Abstract
Focal adhesion kinase (FAK) increasingly has been implicated in cancer growth and progression. 1,2,4,5-Benzenetetraamine tetrahydrochloride (Y15) is a small molecule FAK inhibitor that blocks the Y397 autophosphorylation site. FAK inhibitor, Y15 decreased Y397 FAK in different colon cancer cells lines in a dose-dependent manner. In addition, Y15 decreased phosphorylated Src in SW480 and SW620 cells. Y15 decreased cell viability, increased detachment, and increased apoptosis in SW480 and SW620 cells in vitro. Combination of FAK inhibitor Y15 and Src inhibitor PP2 decreased colon cancer cell viability more effectively than each agent alone. In addition, when combined with 5-FU, oxaliplatin or 5-FU and oxaliplatin, colon cancer viability was decreased further, demonstrating that dual and triple therapy synergistically inhibits cell viability. In vivo, Y15 decreased subcutaneous SW620 tumor growth by 28%. Combination of oral Y15 with 5-FU/or oxaliplatin decreased tumor growth by 48% more effectively than each inhibitor alone. Finally, tumors treated with Y15 expressed less Y397 phosphorylation, Src phosphorylation and had greater apoptosis than controls. Thus, the small molecule FAK inhibitor, Y15, inhibits cell growth in vitro and in vivo and enhances the efficacy of chemotherapy, demonstrating that it can be an effective therapeutic inhibitor for treating colon cancer.
Introduction
Colorectal cancer the third leading cause of cancer death, with over 140 000 new cases and 50 000 deaths expected in 2011.Citation1 Although early stage colorectal cancer can be cured, as many as 20% of patients will have metastatic disease at the time of presentation and then will develop systemic recurrence. Although cytotoxic chemotherapy has traditionally been the mainstay of treatment for systemic disease, these agents are rarely curative. Moreover, toxicity can be dose-limiting. As such, it is imperative to develop new agents to prevent growth and progression of colorectal cancer.
Focal adhesion kinase (FAK) is a nonreceptor tyrosine kinase that was first identified at the sites of adhesion between cells and the extracellular matrix. Compared with normal tissue, both primary and metastatic tumors express high levels of FAK.Citation2 For example, we now know that FAK is upregulated in ovarian, thyroid, head and neck, lung, kidney, brain, hepatocellular, pancreatic, breast, and prostate cancers.Citation3-Citation5 In particular, FAK is overexpressed in metastatic colon cancer, and Y397 phosphorylated FAK is also increased in colorectal tumor cells.Citation2 This is not surprising given that FAK is involved in several processes related to the potential for tumor progression. FAK signaling plays a role in angiogenesis, cell proliferation and survival, motility, and invasion.Citation6 Given its apparent role in tumorigenesis and overexpression in many types of cancers, FAK inhibition is an appropriate approach to targeted cancer treatment.
The mechanism of FAK activation involves a complex set of signaling pathways that begin with tyrosine phosphorylation at the Y397 site. Subsequent increased affinity of the SH2 domain of Src stimulates both Src binding and phosphorylation, as well as phosphorylation of FAK at other tyrosine residues. This can be seen as an important step in tumorigenic FAK signaling because other tyrosine kinases that possess the SH2 domain (e.g., PI3K and Grb2 as well as paxillin and p130) are then actively phosphorylated, stimulating cellular processes like angiogenesis, survival, and metastasis.Citation7,Citation8 Y397 phosphorylation is hence the key to the induction of widespread FAK-induced intracellular signaling that can promote tumorigenesis.Citation9 Moreover, the interaction between FAK and Src can be seen as a key step in the initiation of a potentially malignant survival cascade.
The association between FAK and Src, specifically, has been studied in the past suggesting their role in tumor cell survival. For example, in breast cancer cells, overexpression of FAK stimulates the propogation of signal cascades that hinder apoptosis. Suppression of FAK expression by adenoviral FAK-CD in these cells resulted in apoptosis and decreased adhesion.Citation10 Overexpression of Src repressed the pro-apoptotic effects of FAK inhibition, thus propagating tumor progression.Citation2,Citation8 This suggests that despite appropriate FAK inhibition, cells may still escape death due to constitutive Src signaling and complete eradication of this interaction may enhance our targeted anti-cancer efforts. The colon cancer cell line HT29, which is resistant to the effects of the apoptotic agent staurosporine demonstrated increased apoptosis with the addition of dual FAK-Src inhibition with adenoviral FAK-CD and PP2.Citation8 In this report we present data demonstrating that allosteric FAK inhibition, via blockage of the Y397 site of phosphorylation, does inhibit Src activation and that chemical Src inhibition with PP2 blocks FAK autophosphorylation.
With accumulating data showing that FAK activation plays a central role in carcinogenesis, inhibition of FAK activation has emerged as a potential strategy for preventing cancer growth and progression. Initial efforts to inhibit FAK included downregulation of FAK expression via transfection of cells with dominant negative C-terminal FAK-CD and transfection with FAK-silenced RNA (FAK siRNA).Citation11 More recently, a variety of pharmacological small molecules have been developed that target FAK kinase activity: PF-562,271 and PF-573–228 (Pfizer), which block ATP binding as well as Pyk2 (protein-rich kinase 2).Citation12,Citation13 Similarly, TAE226 (Novartis) blocks ATP binding but also inhibits the insulin-like growth factor-1 receptor (IGF-1R).Citation14,Citation15 PND-1186 is a novel FAK inhibitor which is proposed to block Y397 FAK phosphorylation but similar to the kinase inhibitors, is non-specific and affects several other kinases.Citation16 These molecules inhibit Y397 phosphorylation, but their lack of specificity opens the door to potentially untoward and unintended downstream effects. Solely and selectively targeting the Y397 site is a more specific and less toxic approach to FAK inhibition. The allosteric inhibitor: 1,2,4,5-Benzenetetraamine tetrahydrochloride (Y15) is a small molecule FAK inhibitor that specifically binds the Y397 site of the FAK molecule.Citation9 Unlike earlier small molecule inhibitors, Y15 blocks FAK Y397 autophosphorylation but does not affect ATP binding or IGF-1R kinase activity.Citation6 This molecule decreases Y397 phosphorylation in breast cancer cells and has also been shown to increase detachment, inhibit adhesion, and decrease breast cancer cell growth in vitro and in vivo.Citation9 Similarly, Y15 increases cell detachment and apoptosis and decreases tumorigenesis in pancreatic cells and neuroblastoma cells, which overexpress FAK.Citation17,Citation18
These observations, along with the observation that FAK is overexpressed in colon cancer tissue, led us to hypothesize that inhibition of FAK activation with Y15 would effectively inhibit colon cancer cell viability and tumor growth in vivo. We focused our work on two colorectal cancer cells lines: SW620 cells, which are highly aggressive cells and derived from lymph node metastasis and SW480 cells, which are derived from the primary tumor of the same patient. We looked at the effects of Y15 on colon tumor viability, clonogenicity, and apoptosis in vitro and examined its effects on tumor growth in vivo. In addition, given the proposed link between FAK and Src in tumor progression, we investigated the effect of FAK inhibition with Y15 on Src activation to further delineate the mechanism by which Y15 influences tumor growth. Moreover, we analyzed the effects of Src inhibition on FAK phosphorylation and tested the efficacy of dual FAK/Src inhibition on cell viability using the Src inhibitor PP2. The data show that allosteric inhibition of FAK with Y15 prevents Src activation and can effectively inhibit colon cancer viability and growth. In addition, Y15 blocks colon cancer growth in vitro and in vivo and combination with chemotherapy 5-fluouracil or oxaliplatin synergistically decrease tumor growth.
