Abstract
Prostate cancer is the second highest cause of male cancer deaths in the United States. A significant number of tumors advance to a highly invasive and metastatic stage, which is typically resistant to traditional cancer therapeutics. In order to identify chromosomal structural variants that may contribute to prostate cancer progression we sequenced the genomes of a HPV-18 immortalized nonmalignant human prostate epithelial cell line, RWPE1, and compared it to its malignant, metastatic derivative, WPE1-NB26. There were a total of 34 large (>1 Mbp) and 38 small copy number variants (<100 kbp) in WPE1-NB26 that were not present in the precursor cell line. We also identified and validated 46 structural variants present in the two cell lines, of which 23 were unique to WPE1-NB26. Structural variants unique to the malignant cell line inactivated: (1) the neurofibromin2 (NF2) gene, a known tumor suppressor; (2) its neighboring gene NIPSNAP1, another putative tumor suppressor that inhibits TRPV6, an anti-apoptotic oncogene implicated in prostate cancer progression; (3) UGT2B17, a gene that inactivates dihydrotestosterone, a known activator of prostate cancer progression; and (4) LPIN2, a phosphatidic acid phosphatase and a co-factor of PGC1a that is important for lipid metabolism and for suppressing autoinflammation. Our results illustrate the value of comparing the genomes of defined related pairs of cell lines to discover chromosomal structural variants that may contribute to cancer progression.
Introduction
Prostate cancer is one of the most widely diagnosed cancers among males worldwide. According to the NCI it remains one of highest contributors to cancer-related deaths among males in the US. Most of the patients who die from the cancer do so from metastasis of the cancer.
Genetic variants such as single nucleotide polymorphisms, chromosomal rearrangements, and copy number variants have long been associated with cancer. The identification of the BCR-ABL gene fusion in chronic myeloid leukemia (CML)Citation1 and the success of therapies that target BCR-ABL indicate that such genetic variants contribute to cancer and provide critical targets for therapy. With the emergence of the massively parallel sequencing technologies, several studies have focused on identifying structural variants across the genome using the paired-end and single read sequencing methods in various cancers.Citation2-Citation8 Several recent studies have started to catalog genetic variants in prostate cancer. Using transcriptome-sequencing approaches several gene rearrangements have been reported in prostate cancer,Citation9-Citation11 including ETS family gene fusions (with TMPRSS2-ERG as the most commonly reported fusion). However genomic structural variants acquired as a prostate cancer progresses to advanced, metastatic, androgen-deprivation-refractory disease have not been completely cataloged.
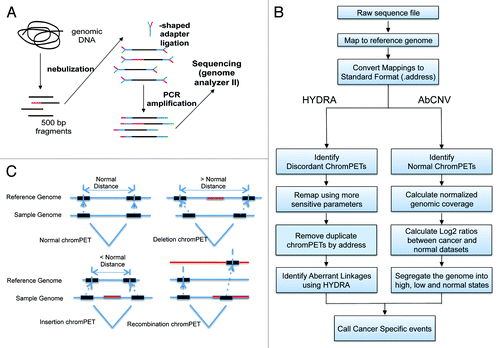
Because genetic studies with prostate tumors have been begun by others and are complicated by intra-tumor heterogeneity and contamination of the malignant cells by normal stromal cells, we took a different and complementary approach. We chose RWPE1, a human papilloma virus 18 (HPV-18) immortalized non-tumorigenic prostate epithelial cell line, and its derivative, WPE1-NB26, which has been mutagenized by N-methyl-N-nitrosourea and selected for high metastatic and invasive potential. These two human cell lines provide an in vitro model for studying prostate carcinogenesis and progression.Citation12 We performed massively parallel paired-end sequencing to systematically identify and characterize structural variants across the genome (). We applied two independent bioinformatic pipelines, HYDRACitation13 and AbCNV (Aberrant Copy Number Variations) () to identify structural variants () that are associated with the phenotypic progression of immortalized prostate epithelial cells to metastatic and invasive prostate cancer cells.
