TC71 cells with full-length APLP2 cDNA prior to western blotting procedures, TC71 cells were seeded in 100 mm diameter dishes at 5 × 106 cells per dish. Once the cells had attached and reached 40–50% confluence (at ~24 h), they were transfected with the pCMV-Tag4A empty vector as a control (by the use of Lipofectamine from Life Technologies, 11668019) or transfected with pCMV-Tag4A-APLP2 (also by the use of Lipofectamine). Transfected cells were collected at 48 h post-transfection. At collection, all cells were washed and lysed to create whole cell lysates to be electrophoresed with 5× SDS loading buffer on a 4‒20% Tris-glycine gel. As described in more detail below, following transfer to blotting membrane, the proteins were probed with antibodies.
To prepare full-length APLP2 cDNA-transfected cells for use in cell cycle analysis experiments, TC71 cells were seeded in 60 mm diameter dishes at 30‒40% confluence. After cells had attached and reached 40–50% confluence, they were transfected (using Lipofectamine) with the pCMV-Tag4A empty vector or with pCMV-Tag4A-APLP2 and incubated overnight. After replacement of the media, the transfected cells were incubated another 24 h. The percentage of sub-G1 DNA content of the cells was then analyzed by flow cytometry in the transfected cells at 24 h post-radiation treatment (20 Gy) relative to no radiation treatment (0 Gy). Before flow cytometric analysis, the cells were harvested, fixed with 70% ethanol, and stained with propidium iodide.
Cytotoxicity assay
Ewing sarcoma target cells were labeled with Na-51Cr for 1 h and then incubated with the LAK cells (generated as described above) for 4 h in triplicate at a series of effector:target ratios. The cells were gently centrifuged and supernatants were counted on a Pharmacia LKB gamma counter. The percent cytotoxicity was calculated from each set of experimental values minus the average of spontaneous release values, divided by the average of total release values minus the average of spontaneous release values, all multiplied by 100. Total release values were obtained by lysing cells in Triton X-100, and spontaneous release values were obtained from 51Cr-labeled target cells incubated alone in media. The chronic myelogenous leukemia K562 cell line served as a positive control. Due to their lack of surface major histocompatibility complex class I molecules, K562 cells can readily be lysed by the NK cells present in LAK cell lines.Citation51
Immunoblots
For preparation of cell lysates, pellets of 1 × 107 cells were resuspended in cell lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% Triton X-100). Resuspended pellets were incubated on ice for 1 h with occasional vortexing and then stored at -80 °C overnight. The next day, the lysates were thawed on ice and then spun in a desktop centrifuge at top speed for 30 min at 4 °C. Supernatants were collected in new tubes and stored at -80 °C. Aliquots of lysates were added to 5× SDS loading buffer and boiled for 5 min prior to electrophoresis on 4→20% Tris-glycine pre-cast gels (Invitrogen, EC60285). Proteins were transferred to Immobilon membranes (Millipore, IPVH000010). Following overnight blocking in 5% w/v nonfat dry milk, the blots were incubated with primary antibodies diluted in 5% nonfat dry milk for 2 h, washed 3 times with 0.05% Tween 20 in PBS and incubated with secondary antibodies diluted in 0.05% Tween 20 for 1 h. Three washes with 0.3% Tween 20 were performed, and then membranes were submerged for 1 min in Pierce ECL substrate from Thermo Scientific (32106) and developed on Kodak BioMax MR film (894-1114). When protein band densities were quantified, the Molecular Imager ChemiDoc XRS system with Quantity One 1-D Analysis Software (Bio-Rad) was used.
Irradiation and cell cycle analysis
Cells were seeded at least 24 h prior to irradiation with a Mark I 68A Cesium-137 Irradiator (J. L. Shepherd and Associates). Cell cycle analysis was performed as previously described.Citation52 Briefly, irradiated cells were cultured at 37 °C, 5% CO2 for an additional 24 h, then stained with propidium iodide and analyzed for DNA content by flow cytometry on a BD FACS Calibur with ModFit software (Verity Software House, Inc.) or Diva software (BD Biosciences). All analyses were performed using at least 20 000 cells.