Results
Colon cancer cells expressed variable but high levels of FAK and phosphorylated Y397 FAK
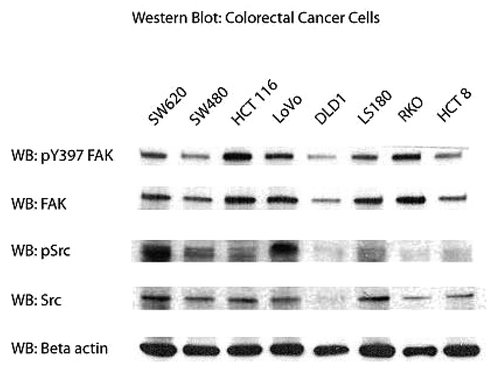
Before examining the efficacy of FAK inhibition on cell viability, we first looked at the expression of total FAK and phosphorylated FAK (pY397 FAK) in a panel of different colon cancer cell lines. The SW480, HCT 116, DLD1, LS-180, HCT 8, and RKO cells were derived from primary colon adenocarcinoma; and SW620 and LoVo cells were derived from lymph node metastases. shows expression of Y397-FAK and pY418-Src and total FAK and Src in all cell lines. Interestingly, there were variable levels of total FAK and activated FAK in these cells. For example, SW480, DLD1, and HCT 8 cells demonstrated visibly less FAK and pY397 FAK than the other cells. And of these cells, HCT116, LoVo, and RKO expressed the highest levels of FAK and pY397 FAK. Src and phosphorylated Src, were differentially expressed as well. The SW620 and LoVo cells, which both derived from lymph node metastases, demonstrated high levels of phosphorylated Src, while the primary tumor cell lines had significantly less of the activated form of activated Src protein. Thus, all colon cancer cell lines express variable but high levels of activated FAK and Src proteins.
Figure 1. Expression of phosphorylated FAK, Src, and total FAk and Src in different colon cancer cell lines. Western blot shows the expression of FAK and Src proteins and their phosphorylated counterparts (pY397 FAK, pY418-Src); β actin was used as a control for equal loading.
Y15 inhibited the viability of several different colon cancer cell lines in a dose-dependent manner
Once we had demonstrated that several different colon cancer cell lines express FAK and phosphorylated FAK we tested the effect of FAK autophosphorylation inhibitor, Y15 on cell viability in a series of MTT assays. Y15 decreased viability of all different colon cancer cells in a dose-dependent manner (). SW620, SW480 and DLD1 cells were most sensitive to Y15, with their viability following treatment with just 1 µM of Y15 being significantly less than in untreated cells (P < 0.05). HCT116 and LS180 cell viability was significantly less than untreated cells following treatment with 2 µM of Y15 (P < 0.05). LoVo cells were less sensitive to FAK inhibition with Y15 (). Thus, all colon cancer cell lines with varying degree responded to Y15 in a dose-dependent manner.
Figure 2. (A) Y15 affected viability in a dose-dependent manner in colon cancer cells. MTT assay of colon cancer cells treated with Y15. 5 × 103 cells were plated onto a 96-well plate, allowed to incubate overnight and then treated with Y15 (1 µM, 2 µM, 4 µM, 8 µM, and 10 µM). Twenty four hours after treatment with Y15, cells were exposed to 20 µl of Cell Titer 96Aqueous One Solution Cell Proliferation Assay (MTT; Promega Corporation) for 1 h. Y15 significantly inhibited viability of all colon cancer cells, except of Lovo at a dose of 1−2 µM (P < 0.05) and continued to inhibit viability further at 10 µM dose. (B) Y15 decreased Y397-FAK in colon cancer cells in a dose-dependent manner. The colon cancer cells were treated with the same doses of Y15 as above and western blot was performed with Y397-FAK and FAK antibodies. Y15 decreases Y397-FAK in colon cancer cells in a dose-dependent manner.
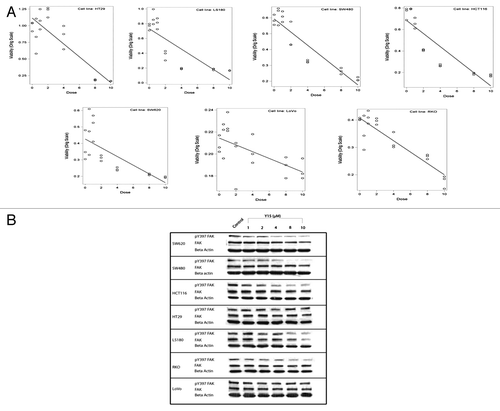
Y15 decreased Y397-FAK in colon cancer cells in a dose-dependent manner
To test the effect of FAK inhibitor on FAK autophosphorylation, we performed western blotting with Y397-FAK and FAK antibodies on the same colon cancer cells, which were used in MTT assay (). Y15 decreased Y397-FAK starting 1–2 µM in many cell lines and decreased more with increasing dose of Y15. Y15 decreased Y397-FAK in most colon cancer cell lines. Lovo cell line that was less sensitive to Y15 had less dramatic decrease of y397-FAK (). Thus, Y15 decreased Y397-FAK in most colon cancer cells in a dose-dependent manner,
Y15 decreased clonogenicity and increased detachment and apoptosis in a dose-dependent manner in SW620 and SW480 colon cancer cell lines
After testing Y15 in vitro on a broad panel of colon cancer cell lines, we focused our work on the related SW480 and SW620 cells to test the effect of Y15 on clonogenicity, detachment and/or apoptosis in vitro. shows that Y15 induced cell detachment in SW480 and SW620 cells in a dose- dependent manner, supporting the effect of Y15 on cell viability. Compared with untreated cells, a significantly higher number of detached cells were seen following treatment with 10 µM of Y15 compared with control (P < 0.05) and at 50 µM, detachment reached 100% in SW480 and 96% in SW620 cells (, P < 0.05). Thus, Y15 increased detachment in colon cancer cells in a dose-dependent manner.