Figure 1. (A) Overview of the protocol followed to extract genomic DNA and prepare it for sequencing on the Illumina platform. (B) Flowchart depicting the data flow and analysis performed. (C) Different kinds of structural variants, and the logic for calling them.
Results
Genomic DNA from each cell line was isolated and nebulized to obtain a library of 500 bp fragments (). This fragment library was then subjected to paired-end DNA sequencing with the Illumina GAII sequencing system, using 38–40 bp reads. A total of 308 234 420 chromPETs were obtained for RWPE1 and 303 838 292 chromPETs for WPE1-NB26 (). The chromPETs were mapped back to the current reference genome assembly (hg19) using NovoalignCitation14 (see Materials and Methods) with default parameters. This resulted in 279 001 461 (90.52%) and 272 883 446 (89.8%) chromPETs with both ends mapping back uniquely and within the expected distance for the immortalized RWPE1 and tumorigenic WPE1-NB26 cell lines, respectively (Table 1). The mapped distances between paired-tags for all intra-chromosomal chromPETs (both tags mapping to the same chromosome) yielded a median insert size of 510 bp for RWPE1 and 511 bp for WPE1-NB26 with a median absolute deviation of 31 bp. This gives us approximately 50× physical coverage for both genomes. All chromPETs with insert sizes within the median ± 6*MAD are classified as normal chromPETs and the rest as discordant chromPETs. The schematic in shows the different classifications that could give rise to discordant chromPETs. Using the data analysis workflow shown in , normal chromPETs were then used to estimate copy number using depth of coverage and the discordant chromPETs were used to identify genomic rearrangements.
Table 1A. Raw sequencing and variant numbers
Genome rearrangements
As shown in we had 2 527 047 discordant chromPETs in WPE1-NB26 and 2 080 954 discordant chromPETs in RWPE1. To identify putative breakpoints we used the HYDRA pipeline as described in Quinlan et al.Citation13 HYDRA has several key strengths. First, it can identify breakpoints in both unique and repetitive genomic regions which allows us to assess structural variant (SV) within the most structurally dynamic parts of the genome, namely at transposons and low copy repeats (LCRs). Second, HYDRA does not require assumptions about variant structure, which allows us to identify complex events often missed by other methods. HYDRA called a total of 1598 rearrangements (with the filtering scheme described in the Materials and Methods) in the two cell lines, with 88 predicted to be somatic breakpoints specific for WPE1-NB26 (). To validate these potential breakpoints we performed PCR on genomic DNA from the two cell lines and normal lung tissue using forward and reverse primers designed within the footprints of each breakpoint call. 50/88 (56.8%) of the predicted breakpoints were detected in at least one of the genomes tested. 23/50 (~46%) of the validated breakpoints were specific for the WPE1-NB26 malignant cell line.
Table 2. HYDRA calls and validation status
An independent measure of the HYDRA breakpoint calls was obtained by evaluating how many of the deletion breakpoints from the HYDRA pipeline were seen as germline deletions in the Thousand Genomes project.Citation18 569/878 (~65%) of the deletion breakpoints predicted in both RWPE1 and WPE1-NB26 cells overlapped with deletion breakpoints identified in the Thousand Genomes project. Such a high amount of overlap indicates that the variants that are common to both cell lines in our pipeline are likely germline variants present in the population and provides additional confidence in the HYDRA algorithm.
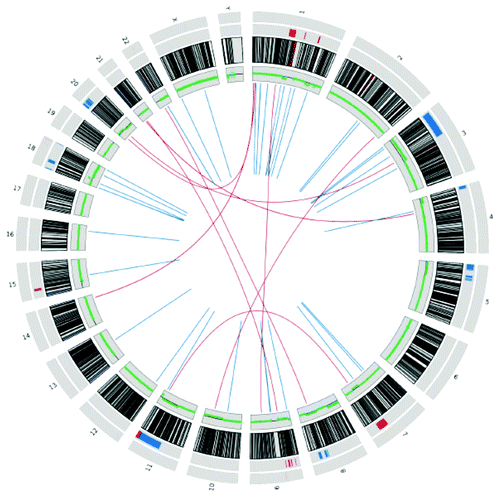
A Circos plotCitation19 visualizing the 46 structural variants seen in WPE1-NB26 malignant cells (23 of which are present in the precursor RWPE1 cells) is shown in .