Flow cytometry
For flow cytometry, cells were gently collected on the morning of analysis and kept on ice throughout the assay. To prepare cytotoxic effector cells for flow cytometry, suspension control effector cells and LAK cells were centrifuged at 125 × g for 8 min without any brake and then the supernatant was discarded. LAK cell pellets were washed twice with 0.2% weight per volume bovine serum albumin in phosphate-buffered saline and incubated with 5 μL of primary antibody per 1 × 106 cells in a 96-well plate for 30 min on ice in the dark. Excess antibodies were removed by centrifugation at 450 × g for 5 min at 4 °C and three washes in 0.2% weight per volume bovine serum albumin in phosphate-buffered saline. Antibody-labeled LAK cells were resuspended to 2.5 × 106 cell/mL and analyzed with a FACS Calibur instrument and Cell Quest software at the UNMC Cell Analysis Facility.
| Abbreviations: | ||
| APLP2 | = | amyloid precursor-like protein 2 |
| LAK | = | lymphokine-activated killer cell |
| APP | = | amyloid precursor protein |
| APLP1 | = | amyloid precursor-like protein 1 |
Acknowledgments
The authors thank Drs Eugenie Kleinerman, Vinod Labhasetwar, and Ted Hansen for providing cell lines for this project. We also gratefully acknowledge Dr Xiaojian Wang and Brittney Smith for technical support, Dr Janina Baranowska-Kortylewicz for assistance in the irradiation experiments, and the assistance of the personnel of the University of Nebraska Medical Center Cell Analysis Facility in the flow cytometry experiments. This work was supported by an Edna Ittner Pediatric Research Support Fund Grant (to SJ), an Eppley Cancer Center Pediatric Research Grant and NIH R03CA176557 (to JCS), NIH COBRE (P20RR018759/P20GM103489) and SPORE (P50CA127297) Developmental Grants (to JCS), Nebraska DHHS-LB506 Grants (to YY and JCS), a Structural Biology and Biophysics Training Program Fellowship from the Department of Education GAANN Program (to HLP), an NIH Training Grant T32CA009476 Fellowship (to HLP) and a UNMC Graduate Studies Office Emley Fellowship/Regents Tuition Fellowship (to HLP).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Notes
†Current affiliation: Department of Experimental Therapeutics; University of Texas MD Anderson Cancer Center; Houston TX USA
‡Current affiliation: Department of Internal Medicine; University of Nebraska Medical Center; Omaha, NE USA
§Current affiliation: Biology Department; University of Nebraska at Omaha; Omaha, NE USA
References
- Applebaum MA, Goldsby R, Neuhaus J, DuBois SG. Clinical features and outcomes in patients with Ewing sarcoma and regional lymph node involvement. Pediatr Blood Cancer 2012; 59:617 - 20; http://dx.doi.org/10.1002/pbc.24053; PMID: 22184129
- Stahl M, Ranft A, Paulussen M, Bölling T, Vieth V, Bielack S, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011; 57:549 - 53; http://dx.doi.org/10.1002/pbc.23040; PMID: 21442722
- Meltzer PS. Is Ewing’s sarcoma a stem cell tumor?. Cell Stem Cell 2007; 1:13 - 5; http://dx.doi.org/10.1016/j.stem.2007.05.011; PMID: 18371327
- Tirode F, Laud-Duval K, Prieur A, Delorme B, Charbord P, Delattre O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007; 11:421 - 9; http://dx.doi.org/10.1016/j.ccr.2007.02.027; PMID: 17482132
- Riggi N, Stamenkovic I. The biology of Ewing sarcoma. Cancer Lett 2007; 254:1 - 10; http://dx.doi.org/10.1016/j.canlet.2006.12.009; PMID: 17250957
- Herrero-Martín D, Osuna D, Ordóñez JL, Sevillano V, Martins AS, Mackintosh C, et al. Stable interference of EWS-FLI1 in an Ewing sarcoma cell line impairs IGF-1/IGF-1R signalling and reveals TOPK as a new target. Br J Cancer 2009; 101:80 - 90; http://dx.doi.org/10.1038/sj.bjc.6605104; PMID: 19491900
- Soldatenkov VA, Trofimova IN, Rouzaut A, McDermott F, Dritschilo A, Notario V. Differential regulation of the response to DNA damage in Ewing’s sarcoma cells by ETS1 and EWS/FLI-1. Oncogene 2002; 21:2890 - 5; http://dx.doi.org/10.1038/sj.onc.1205393; PMID: 11973649
- Sharib JM, Cyrus J, Horvai A, Gray Hazard FK, Neuhaus J, Matthay KK, et al. Predictors of acute chemotherapy-associated toxicity in patients with Ewing sarcoma. Pediatr Blood Cancer 2012; 59:611 - 6; http://dx.doi.org/10.1002/pbc.24031; PMID: 22180320
- Hattangadi J, Esty B, Winey B, Duigenan S, Huang M, Yock T. Radiation recall myositis in pediatric Ewing sarcoma. Pediatr Blood Cancer 2012; 59:570 - 2; http://dx.doi.org/10.1002/pbc.23374; PMID: 22021129
- Ewing J. Diffuse endothelium of bone. Proc NY Path Soc 1921; 21:17-24.
- Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA, et al. Ewing’s sarcoma family of tumors: current management. Oncologist 2006; 11:503 - 19; http://dx.doi.org/10.1634/theoncologist.11-5-503; PMID: 16720851
- Paulussen M, Bielack S, Jürgens H, Casali PG, ESMO Guidelines Working Group. Ewing’s sarcoma of the bone: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 2009; 20:Suppl 4 140 - 2; http://dx.doi.org/10.1093/annonc/mdp155; PMID: 19454436
- Kovar H, Pospisilova S, Jug G, Printz D, Gadner H. Response of Ewing tumor cells to forced and activated p53 expression. Oncogene 2003; 22:3193 - 204; http://dx.doi.org/10.1038/sj.onc.1206391; PMID: 12761489
- Veeraraghavan J, Natarajan M, Herman TS, Aravindan N. Curcumin-altered p53-response genes regulate radiosensitivity in p53-mutant Ewing’s sarcoma cells. Anticancer Res 2010; 30:4007 - 15; PMID: 21036715
- Soldatenkov V, Notario V, Dritschilo A. Expression of the human Bcl-2 increases resistance of Ewing’s sarcoma cells to apoptosis and inhibits poly(ADP-ribose) polymerase cleavage induced by radiation. Int J Oncol 1996; 9:547 - 51; PMID: 21541549
- Attawia MA, Borden MD, Herbert KM, Katti DS, Asrari F, Uhrich KE, et al. Regional drug delivery with radiation for the treatment of Ewing’s sarcoma. In vitro development of a taxol release system. J Control Release 2001; 71:193 - 202; http://dx.doi.org/10.1016/S0168-3659(01)00217-6; PMID: 11274751
- Atzpodien J, Gulati SC, Shimazaki C, Bührer C, Oz S, Kwon JH, et al. Ewing’s sarcoma: ex vivo sensitivity towards natural and lymphokine-activated killing. Oncology 1988; 45:437 - 43; http://dx.doi.org/10.1159/000226661; PMID: 3263598
- Chin T, Toy C, Vandeven C, Cairo MS. Lymphokine-activated killer cytotoxicity in neonatal mononuclear cells: in vitro responses to tumor cell lines from pediatric solid tumors. Pediatr Res 1989; 25:156 - 60; http://dx.doi.org/10.1203/00006450-198902000-00016; PMID: 2537488
- Kondo S, Miyatake S, Kikuchi H, Oda Y, Iwasaki K, Ohyama K, et al. Mechanism of interferon gamma-induced protection of human gliosarcoma cells from lymphokine-activated killer lysis: division of lymphokine-activated killer cells into natural killer- and T-like cells. Neurosurgery 1992; 31:534 - 40; http://dx.doi.org/10.1227/00006123-199209000-00016; PMID: 1407434
- Joshi AD, Clark EM, Wang P, Munger CM, Hegde GV, Sanderson S, et al. Immunotherapy of human neuroblastoma using umbilical cord blood-derived effector cells. J Neuroimmune Pharmacol 2007; 2:202 - 12; http://dx.doi.org/10.1007/s11481-006-9038-y; PMID: 18040845
- Foreman NK, Rill DR, Coustan-Smith E, Douglass EC, Brenner MK. Mechanisms of selective killing of neuroblastoma cells by natural killer cells and lymphokine activated killer cells. Potential for residual disease eradication. Br J Cancer 1993; 67:933 - 8; http://dx.doi.org/10.1038/bjc.1993.173; PMID: 8494726
- Belova OB, Vinnichuk UD, Shlakhovenko VA, Berezhnaya NM. Efficacy of different immunotherapy approaches toward treatment of doxorubicin-resistant and doxorubicin-sensitive transplantable rhabdomyosarcoma. Exp Oncol 2007; 29:272 - 6; PMID: 18199982