Figure 3. (A) Y15 increased detachment in SW480 and SW620 cells in a dose-dependent manner. Y15 increased cell detachment in a dose-dependent manner, with 96% of SW620 and 100% of SW480 cells detached at 50 µM of Y15 treatment. (B) Y15 also inhibited clonogenicity in SW480 and SW620 cell lines in a dose-dependent manner. 5 × 102 cells were seeded onto a 6-well plate and were then treated with different doses of Y15 for 14 d. There was a dose-dependent decrease in clonogenicity in both cell lines. At 1 µM dose Y15 significantly inhibited clonogenicity (P < 0.05), and this effect became more significant as the dose was increased to 10 µM. (C and D) Y15 induced apoptosis in SW620 (C) and SW480 (D) cells. Cells were treated with different doses of Y15 for 24, 48 or 72 h. TAE226 (10 µM), a FAK inhibitor known to induce apoptosis was used as a control. Cells were examined at 100× magnification using the Zeiss Observer.A1 microscope after being fixed and stained with Hoechst. Y15 induced apoptosis in a dose and time-dependent manner. (E) Two representative photomicrographs show nuclei of untreated SW620 cells and cells treated with just 2 µM of Y15. Fragmented apoptotic nuclei are shown by white arrows.
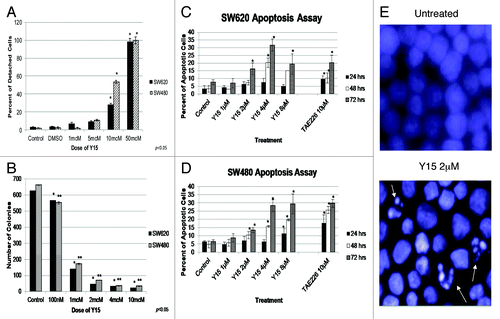
In a similar manner, Y15 decreased colony formation in both cell lines (). Colony formation in cells treated with just 100 nM of Y15 was significantly less than control (P < 0.05), and this effect became more pronounced as the dose from 2 µM to 10 µM (P < 0.05; ). Y15 decreased clonogenicity in a dose-dependent manner in colon cancer cells.
Finally, we studied the levels of apoptosis in cells treated with Y15. We observed a dose dependent increase in apoptosis in SW620 and SW480 cells by Y15. The TAE226 inhibitor (Novartis) was used as control. show that Y15 increased apoptosis in dose- and time-dependent manner in SW620 cells and in SW480 cells, respectively. The image of fragmented apoptotic nuclei is shown in SW620 cells treated with 2 µM of Y15 (). Therefore, Y15 effectively and significantly induces apoptosis in dose- and time-dependent manner in SW620 and SW480 cells.
Y15 inhibited tumor growth in vivo
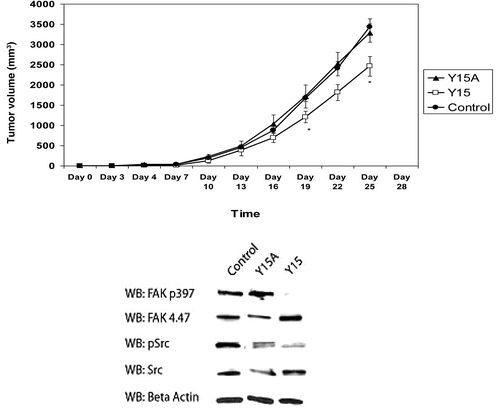
Next, we sought to determine whether Y15 will inhibit tumor growth in vivo in nude mouse xenograft model. We used SW620 colon cancer cells because SW480 does not grow in a suitable fashion in this model.Citation19,Citation20 We treated mice with either Y15, or with a chemical derivative of Y15 (Y15A), which has shown minimal activity in inhibiting viability in this cell line and thus served as a negative control (data not shown) or with 1× PBS (control). On the day of tumor inoculation, mice were started on a daily intraperitoneal dosing regimen of Y15, Y15A or 1× PBS. After 19 d of treatment, the volume of tumors in mice treated with Y15 was significantly less than that of mice treated with Y15A or PBS (P < 0.05; , upper panel). We detected decreased Y397-FAK in the tumors treated with Y15 in contrast to tumors treated with Y15A that did not decrease Y397-FAK (, lower panel). Thus, Y15 effectively decreased colon tumor growth and decreased Y397-FAK in mice xenografts in vivo.
Figure 4. Y15 inhibited in vivo tumorigenesis and pY397 FAK expression. Upper panel: Mice were inoculated with 2 × 106 SW620 cells and treated 5 d per week with Y15, Y15A (Y15 derivative), or PBS. Tumor volumes were calculated every 3 d. After 19 d of treatment, Y15 significantly decreased tumor volume compared with 1× PBS or Y15A. Lower panel: Following sacrifice, protein lysates from select tumor samples were examined by western blot. pY397 FAK and pSrc expression were decreased in tumors treated with Y15.
Y15 decreased activated FAK and Src in a dose-dependent manner
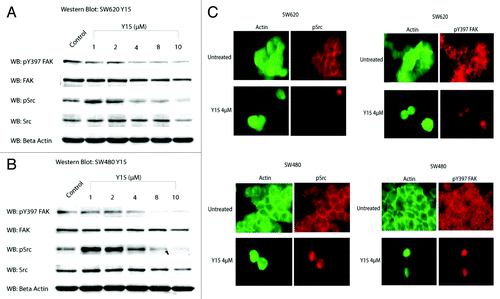
Because Y15 is an allosteric FAK inhibitor, that targets and blocks the Y397 site of autophosphorylation, which is also site of Src binding at the SH2 domain, we hypothesized that it would not only inhibit phosphorylated Y397 FAK, but also Src phosphorylation. We analyzed the effect of Y15 on dose-dependent cellular expression of FAK and pY397 FAK by treating our cells with increasing doses of Y15 for 24 h (). show decreased FAK and pY397 FAK in both SW620 and SW480 cells following treatment with Y15, respectively. Y15 also decreased phosphorylated (pSrc) and total Src in SW480 and SW620 cell lines (). Immunostaining for pY397 FAK and p-Src was done to confirm the results of western blot. In both SW480 and SW620 cells, treatment with Y15 caused a visible decrease in cytoplasmic pY397 FAK and p-Src and displacement from focal adhesions ().