Figure 2. Circos plot for 46 structural variants seen in the WPE1-NB26 malignant cells. Innermost lines depict the breakpoints (intra-chromosomal in blue, inter-chromosomal in red). The next track in green depicts the log ratio of read depth (WPE1-NB26:RWPE1) across the genome. The next track is the ideogram of the chromosomes, followed by the validated copy number changes across the genome (large changes inner to the small changes, amplifications in blue and deletions in red).
The 46 somatic breakpoints identified in WPE1-NB26 intersected with 34 refseq transcripts (). The genes involved (with a positive validation by PCR analysis) include, NF2, NIPSNAP1, FAM118B, WWP2, TBX15, PDE4DIP, SLC2A5-BTBD7, COL24A1-C9orf156, LRP1B, FMNL2, SCAP, ITPR1 (), and RHOH. We used g:ProfilerCitation20,Citation21 to look for a functional enrichment of the genes with breakpoint in a particular functional group. We only found an enrichment of Rho GTPase binding gene ontology (Molecular Function—GO:0017048, ROCK1, RHOH, FMNL2, and DIAPH2) with a P value of 1.14e−03 (or ROCK1, RHOH, and DIAPH2 with a P value of 9.28e−03). Although not highly significant statistically, the enrichment suggests that breaks in genes regulating Rho GTPase are selected for during progression to malignancy, metastasis, and invasion. We also found that the genes with breakpoints were enriched in a network of interacting proteins from BioGRID interaction database: NF2, FAM118B, GNB2, PRKG1, and ITPR1 with a P value of 7.72e−04 (Fig. S2A). The same pathway is also found enriched among the genes that have breakpoints only in WPE1-NB26: NF2, GNB2, and FAM118B, with a P value of 3.07e−02 as shown in Figure S2B.
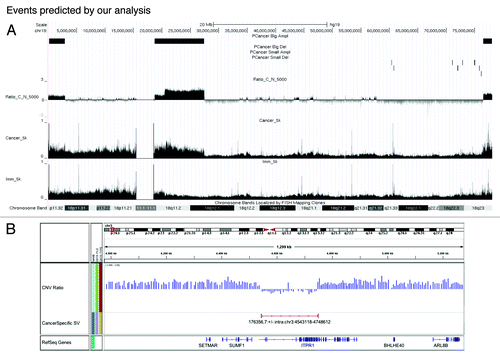
Figure 3. Example of events predicted by our analysis. (A) AbCNV results from chr18. UCSC browser snapshot of chr18. Topmost plots (PCancer Big Del, PCancer Big Ampl, PCancer Small Ampl, and PCancer Small Del) show the segments with copy number variants flagged by AbCNV. Ratio_C_N_5000 is for the log2 ratio of the read depths (WPE1-NB26:RWPE1) over 5000 base windows. The final two plots show the read depth for Cancer (WPE1-NB26) and the Immortalized (RWPE1) cell lines. (B) WPE1-NB26 specific deletion predicted by HYDRA. This deletion on chr3 removes the 3′ end of the ITPR1 gene.
Copy number variations
The uniquely mapped chromPETs that were classified as reporting non-rearranged genomic architecture were used to estimate copy number variant of genomic segments using depth of coverage.Citation15,Citation17,Citation22,Citation23 A total of 279 001 461 and 272 883 446 concordant chromPETs were obtained for RWPE1 and WPE1 NB26, respectively. We used a window size of 15 000 bp to analyze the data, and this determines the minimum resolution for our CNV calls.
We developed the AbCNV program to identify regions with copy number variants (). This involves (1) creating a normalized profile of coverage across the genome, (2) calculating the log2 ratio of sequence coverage between the WPE1-NB26 and the RWPE1 cell lines (Ratio_C_N), and (3) using a segmentation algorithm to identify regions with high/low Ratio_C_N. For each of these regions we compared the raw values of coverage relative to adjoining regions in RWPE1 cells to eliminate regions that were amplified or deleted in these control cells. This eliminated false positives stemming from a change in gene copy number that was specific to RWPE1 cells.