- Berdeja JG, Hess A, Lucas DM, O’Donnell P, Ambinder RF, Diehl LF, et al. Systemic interleukin-2 and adoptive transfer of lymphokine-activated killer cells improves antibody-dependent cellular cytotoxicity in patients with relapsed B-cell lymphoma treated with rituximab. Clin Cancer Res 2007; 13:2392 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-06-1860; PMID: 17438098
- Bertelli R, Neri F, Tsivian M, Ruhrman N, Cavallari G, Beltempo P, et al. Endolymphatic immunotherapy in inoperable hepatocellular carcinoma. Transplant Proc 2008; 40:1913 - 5; http://dx.doi.org/10.1016/j.transproceed.2008.05.049; PMID: 18675087
- Hirooka Y, Itoh A, Kawashima H, Hara K, Nonogaki K, Kasugai T, et al. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas 2009; 38:e69 - 74; http://dx.doi.org/10.1097/MPA.0b013e318197a9e3; PMID: 19276867
- Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL, et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother 2009; 32:914 - 9; http://dx.doi.org/10.1097/CJI.0b013e3181b2910f; PMID: 19816190
- Correale P, Marra M, Remondo C, Migali C, Misso G, Arcuri FP, et al. Cytotoxic drugs up-regulate epidermal growth factor receptor (EGFR) expression in colon cancer cells and enhance their susceptibility to EGFR-targeted antibody-dependent cell-mediated-cytotoxicity (ADCC). Eur J Cancer 2010; 46:1703 - 11; http://dx.doi.org/10.1016/j.ejca.2010.03.005; PMID: 20399639
- Yin X, Ouyang S, Xu W, Zhang X, Fok KL, Wong HY, et al. YWK-II protein as a novel G(o)-coupled receptor for Müllerian inhibiting substance in cell survival. J Cell Sci 2007; 120:1521 - 8; http://dx.doi.org/10.1242/jcs.001230; PMID: 17452623
- Peters HL, Tuli A, Wang X, Liu C, Pan Z, Ouellette MM, et al. Relevance of amyloid precursor-like protein 2 C-terminal fragments in pancreatic cancer cells. Int J Oncol 2012; 41:1464 - 74; PMID: 22797723
- Tuli A, Sharma M, McIlhaney MM, Talmadge JE, Naslavsky N, Caplan S, et al. Amyloid precursor-like protein 2 increases the endocytosis, instability, and turnover of the H2-K(d) MHC class I molecule. J Immunol 2008; 181:1978 - 87; PMID: 18641335
- Tuli A, Sharma M, Capek HL, Naslavsky N, Caplan S, Solheim JC. Mechanism for amyloid precursor-like protein 2 enhancement of major histocompatibility complex class I molecule degradation. J Biol Chem 2009; 284:34296 - 307; http://dx.doi.org/10.1074/jbc.M109.039727; PMID: 19808674
- Tuli A, Sharma M, Wang X, Simone LC, Capek HL, Cate S, et al. Amyloid precursor-like protein 2 association with HLA class I molecules. Cancer Immunol Immunother 2009; 58:1419 - 31; http://dx.doi.org/10.1007/s00262-009-0657-z; PMID: 19184004
- Peters HL, Tuli A, Sharma M, Naslavsky N, Caplan S, MacDonald RG, et al. Regulation of major histocompatibility complex class I molecule expression on cancer cells by amyloid precursor-like protein 2. Immunol Res 2011; 51:39 - 44; http://dx.doi.org/10.1007/s12026-011-8238-6; PMID: 21826533
- Wu W, Song W, Li S, Ouyang S, Fok KL, Diao R, et al. Regulation of apoptosis by Bat3-enhanced YWK-II/APLP2 protein stability. J Cell Sci 2012; 125:4219 - 29; http://dx.doi.org/10.1242/jcs.086553; PMID: 22641691
- Covell DG, Wallqvist A, Rabow AA, Thanki N. Molecular classification of cancer: unsupervised self-organizing map analysis of gene expression microarray data. Mol Cancer Ther 2003; 2:317 - 32; PMID: 12657727
- Mauri P, Scarpa A, Nascimbeni AC, Benazzi L, Parmagnani E, Mafficini A, et al. Identification of proteins released by pancreatic cancer cells by multidimensional protein identification technology: a strategy for identification of novel cancer markers. FASEB J 2005; 19:1125 - 7; PMID: 15985535
- Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 1975; 45:321 - 34; PMID: 163658
- McLoughlin DM, Miller CC. The FE65 proteins and Alzheimer’s disease. J Neurosci Res 2008; 86:744 - 54; http://dx.doi.org/10.1002/jnr.21532; PMID: 17828772
- Orcholski ME, Zhang Q, Bredesen DE. Signaling via amyloid precursor-like proteins APLP1 and APLP2. J Alzheimers Dis 2011; 23:689 - 99; PMID: 21178287
- Chong AS, Scuderi P, Grimes WJ, Hersh EM. Tumor targets stimulate IL-2 activated killer cells to produce interferon-gamma and tumor necrosis factor. J Immunol 1989; 142:2133 - 9; PMID: 2493506
- Mingari MC, Ferrini S, Pende D, Bottino C, Prigione I, Moretta A, et al. Phenotypic and functional analysis of human CD3+ and CD3- clones with “lymphokine-activated killer” (LAK) activity. Frequent occurrence of CD3+ LAK clones which produce interleukin-2. Int J Cancer 1987; 40:495 - 8; http://dx.doi.org/10.1002/ijc.2910400411; PMID: 3117711
- Verhoeven DH, de Hooge AS, Mooiman EC, Santos SJ, ten Dam MM, Gelderblom H, et al. NK cells recognize and lyse Ewing sarcoma cells through NKG2D and DNAM-1 receptor dependent pathways. Mol Immunol 2008; 45:3917 - 25; http://dx.doi.org/10.1016/j.molimm.2008.06.016; PMID: 18657862
- Cho D, Shook DR, Shimasaki N, Chang YH, Fujisaki H, Campana D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin Cancer Res 2010; 16:3901 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-10-0735; PMID: 20542985
- Handa K, Suzuki R, Matsui H, Shimizu Y, Kumagai K. Natural killer (NK) cells as a responder to interleukin 2 (IL 2). II. IL 2-induced interferon gamma production. J Immunol 1983; 130:988 - 92; PMID: 6294182
- Stante M, Minopoli G, Passaro F, Raia M, Vecchio LD, Russo T. Fe65 is required for Tip60-directed histone H4 acetylation at DNA strand breaks. Proc Natl Acad Sci U S A 2009; 106:5093 - 8; http://dx.doi.org/10.1073/pnas.0810869106; PMID: 19282473
- Hallett WH, Murphy WJ. Natural killer cells: biology and clinical use in cancer therapy. Cell Mol Immunol 2004; 1:12 - 21; PMID: 16212916
- Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol 2008; 9:495 - 502; http://dx.doi.org/10.1038/ni1581; PMID: 18425106
- Shresta S, MacIvor DM, Heusel JW, Russell JH, Ley TJ. Natural killer and lymphokine-activated killer cells require granzyme B for the rapid induction of apoptosis in susceptible target cells. Proc Natl Acad Sci U S A 1995; 92:5679 - 83; http://dx.doi.org/10.1073/pnas.92.12.5679; PMID: 7777569
- Shresta S, Pham CT, Thomas DA, Graubert TA, Ley TJ. How do cytotoxic lymphocytes kill their targets?. Curr Opin Immunol 1998; 10:581 - 7; http://dx.doi.org/10.1016/S0952-7915(98)80227-6; PMID: 9794837
- Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 2002; 20:323 - 70; http://dx.doi.org/10.1146/annurev.immunol.20.100201.131730; PMID: 11861606
- Grossman D, Kim PJ, Schechner JS, Altieri DC. Inhibition of melanoma tumor growth in vivo by survivin targeting. Proc Natl Acad Sci U S A 2001; 98:635 - 40; http://dx.doi.org/10.1073/pnas.98.2.635; PMID: 11149963
- Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene 2005; 24:3285 - 96; http://dx.doi.org/10.1038/sj.onc.1208492; PMID: 15735702