Figure 5. Y15 decreases pY397 FAK and pY418 Src expression in a dose-dependent manner. (A) Upper panel: SW620 cells were treated with different doses of Y15. pY397 FAK expression was markedly decreased with increasing dose of Y15, but total FAK expression was less affected. Similarly, Y15 decreased phosphorylated Src (pY418 Src), p-Src expression and total Src expression was less affected. Y15A had no effect of protein expression. (B) SW480 cells expressed less pY397 FAK and pSrc with increasing dose of Y15, while total FAK and Src were less affected by Y15. The Y397-FAK, FAK, and β-actin lanes are from image. (C) Immunohistochemistry shows decreased pY397 FAK (red) and p-Src (red) in SW480 and SW620 cells treated with 4 µM Y15. Following treatment, FAK expression is decreased and cell morphology changed to more rounded. FAK was displaced from the cytoplasm and focal adhesions. Green shows actin staining with Phalloidin-FITC.
Y15 enhanced the effect of the selective Src inhibitor, PP2
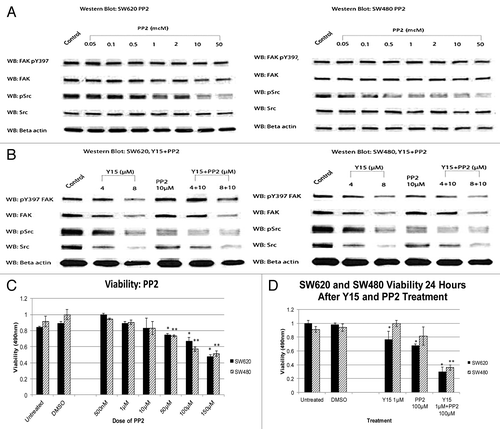
Since Y15 decreased both FAK and Src phosphorylation, we next sought to determine if Src inhibition would conversely decrease FAK activation, and if the addition of Y15 enhances the effects of a selected Src inhibitor. We treated cells with different doses of PP2 (Src inhibitor) and performed western blots on SW620 and SW480 cells with pY397 FAK, phosphorylated Src, and total FAK and Src antibodies (). As would be expected, PP2 specifically decreased p-Src dose-dependently, with a minimal effect of total Src, as well as total and phosphorylated FAK (). shows western blot expression of FAK and Src in SW480 and SW620 cells following treatment with Y15, PP2 or a combination of the two inhibitors. As would be expected, Y15 inhibited both FAK and Src phosphorylation at a dose of 8 µM and while P22 inhibited Src phosphorylation, it did not affect FAK phosphorylation. Combination of Y15 and PP2 did not decrease pY397 FAK compared with cells treated with Y15 alone, but it significantly decreased pSrc compared with cells treated with Y15 or PP2 alone (). This shows that Y15, which blocked FAK autophosphorylation, also blocked Src activation and enhanced the effect of the Src inhibitor PP2.
Figure 6. (A) Combination of Y15 and Src inhibitor PP2 decreased phosphorylation of FAK and Src and cancer cell viability more effectively than each inhibitor alone. SW620 (left panel) and SW480 (right panel) cells treated with Src inhibitor PP2 demonstrated dose-dependent decrease of pSrc compared with untreated cells, while FAK or Y397-FAK were less affected by PP2. (B) Combination of Y15 and PP2 decreased Y397-FAK and p-Src more effectively than each inhibitor alone in SW620 cells. Left panel: SW620 cells. Western blotting was performed on Y15, PP2, and Y15+PP2-treated cells with Y397FAK, p-Src, FAK, and Src antibodies. Beta-actin was used as a control. Right panel: The same effect was observed in SW480 cells (right panel). (C) PP2 decreased viability of SW620 and SW480 cells in a dose-dependent manner. (D) Combination of Y15 and PP2 decreased more significantly viability of SW620 and SW40 cells than each inhibitor alone. *,**P < 0.05 vs. Y15, PP2, and untreated cells.
Combination of Y15 and PP2 synergistically decreased cancer cell viability
To test combination of Y15 and PP2 on cancer cell viability, we first performed MTT assays on SW480 and SW620 cells treated with PP2 as a single agent (). PP2 decreased viability in a dose-dependent manner, with viability significantly less than untreated cells at a dose of 50−150 µM (P < 0.05). When we added FAK inhibitor, we saw a more significant decrease in viability at 1 µM Y15 and 100 µM PP2 (P < 0.05) (). Thus, dual FAK/Src inhibition with Y15 and PP2 synergistically inhibited cancer cell viability.
Y15 enhanced the efficacy of chemotherapy in vitro
The next step in testing Y15’s clinical utility by combining it with chemotherapy to test for a synergistic anti-tumor effect. That is, since colon cancer is often treated with combination cytotoxic therapy (e.g., 5-fluorouracil [5-FU], oxaliplatin, leukovorin), we looked at the potential for Y15 to enhance the effect of some of these agents on colon cancer cell viability and growth.
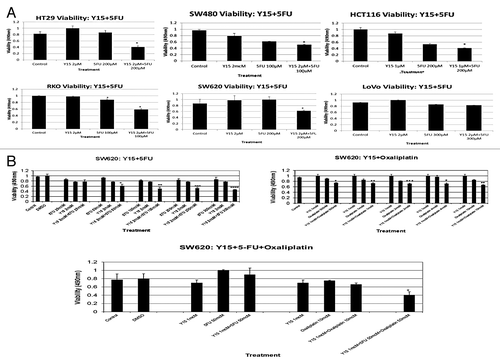
shows that the addition of Y15 to 5-FU significantly decreased the viability of most of the colon cancer cell lines: SW620, SW480, HCT 116, LoVo, RKO, and HT29 cells (P < 0.05) (). The metastatic cell line SW620, expressed enhanced in vitro inhibition of viability when Y15 was added to 5-FU and oxaliplatin (P < 0.05; , left upper panel). Adding 2 µM of Y15 to 50 µM of 5-FU significantly decreased cell viability, and this response became more pronounced at the dose of 5-FU was increased to 300 µM. Similar synergy was seen when Y15 was combined with oxaliplatin (, right upper panel). Adding 1 µM of Y15 to 500nM of oxaliplatin significantly decreased viability and again, increasing the dose of oxaliplatin to 100 µM enhanced this effect. The triple therapy inhibited SW620 viability better than single treatment with Y15 or chemotherapy, or dual treatment with Y15 and chemotherapy (, lower panel). Thus, combining just 1 µM of Y15 to 50 µM of 5-FU and 10 µM of oxaliplatin significantly decreased cell viability (P < 0.05).
Figure 7. (A) Combination of Y15 and 5-FU decreases viability in most colon cancer cells more significantly than each agent alone. Different colon cancer cells were treated with Y15, 5-FU or combination of Y15 and 5-FU for 24 h and MTT assay was performed. The viability of most colon cancer cells, except of Lovo was significantly less than Y15 and 5-FU-treated cells alone. *, P < 0.05, Y15 + 5FU vs. Y15 and 5-FU or 5-FU vs. untreated. (B) Combination of Y15 with 5-FU or with oxaliplatin or all three drugs together is more significant than single or dual dose-treatment, respectively in decreasing colon cancer cell viability in vitro. SW620 cells were treated with 2 µM Y15 and different doses of 5-FU (left upper panel) or Y15 1 µM and different doses of oxaliplatin (right upper panel) or with all three inhibitors together (lower middle panel) for 24 h and MTT assay was performed. *P, **P < 0.05 vs. untreated and single or dual dose, respectively.