In total we had 20 large (>1 Mbp) amplifications and 14 large (>1 Mbp) deletions that were specific for WPE1-NB26 (). Similarly we also observed 9 small amplifications and 29 small deletions specific for WPE1-NB26. We used quantitative PCR (qPCR) of genomic DNA from the two cell lines and normal lung to validate these copy number changes. Table 3 lists all the sites and shows that our validation rates were 83% (10/12 regions tested) for the large CNVs and 66.7% (6/9 regions tested) for the small CNVs. For the small CNVs, we only validated the sites that involved known genes. Copy number variant (CNV) data for the whole genome is also shown in the Circos plot in .
Table 1B. Raw sequencing and variant numbers
Examples of our analysis are shown in and . The Ratio_C_N identifies a 50 Mb region that is enriched in WPE1-NB26 relative to RWPE1 (). Examination of the raw sequence coverage of this region relative to its neighbors in both cell lines (tracks marked Imm_Sk or Cancer_Sk) confirms that the copy number is normal in RWPE1 but is amplified in WPE1-NB26 cells. This is confirmed by q-PCR of genomic DNA (, big amplifications, line 1)
Figure 4. Deletion of 11q24–25. (A) UCSC genome browser snapshot of 11q24–25 locus. The top track shows the deletion predicted by AbCNV. The next two tracks show the normalized chromPET coverage from Cancer (WPE1-NB26) and Immortalized (RWPE1) cell lines. Ratio_C_N_5000 is as described in . (B) qPCR validation of the deletion. (C) UCSC genome browser snapshot depicting the small deletion in the UGT2B17 locus on chr4. The tracks are as indicated in and (A).
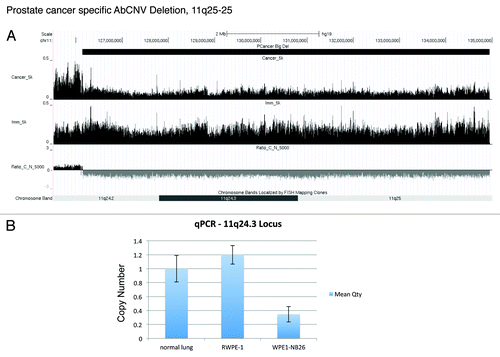
Table 3A. AbCNV predictions and validations
A similar analysis is shown for a 200 kb deletion involving the ITPR1 gene (). Here we also have independent validation of the deletion from the HYDRA calls, because a breakpoint is identified spanning the deletion segment.
A 9 Mb deletion in 11q24–25 is identified in (, big deletions, line 1). Here we also show q-PCR validation of the decrease in DNA copy number in WPE1-NB26 relative to RWPE1 or normal lung genome ().
Finally, we highlight a 50 kb deletion across the UGT2B17 gene (). This deletion was validated by PCR of genomic DNA (). For independent validation we show a reverse-transcription and PCR of RNA from RWPE1 and WPE1-NB26 cells (). As expected, the UGT2B17 mRNA is absent in WPE1-NB26. Similar to this we saw several small deletions in the genic or promoter regions of several genes, such as NF2, NME7, FILIP1L, UGT2B14/17, and DIAPH2 ().
Table 3B. AbCNV predictions and validations
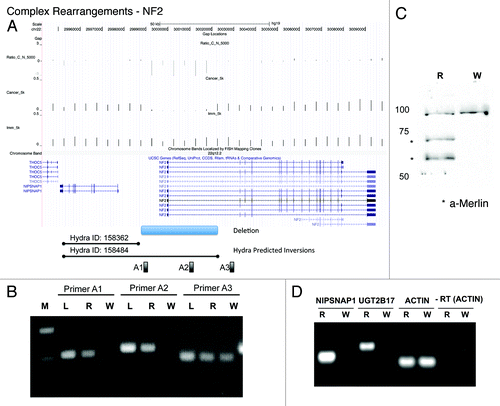
Figure 5. Complex rearrangements delete the NF2 gene. (A) UCSC genome browser snapshot of the NF2/NIPSNAP1 locus depicting the read depths for Cancer (WPE1-NB26) and Immortalized (RWPE1) and Ratio_C_N_5000 that shows the small deletion in the promoter region of NF2. The AbCNV deletion is shown at the bottom followed by the two HYDRA predicted inversion breakpoints. A1, A2 and A3 are the primer pairs used in (B). (B) PCR validation of the NF2 copy number deletion in genomic DNA. M:MW marker, L:normal lung, R:RWPE1, W:WPE1-NB26. PCR primer pairs shown at the top and (A). (C) Western blot for Merlin (NF2) in R (RWPE1) and W (WPE1-NB26). The * show the two different isoforms of Merlin. The uppermost band is a cross-reactive band that serves as loading control. (D) RT-PCR to show loss of expression of NIPSNAP1 and UGT2B17 mRNA in WPE1-NB26 (W) compared with RWPE1 (R). Actin shows that equal amounts of mRNA were input from the two cell lines. -RT: Actin PCR without the reverse-transcription step.
Complex rearrangements
A striking complex rearrangement in WPE1-NB26 cells is on 22q12.2. We found three HYDRA breakpoint calls in this region, suggesting deletions spanning from the 3′ region of NIPSNAP1 (non-neuronal SNAP25-like protein homolog 1) gene to the 5′ region of NF2 (neurofibromatosis type 2) gene, and also an inversion of the NIPSNAP1 gene (). By the AbCNV analysis we also found a small deletion of 45 Kb that removes the promoter and the 5′ UTR of the tumor suppressor NF2 gene in WPE1-NB26 cells (). As our model in suggests, in the first step segments B and C containing the NIPSNAP1 gene were inverted to create the junction (reported by HYDRA breakpoints 158362 and 158424) joining A to C. In the second step the B and D segments containing the 5′ ends of NIPSNAP1 and NF2, respectively, are deleted to create the junction joining C to E. PCR with primer pairs located in the deletion (A1 and A2) and outside (A3) confirmed the deletion involving NF2 in WPE1-NB26 (). This model suggested that NIPSNAP1 and NF2 gene expression should be suppressed in WPE1-NB26. Western blot shows that the NF2 protein is indeed not expressed in the WPE1-NB26 malignant cells (). Reverse-transcription PCR on cellular RNA confirms that NIPSNAP1 mRNA is also not expressed in WPE1-NB26 cells ().
We identified another complex rearrangement event involving the EMILIN2/LPIN2 and ROCK1 genes on chr18 (). The individual breakpoints predicted by HYDRA (ID: 104993, 104965, and 104992 in and ) have been confirmed by PCR across the breakpoints (). To elucidate the genomic structure of this locus, we sequenced the PCR amplified fragments () and also measured the DNA copy number at selected sites across the entire region (results summarized in ). Sequencing of the PCR fragments across the breakpoints indicated that HYDRA ID:104993 and ID:104965 were reporting on the same breakpoint. A deletion of 19 005 bases between chr18:2 908 934 (end of segment A) and 2 927 939 (beginning of segment E) creates the A–E adjacency reported by HYDRA ID:104965. Unexpectedly, a 245 base DNA fragment G from chr18:18 691 959–18 692 204 was inserted between fragments A and E to create the A–G adjacency reported by HYDRA ID:104993 and confirmed by the sequencing of the PCR products in . Intriguingly, part of the BCD segment is not lost completely as evidenced both by the copy number measurements () and the C–H adjacency reported by HYDRA ID:104992.
Figure 6. Complex rearrangements in EMILIN2-LPIN2-ROCK1 locus on chr18. (A) Cartoon showing the chr18: 2 900 000–19 500 000 locus divided into named segments from A–H. The track underneath shows the three genes of interest (EMILIN2, LPIN2, and ROCK1). The next tracks show the location of the footprint of HYDRA breakpoint IDs: 104992, 104993, and 104965. The next track shows the measured copy number across this whole region. (B) Cartoon showing the sequencing results obtained for the three HYDRA breakpoints (IDs: 104992, 104993, and 104965) in the terms of the same named segments as defined in (A). (C) Model depicting our hypothesis for the complex event that takes place on chr18 to create the deletion and the HYDRA breakpoints reported in (A).