Y15 enhanced the efficacy of chemotherapy on in vivo tumor growth
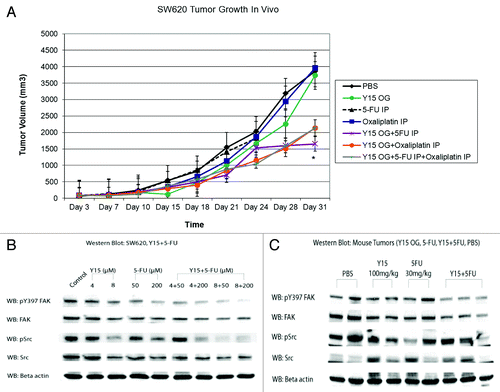
Based on our data in vitro, we repeated our in vivo study using the nude mouse xenograft model to test the efficacy of triple therapy on in vivo tumor growth. We used SW620 colon cancer cells, but in this experiment, we treated established tumors (). On day 3 after tumor inoculation, when all mice had visible established tumors, we randomized them to groups of equivalent tumor volume and started treatment with daily Y15 (100 mg/kg oral gavage). Oxaliplatin (1 mg/kg) was given by intraperitoneal injection twice a week and 5-FU (30 mg/kg) was administered once a week by intraperitoneal injection. After 28 d of treatment, tumor volume in mice treated with combination Y15+5FU, Y15+oxaliplatin or Y15+5FU and oxaliplatin was significantly less than that of mice treated with either agent alone or 1× PBS (P < 0.05; ). This significant difference, 48% decrease in tumor volume persisted until day 31, at which point the trial was terminated due to tumor burden in the control and single treatment groups (). Interestingly, triple therapy with Y15+5FU+oxaliplatin was not significantly better than dual treatment with Y15+5FU or Y15+oxaliplatin (). This suggests that adding Y15 to chemotherapy in vivo may ameliorate the need for multiple cytotoxic agents.
Figure 8. (A) Combination of Y15 and 5FU, Y15 and oxaliplatin, and Y15 plus 5-FU plus oxaliplatin more significantly decreases tumor growth in vivo than each agent alone. The tumor growth experiment was performed on established with oral Y15 and 5-FU and oxaliplatin, as described in Materials and Methods. Combination of Y15 and 5-FU or Y15 plus oxaliplatin decreases colon cancer tumor growth in vivo more significantly then each agent alone and combination of all three agents together is the same as dual inhibition and is more significant than single dose of each inhibitor/chemotherapy drug. *P < 0.05, the Student t test. (B) Combination of Y15 and 5-FU decreases Y307-FAK and p-Src more significantly than each agent alone after treatment of SW620 cells with Y15, 5-FU, and Y15 plus 5-FU in vitro. Western blotting with Y397-FAK and Y418-Src and total FAK and Src antibodies was performed after treatment of SW620 cells with Y15, 5-FU, or Y15+5FU. β-actin was used as a control. Combination treatment of Y15 plus 5-FU decreases more significant Y397-FAK and p-Src than each agent alone. (C) Combination of Y15 and 5-FU decreases Y397-FAK and p-Src more significantly than each agent alone in tumor xenografts after treatment with these agents alone or in combination. The tumors from (A) were collected and analyzed by western blot with pY397-FAK and p-Src and total FAK and p-Src antibodies. β-actin was used as a control. The combination treatment more significantly decreased Y397-FAK, p-Src than each agent alone in xenograft tumors.
Y15 with chemotherapy decreased Y397-FAK and p-Src more significantly than each agent alone
To study the mechanism for the synergistic effect of Y15 and chemotherapy, we performed western blots on SW620 cells treated for 24 h with Y15 (4 µM or 8 µM), 5-FU (50 µM or 200 µM), or a combination of Y15+5FU with the same doses. shows that when FAK inhibition is combined with chemotherapy, there is a more significant decrease of pY397 FAK and pSrc than with each treatment alone. Specifically, 8 µM of Y15 + 200 µM of 5FU nearly eliminated the activated form of FAK (pY397 FAK) and 8 µM of Y15 + 50 µM of 5FU nearly eliminated activated Src (pSrc) (Fugure 8B). We also analyzed pY397 FAK and pSrc in proteins isolated from the tumors harvested from the mice xenografts (). The tumors treated with Y15+5-FU expressed less pY397 FAK and pSrc similarly to in vitro data (). This shows that Y15 and 5-FU combination treatment inhibits FAK and Src activation to a greater degree and corresponds to the synergistic inhibition on tumor growth observed in vivo.
Discussion
FAK is a tyrosine kinase that is involved in normal and malignant cell processes.Citation21 In colorectal cancer, signaling initiated by FAK and Src are central to tumor growth, proliferation, and metastasis and unilateral FAK inhibition with FAK-CD in previous studies has demonstrated that the reciprocal nature of these two molecules can limit the therapeutic efficacy of targeting just one facet of the cancer cell. The highly malignant metastatic SW620 colon cancer cell line is a good way to examine the efficacy of targeted therapeutics on a disease that can be difficult to treat with traditional chemotherapeutic regimens. In the present study, the effect of Y15 on FAK and Src activation was evident by decreased phosphorylated pY397 FAK and pY418Src expression by western blot and immunostaining while total FAK and Src expression were relatively unchanged following treatment with Y15 in vitro and in vivo. Essentially, selective pY397 inhibition did not reduce the overall presence of Src and FAK in the cell, but it did block their activation and subsequent downstream signaling effect. Indeed, compared with breast cancer cells, colon cancer cell lines express high levels of both FAK and Src and are more resistant to inhibition of FAK expression at the genetic level. However, inhibiting FAK at its Y397 phosphorylation site blocked both FAK and Src phosphorylation and effectively induced apoptosis and inhibited viability and in vivo tumor growth. In addition, inhibiting both FAK and Src with dual targeted therapy enhanced this antitumor effect. Furthermore, inhibition of FAK and chemotherapy 5-FU synergized to decrease colon cancer viability and tumor growth in vivo.
The data presented herein indicate that Y15 decreases cell viability by inducing apoptosis. Moreover dual inhibition of FAK and Src decreased more viability of colon cancer cells than each agent alone. The data are consistent with data with data on neuroblastoma, where dual FAK-Src inhibition induced apoptosis as evidenced by decreased PARP and caspase-9 expression, with the amount of phosphorylated Src directly correlated with induced apotosis.Citation22 Immunohistochemistry has demonstrated increased FAK expression in invasive colon tumorsCitation4 and colorectal cancers overexpressed both FAK and Src.Citation8 Colon cancer cells have been more resistant to apoptosis by sole FAK inhibition than breast cancer cells which express relatively lower levels of Src.Citation8 However, because dual FAK-Src inhibition (adenoviral FAK-CD and PP2) increased cell detachment and apoptosis in resistant colon cancer cell lines, the increase in apoptosis seen in the present study is explained by the ability of Y15 to inhibit the phosphorylation of both molecules.Citation8
The central role of FAK in cell signaling promotes cell survival, migration and motilityCitation23 and FAK and Src have been implicated in blunting the apoptotic response of normal cells promoting transformation and tumor growth. FAK and Src activation are theorized to occur as a result of either integrin clustering or stimulation of tyrosine kinase receptors like PDF-R, EGF-R, FGF-R, HGF-R CSF-1R, or SCF-R.Citation7 Previous work has shown that Y397 FAK phosphorylation increases the binding affinity of the SH2 domain of Src and thus induces downstream phosphorylation cascades.Citation7,Citation8 As demonstrated in the current study, when we block the Y397 phosphorylation site of FAK, Src phosphorylation is compromised, subsequent downstream phsophorylations are reduced, and tumor cell viability and growth are inhibited. The corollary, however, is not true; selective Src inhibition with PP2 does not seem to block Y397 FAK phosphorylation. The addition of FAK inhibition to selective Src inhibition however, enhances the antitumor effect of PP2. This suggests that allosteric FAK inhibition with Y15 blocks the Src activating activity of the molecule, but blocking Src activation does not inhibit FAK activation. These data illustrate the intimate relationship between FAK and Src and suggests that dual FAK and Src inhibition with Y15 inhibitor is a promising therapeutic modality.
Several Src inhibitors have been studied and applied to the clinical arena. PP1 and PP2 were the first anti-Src drugs synthesized, and PP2 was shown to be highly selective for Src-family kinases. Most notably, when one of the pioneering targeted therapy agents imatinib, was found to have resistance in certain Bcr-Abl leukemias, treatment with dasatinib (a Src-Abl inhibitor) induced a hematologic and cytogenic response.Citation24 Dasatinib also inhibits Src family kinases Fyn and Lck, c-kit, PDF-R, and ephrins.Citation7 Bosutinib is another Src-Abl inhibitor that is currently in phase II trials for imatinib-resistant CML.Citation7 Similarly, several FAK inhibitors have been studied and some have progressed to clinical trials. GSK2256098 inhibits FAK activation by blocking ATP binding.Citation25 PF-562,271 and PF-573,228 (Pfizer) are interesting in that they block Src-activating Pyk2 and FAK kinase activity. However, while this dual inhibition is promising, the non-selective approach of preventing ATP-binding raises concern for potential toxicity.Citation25 Y15 is unique and attractive in this setting because it selectively binds FAK at the Y397 autophosphorylation site and does not affect ATP binding.Citation9 While selectively binding this site of the FAK molecule also affects Src activation, it seems that dual inhibition is not a result of ubiquitous molecular inhibition, but providential alteration of the Src-activating site on the FAK molecule. This is supported by earlier findings that FAK inhibition at the genetic level did not halt the anti-apoptotic activities of Src.Citation8 This interaction however, does not swing both ways since we have also shown that selective Src inhibition does not prevent FAK activation. These data show that FAK activation of Src and the resultant activation of other tyrosine residues on FAK by Src are both essential to propogation of the tumorigenic signal cascade. While tumorigenesis is indeed a complex process, perhaps dual or multiple, selective agents are the optimal therapeutic approach.
In addition, we demonstrate that dual inhibition with Y15 and chemotherapy (5-FU and oxaliplatin) decreased more colon cancer viability than each agent alone. While the triple therapy had the same effect as dual therapy indicating that dual inhibition is enough to maximally suppress colon cancer growth. It is important that dual inhibition of Y15 and 5-FU decreased Y397FAK and p-Src in cells and tumors that explains the mechanism of decreased tumor growth through inhibition of these cross-linked survival signaling.
The unique and appealing feature of Y15 is that by exclusively binding the Y397 site of FAK, it selectively inhibits FAK activation and thus, may have less toxic effects than agents that are less specific. Moreover, in selectively binding the Y397 phosphorylation site, it prevents the propagation of signaling by prohibiting down-stream Src activation. When both molecules are targeted, we see there is a unique relationship between FAK and Src, and while we have demonstrated the anticancer activity of FAK inhibition with Y15 in a malignant phenotype, the complex nature of malignant disease suggests a multi-targeted approach is a promising modality in eradicating the many protective mechanisms of cancer cells.
Materials and Methods
Cell lines and culture conditions
The colon cancer cell lines: SW620 and SW480 were used and described in reference Citation26. Cells were purchased from ATCC and maintained according to protocol in Dulbecco’s modified Eagle medium (D-MEM) plus 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (37 °C, in 5% CO2). Other colon cancer cell lines were: DLD1, Lovo, HT-8, HT-29 (maintained in RPMI plus 10% FBS and 1% penicillin/streptomycin); LS180, RKO (maintained in Eagles Minimum Essential Medium plus %10 FBS and 1% penicillin/streptomycin); HCT116, maintained in McCoy’s 5a Medium Modified plus 10FBS and 1% pencicillin/streptomycin).
Antibodies
Monoclonal anti-FAK (clone 4.47) antibody (Millipore) and polyclonal anti-phospho-Tyr397 FAK antibody (Stressgen) were used for detection of total and activated FAK. In addition, anti-v-Src monoclonal antibody (Calbiochem), anti-phospho-pY418-Src polyclonal antibody (Invitrogen), were used to detect total and phosphorylated Src, respectively. Monoclonal anti-β-actin antibody was used as control (Sigma). Secondary mouse (from sheep) and rabbit (from donkey) horseradish peroxidase linked IgG (GE Healthcare UK Unlimited) were used as secondary antibodies as indicated.
Small molecule inhibitors
Y15 is an allosteric FAK inhibitor and was ordered from Sigma. The four chemical derivatives of Y15A (1,4-benzenediamine dihydrochloride) was purchased from Sigma and obtained from Dr Ravindra Pandey (Roswell Park Cancer Institute). Each compound was solubilized in 1× PBS at a concentration of 25 mM and stored at −20 °C.