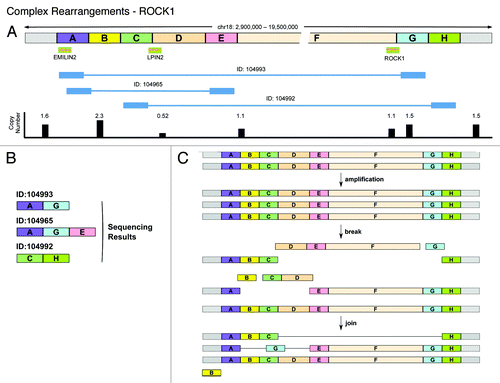
We suggest that initially there was an increase of one copy of the entire region, accounting for the 1.5× copy number of the ends of the region (). Two deletions occur in two different copies of the locus: one removes DEFG to create the C–H junction, while the other removes BCD, but incorporates G from the first deletion as a genome shard in the A–E junction. The net result will be that WPE1-NB26 will have 3 copies of G (1.5×), 1 copy of D (0.5×), and 2 copies of B, C, E, and F (1×). However, since there are >2× copies of B, this segment may have amplified independently or inserted elsewhere in the genome as genome shards. The loss of segment G in one allele and its insertion as a genome shard in a deletion in another allele raises the possibility that these alleles were close together (perhaps due to binding to a transcription factory) when the break and joining events occurred in the two alleles. The allele containing the C–H junction may lack a centromere and therefore might be associated with another chromosome.
Conclusion
There have been several previous studies that have studied gene rearrangements in prostate cancer. In 2006, Tomlins et al.Citation24 found gene fusions between TMPRSS2 and ETS family members ETV and ERG, resulting in an overexpression of the resulting transcript. Since then the TMPRSS2:ERG gene fusion has been the found out to be one of the most prevalent gene fusion found in prostate cancers.Citation25 Chinnaiyan et al. and others have shown, using microarray data and exome sequencing data that 50–70% of cancer samples have members of the ETS and ETV family of transcription factors involved in gene rearrangements.Citation24-Citation29 Pflueger et al.Citation7 also discovered N-Myc downstream regulated gene 1 (NDRG1) to be fused with ERG in prostate cancers. However, recent transcriptome sequencing experiments have also discovered gene fusions not involving any ETS family of transcription factors. Using ETS rearrangement negative prostate cancers, Palanisamy et al.Citation30 found RAF kinase family of genes—BRAF and RAF1 to be involved in gene fusions. They also screened a large cohort of patients and found RAF pathway genes were involved in gene fusions in advanced prostate cancers. In 2011, Berger et al.Citation31 sequenced and analyzed tumor and matched normal DNA from seven patients with high-risk primary prostate cancer. Only 3 of the patients harbored the TMPRSS2-ERG gene fusion. On an average they found ~90 arrangements per genome, which compares favorably with the 46 rearrangements found in WPE1-NB26 in our study. In total they found 16 genes that had rearrangements in more than one patient sample, including ZNF407, CHD1, PTEN, C21ORF45, CSMD3, CADM2, ERG, and TMPRSS2.
Prostate cancer is indolent in most patients, but progresses to a more invasive and metastatic stage in a small fraction of the patients. In this paper we attempted to identify rearrangements and copy number variants in the context of prostate cancer progression. By using a prostate epithelial cell line and its metastatic and invasive form and comparing it to an immortalized but non-metastatic precursor, we hope to identify genetic changes that contribute to prostate cancer progression. Although we did not detect the more widely known ETS family of gene DNA fusions seen in prostate cancer, it is entirely possible that such fusions can be achieved in the cancer cells by trans-splicing of RNAs.Citation32
Interestingly, several of the predicted regions containing a CNV have also been implicated in either prostate cancer or tumorigenesis in general. For example, AbCNV predicted a deletion at 11q24–25. By qPCR we discovered that this 8.8 Mbp deletion has normal copy number in immortalized cell line (RWPE1) and only half the copy number in cancer cell line (WPE1-NB26). This region harbors the OPCML gene that has been demonstrated to act as a broad tumor suppressor in prostate cancer cell lines.Citation33 The authors showed the gene is epigenetically silenced by CpG methylation. Thus the presence of this deletion in WPE1-NB26 is consistent with deletion of the tumor suppressor OPCML during progression.