Western blot analysis
Cells were washed with 1× PBS then incubated in lysis buffer [50 mM TRIS-HCl (pH 7.5), 150 mM sodium chloride, 1% Triton X-100. 0.5% sodium deoxycholate, 0.1% SDS, 10% glycerol] and protease inhibitors (10 µg/mL leupeptin, 10 µg/mL PMSF, 1 µg/mL aprotinin, 1 mM sodium vanate and 5 mM sodium fluoride) for 30 min at 4 °C. After centrifugation of lysed cells, protein samples were collected and stored at −80 °C. Protein samples (50 µg) were separated by electrophoresis and transferred onto either a Hybond-ECL nitrocellulose membrane (Amersham Biosciences) or Immobilon-P PVDF membrane (Millipore Corporation), then incubated for one hour in 1% BSA plus 0.01% Tween-20. After washing with PBS with 0.01% Tween-20, the membranes were incubated with primary antibody. Following 1.5 h of incubation with primary antibody, membranes were washed with PBS with 0.01% Tween-20 and then incubated in secondary antibody for 1 h. Immunoblots were then washed and developed with Western Lightning Plus-ECL Enhanced Luminol Reagent Plus (PerkinElmer).
Immunostaining
Immunostaining was performed to assess and localize proteins in the cells cultured with Y15. Briefly, circular coverslips were placed in each well of a 24-well plate and then seeded with 1× 105 cells per well. After 24 h, the cells were washed and fixed in 0.5mL of 3.7% formaldehyde. After washing, the plates were placed on ice and 0.5 mL of permeabilizing solution added to each well, incubated for 3 min and washed. Cells were then blocked for 20 min at room temperature with 25% normal goat serum. Cells were then incubated with primary anti-FAK or anti-pY397 FAK antibody (1:200; Invitrogen) for 30 min, washed, then incubated with Texas Red or Alexa Fluor 594 secondary antibodies (1:200) for 30 min under dark conditions. Cells were counterstained with a FITC conjugated phalloidin (1:25; Molecular Probes) to label actin. The coverslips were then transferred to a slide and examined using a Zeiss Observer.A1 microscope at 100× magnification.
Viability
Viability was assessed using the MTS assay using kit from (Promega Corporation). Cells were seeded onto 96-well plates (5000 cells per well in 100 µL of D-MEM plus 10% FBS and 1% penicillin/streptomycin). Twenty-four hours following treatment with different inhibitors, 20 µL of Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega Corporation) was added to each well. After 1 h of incubation with reagent, the plate was read with the Gen 5 1.07 microplate reader at 490 nm.
Clonogenicity
Two 6-well plates were seeded with SW480 and SW620 cells at a concentration of 500 cells per well in 2mL of media. After overnight incubation (37 °C, in 5% CO2), each well was treated with a different dose of Y15 and left to incubate undisturbed for 14 d. Then cells were stained with crystal violet blue to detect the number of colonies in each well. Specifically, media was aspirated from each well and cells were fixed for 10 min in 10% neutral buffered formalin. The formalin was then removed from each well, and 0.1% crystal violet blue in 20% methanol was added to each well and plates were placed on a shaker for 5 min. The number of colonies in each well was counted in duplicate plates.
Detachment
Six-well plates were seeded with either SW480 or SW620 cells in D-MEM at a concentration of 2 × 106 and allowed to incubate overnight to attach to the floor of the plate. After 24 h, each well was treated with a different dose of Y15. The following day, the media from each well was collected and the number of unattached cells for each treatment condition was counted three times with a hemocytometer. The cells attached to the plate were collected with trypsin-EDTA and similarly counted. The average percent of detached cells for each treatment condition was calculated by dividing the number of detached cells by the number of total (attached plus detached) cells.
Apoptosis
Apoptosis assay was done with Hoechst stain to identify apoptotic nuclei. Sixty millimeter plates were seeded with SW480 or SW620 cells at a concentration of 2 × 106 cells in D-MEM. After overnight incubation, plates were treated with one of several doses of Y15 or control TAE 226 (Novartis), a FAK inhibitor that induces apoptosis. After another 24 h of incubation, cells were collected with trypsin, and following centrifugation at 900 rpm, were washed with 1× PBS. Again, cells were centrifuged at a low speed, PBS was removed and cells were fixed for 10 min in 10% formalin. Then, 1 µg/mL of Hoechst stain in PBS was added to each sample and cells were allowed to sit at room temperature for 10 min. After 10 min, samples were centrifuged, washed and then plated on microscope slides for viewing using a Zeiss Observer.A1 microscope at 100 × magnification.
The apoptosis assay using Hoechst staining was confirmed by Annexin V Cell Cycle Assays on SW480 and SW620 cells treated with the same doses of Y15 as above. For this assay, we used the PE Annexin V Apoptosis Detection Kit (BD) and followed manufacturer’s protocol.
Tumor growth in vivo
Eighteen female nude balb/c mice were divided into three groups of six and the right flank of each mouse was injected with 0.1 mL of SW620 cells at a concentration of 2 × 106 cells/mL. Three treatment groups were formed to test the in vivo effects of Y15 on SW620 tumorigenesis. The mice were treated with daily intraperitoneal injections of Y15 (30 mg/kg), Y15A (30 mg/kg), or 1× PBS (untreated control). Treatment was started on the day of tumor inoculation and was administered 5 d per week. Tumors were measured with calipers every 3 d and tumor volumes calculated (volume [mm3] = width2 × length/2). After 26 d, mice were sacrificed because the tumor burden in the control groups had reached the predetermined humane limit of 2.0 cm. Tumors were harvested for biochemical analysis.
For Y15 and chemotherapy experiment, 44 female nude balb/c mice were injected with 2 × 106 SW620 cells/mL, as in the previous trial. To test the efficacy of our agent on established tumors, we observed the mice until tumor volume reached approximately 80 mm3. Treatment was began on day 3, and mice were given either daily 1× PBS (oral gavage), daily Y15 (100 mg/kg oral gavage), once weekly 5-FU (30 mg/kg intraperitoneal injection), twice weekly oxaliplatin (1 mg/kg intraperitoneal injection) or a combination regimen: daily Y15 (oral gavage) with once weekly 5-FU (intraperitoneal injection), daily Y15 (oral gavage) with twice weekly oxaliplatin (intraperitoneal injection), or daily Y15 (oral gavage) with once weekly 5-FU (intraperitoneal injection) and twice weekly oxaliplatin (intraperitoneal injection). Similar to the first study, tumors were measured with calipers and volumes were calculated. Mice were sacrificed due to tumor burden and tumors were harvested for biochemical analysis.
Statistical analysis
The Student t test was performed for analysis, and P value less than 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
Dr Golubovskaya and Dr Cance are Co-Founders and shareholders of CureFAKtor Pharmaceuticals LLC.
Acknowledgments
We thank Drs Manivannan Ethirajan and Ravindra Pandey for providing Y15A derivative. This work was supported by Roswell Park Cancer Institute and National Cancer Institute (NCI) #P30; RO-1 CA016056 and the NCI T32 Surgical Oncology Training Grant to the Roswell Park Cancer Institute 5T32 CA 108456 to WGC.