Similarly, a deletion in a suspected tumor suppressor locus chromosome 7q31, and a known fragile site (FRA7G) has been identified as a loss of heterozygosity region (LOH) in primary prostate cancer and is associated with tumor aggressiveness and progression.Citation34 Caveolin-1 (Cav-1) is one of candidate tumor suppressor genes in this locus and it has been shown to be downregulated in prostate tumors.Citation35 Cav-1 null mice develop a carcinogen-induced tumor susceptibly.Citation36 Also exogenous expression of Cav-1 in MCF7 human breast adenocarcinoma cells inhibited anchorage-independent growth and matrix invasiveness.Citation37
We discovered a complex event affecting two significant genes, NF2 and NIPSNAP1. NF2 gene was identified as mutated in neurofibromatosis type 2 and the encoded protein belongs to the Band 4.1 superfamily. Inherited NF2 mutation predisposes the patient to schwannomas, meningiomas, and ependymomas. Somatic NF2 mutations have also been found in other types of sporadic tumors. In several prostate tumor cell lines, NF2 expression was low or its activity was suppressed by PAK-mediated constitutive phosphorylation.Citation38,Citation39 Many lines of evidence suggest its tumor suppressor properties involved in integrating and regulating the extracellular and intracellular signaling pathways that regulate cell proliferation, and survival. These studies, along with our results, indicate a potential role of NF2 as a tumor suppressor that is inactivated in prostate cancer progression.
NIPSNAP1 belongs to a highly conserved family of proteins with unknown function. This protein was suggested to interact with the transient receptor potential vanilloid channels 5 and 6 (TRPV5/6) and inhibit their activity.Citation40 TRPV6 was demonstrated to confer resistance to apoptosis induced via Ca2+/NFAT-dependent pathways.Citation41 Therefore, NIPSNAP1 might inhibit TRPV6 to promote apoptosis and thus have tumor suppressor activity. NIPSNAP1 deletion during the transition to malignancy will now allow TRPV6 to confer resistance to apoptosis.
UGT2B17 is of interest, because several previous studies have suggested that a deletion polymorphism in UGT2B17 may significantly contribute to prostate cancer susceptibility in men. UGT2B17 catalyzes the transfer of glucuronic acid from uridine diphosphoglucuronic acid to substrates. In the human prostate, natural androgen, dihydrotestosterone (DHT) has the highest affinity for the androgen receptor. UGT2B17 class of enzymes is responsible for DHT glucuronidation and inactivation.Citation42 In other words, downregulation of UGT2B17 will increase the levels of functional DHT. Thus, the mutation of UGT2B17 may explain why WPE1-NB26 cells are much less sensitive to androgen depletion than the parental cell line RWPE1.
Not all deletions could be construed as a simple loss of a tumor suppressor. ROCK1 (and ROCK2) is a serine/threonine kinase that functions downstream of RhoA and RhoC. It phosphorylates a myosin light chain (MLC) and regulates acto-myosin contractility,Citation43 which contributes to invasive and metastatic behavior in cancer. A preclinical study has shown ROCK inhibition suppressed both Rho-mediated activation of actomyosin and invasive activity of rat MM1 hepatoma cells implanted into the peritoneal cavity of syngeneic rats.Citation44 Thus, it is difficult to explain why WPE1-NB26 would select for complex rearrangement that is accompanied by deletion of this oncogene.