References
- American Cancer Society. Colorectal Cancer Facts & Figures 2011-2013. In: Society AC, ed. Atlanta, 2011.
- Golubovskaya VM, Kweh FA, Cance WG. Focal adhesion kinase and cancer. Histol Histopathol 2009; 24:503 - 10; PMID: 19224453
- Figel S, Gelman IH. Focal adhesion kinase controls prostate cancer progression via intrinsic kinase and scaffolding functions. Anticancer Agents Med Chem 2011; 11:607 - 16; http://dx.doi.org/10.2174/187152011796817646; PMID: 21355844
- Cance WG, Harris JE, Iacocca MV, Roche E, Yang X, Chang J, et al. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: correlation with preinvasive and invasive phenotypes. Clin Cancer Res 2000; 6:2417 - 23; PMID: 10873094
- Golubovskaya VM, Cance WG. Focal adhesion kinase and p53 signaling in cancer cells. Int Rev Cytol 2007; 263:103 - 53; http://dx.doi.org/10.1016/S0074-7696(07)63003-4; PMID: 17725966
- Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem 2008; 51:7405 - 16; http://dx.doi.org/10.1021/jm800483v; PMID: 18989950
- Bolós V, Gasent JM, López-Tarruella S, Grande E. The dual kinase complex FAK-Src as a promising therapeutic target in cancer. Onco Targets Ther 2010; 3:83 - 97; http://dx.doi.org/10.2147/OTT.S6909; PMID: 20616959
- Golubovskaya VM, Gross S, Kaur AS, Wilson RI, Xu LH, Yang XH, et al. Simultaneous inhibition of focal adhesion kinase and SRC enhances detachment and apoptosis in colon cancer cell lines. Mol Cancer Res 2003; 1:755 - 64; PMID: 12939401
- Golubovskaya VM, Nyberg C, Zheng M, Kweh F, Magis A, Ostrov D, et al. A small molecule inhibitor, 1,2,4,5-benzenetetraamine tetrahydrochloride, targeting the y397 site of focal adhesion kinase decreases tumor growth. J Med Chem 2008; 51:7405 - 16; http://dx.doi.org/10.1021/jm800483v; PMID: 18989950
- Xu L-H, Yang X, Bradham CA, Brenner DA, Baldwin AS, Craven RJ, et al. The focal adhesion kinase suppresses transformation-associated anchorage-independent apoptosis in human breast cancer cells. J Biol Chem 2000; 275:30597 - 604; http://dx.doi.org/10.1074/jbc.M910027199; PMID: 10777476
- Golubovskaya VM, Zheng M, Zhang L, Li JL, Cance WG. The direct effect of focal adhesion kinase (FAK), dominant-negative FAK, FAK-CD and FAK siRNA on gene expression and human MCF-7 breast cancer cell tumorigenesis. BMC Cancer 2009; 9:280 - 8; http://dx.doi.org/10.1186/1471-2407-9-280; PMID: 19671193
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 1996; 383:547 - 50; http://dx.doi.org/10.1038/383547a0; PMID: 8849729
- Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res 2008; 68:1935 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-07-5155; PMID: 18339875
- Halder J, Lin YG, Merritt WM, Spannuth WA, Nick AM, Honda T, et al. Therapeutic efficacy of a novel focal adhesion kinase inhibitor TAE226 in ovarian carcinoma. Cancer Res 2007; 67:10976 - 83; http://dx.doi.org/10.1158/0008-5472.CAN-07-2667; PMID: 18006843
- Wang ZG, Fukazawa T, Nishikawa T, Watanabe N, Sakurama K, Motoki T, et al. TAE226, a dual inhibitor for FAK and IGF-IR, has inhibitory effects on mTOR signaling in esophageal cancer cells. Oncol Rep 2008; 20:1473 - 7; PMID: 19020730
- Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer Biol Ther 2010; 9:764 - 77; http://dx.doi.org/10.4161/cbt.9.10.11434; PMID: 20234191
- Beierle EA, Ma X, Stewart J, Nyberg C, Trujillo A, Cance WG, et al. Inhibition of focal adhesion kinase decreases tumor growth in human neuroblastoma. Cell Cycle 2010; 9:1005 - 15; http://dx.doi.org/10.4161/cc.9.5.10936; PMID: 20160475
- Hochwald SN, Nyberg C, Zheng M, Zheng D, Wood C, Massoll NA, et al. A novel small molecule inhibitor of FAK decreases growth of human pancreatic cancer. Cell Cycle 2009; 8:2435 - 43; http://dx.doi.org/10.4161/cc.8.15.9145; PMID: 19571674
- Topley P, Jenkins DC, Jessup EA, Stables JN. Effect of reconstituted basement membrane components on the growth of a panel of human tumour cell lines in nude mice. Br J Cancer 1993; 67:953 - 8; http://dx.doi.org/10.1038/bjc.1993.176; PMID: 8494729
- Trainer DL, Kline T, McCabe FL, Faucette LF, Feild J, Chaikin M, et al. Biological characterization and oncogene expression in human colorectal carcinoma cell lines. Int J Cancer 1988; 41:287 - 96; http://dx.doi.org/10.1002/ijc.2910410221; PMID: 3338874
- Parsons JT, Slack-Davis J, Tilghman R, Roberts WG. Focal adhesion kinase: targeting adhesion signaling pathways for therapeutic intervention. Clin Cancer Res 2008; 14:627 - 32; http://dx.doi.org/10.1158/1078-0432.CCR-07-2220; PMID: 18245520
- Beierle EA, Ma X, Trujillo A, Kurenova EV, Cance WG, Golubovskaya VM. Inhibition of focal adhesion kinase and src increases detachment and apoptosis in human neuroblastoma cell lines. Mol Carcinog 2010; 49:224 - 34; PMID: 19885861
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol 1999; 71:435 - 78; http://dx.doi.org/10.1016/S0079-6107(98)00052-2; PMID: 10354709
- Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354:2531 - 41; http://dx.doi.org/10.1056/NEJMoa055229; PMID: 16775234
- Heffler M, Golubovskaya VM, Cance WG, Bullard Dunn KM. Evolving molecular therapeutics and their applications to surgical oncology. American College of Surgeons: Principles and Practice. Chicago (IL): American College of Surgeons, 2011.
- Lai E, Singh R, Teng B, Zhao Y, Sharratt E, Howell G, et al. Inhibition of hyaluronan synthase-3 decreases subcutaneous colon cancer growth in mice. Dis Colon Rectum 2010; 53:475 - 82; http://dx.doi.org/10.1007/DCR.0b013e3181c87084; PMID: 20305449