In summary, our results with pure populations of cells present as cancer cell lines underlines the complexity of the genetic changes that one can observe during progression of prostate cancer. Not all the genetic changes are likely to be driver mutations, but prior knowledge about some of the tumor suppressors and oncogenes allows us to make hypotheses about why some of the genes are lost or amplified as the immortalized RWPE1 prostate epithelial cells progress to the highly malignant and metastatic WPE1-NB26 cells. Our results suggest, for example, that loss of tumor suppressors OPCML, Cav-1, NF2, and UGT2B17 by genetic or epigenetic mechanisms can be anticipated as prostate cancers progress from the indolent to the more aggressive form of the disease. The large number and variety of structural variants seen in this relatively simple model of prostate cancer also indicates that many more genetic changes are waiting to be discovered in clinical prostate cancer as it progresses to the aggressive disease.
Materials and Methods
Reagents
Reagents used were DNAZol reagent (Invitrogen, 10503-027), End-It DNA End Repair Kit (Epicenter, ER0720), human adult normal lung genomic DNA (BioChain, D1234152), MinElute Reaction Cleanup Kit (Qiagen, 28204), QIAquick Gel Extraction Kit (Qiagen, 28704), QIAquick PCR Purification Kit (Qiagen, 28104), Quick Ligation Kit (NEB, M2200S), Phusion High-Fidelity DNA Polymerase (Finzymes, F530), Taq DNA Polymerase (Roche, 11146165001), and TaKaRa Ex Taq DNA Polymerase (Takara, TAK RR001A).
Cell lines
RWPE1 cells (CRL-11609) and WPE1-NB26 cells (CRL-2852) were purchased from ATCC and cultured according to ATCC instructions.
Paired-end sequencing
All chromPET libraries were constructed according to the protocol supplied by Illumina with minor modifications. Genomic DNA was extracted with DNAZol reagent and 2 μg of DNA was sheared by a Nebulizer. The ends of DNA fragments were polished by an End-It DNA End Repair Kit and A-tail added to the 3′ end by Taq DNA polymerase. The Y-shaped adaptor was ligated to both ends of DNA fragments by a Quick Ligation Kit and 600–700 bp DNA fragments were purified by 2.0% agarose gel electrophoresis and a QIAquick Gel Extraction Kit. Y-shaped adaptor ligated DNA was amplified by PCR primer PE1.0 and 2.0 and was again purified. Paired-end high-throughput sequencing was performed according to the manufacturer’s protocol (Illumina).
Correction of sequencing artifacts
PCR bias may lead to artifacts that result in duplication of the chromPETs that can contribute to false positives in our analysis pipeline. Since such chromPETs would map to the same genomic location, we removed all but one of multiple chromPETs with the exact same mapping locations on both sides. Also all chromPETs that have both tags mapping into simple sequence repeats (SSRs) were removed.
Mapping
The tags are mapped back to the hg19 version of the human genome (downloaded from the UCSC genome browser) using Novocraft’s Novoalign software.Citation14 The hg19 human genome was first indexed using the Novoindex software (-k15 and -s1). The initial mapping is done using the default parameters for Novoalign (novoalign -r All -e 50 -c 3). The second mapping process was done using more sensitive parameters that also allows for multiple mappings (novoalign -c 12 -r E 25 -e 100 -t 150).
Correction of AT bias
Illumina genome analyzer GAII data has been shown to have an AT bias in sequencing coverage. To estimate the effect of AT content on sequence coverage, we divided the genome into 7500 bp non-overlapping windows. For each window, we calculated AT content and plotted it with coverage for that particular window. To correct for the AT bias, we binned the non-overlapping windows into 100 bins based on increasing AT content.Citation15 For example, a window with AT content 45.5% goes into bin 45, and a window with AT content 48.6% goes into bin 48. We then convert the coverage scores for all windows in a bin to their respective Z-scores. This results in an average coverage score of zero in each window, thereby correcting for the AT bias.
HYDRA pipeline for detecting chromosomal breaks
After a second alignment of discordant chromPETs back to the human genome with more sensitive parameters, and excluding any “concordant” chromPET that map with the expected size and orientation, we processed all the resulting “discordant” chromPETs (including multiple mappings) using the HYDRA pipeline.Citation13,Citation16 Duplicate alignments were removed and then fed to the HYDRA pipeline with default parameters. To filter for a high confidence set of breakpoint calls, we selected for breakpoints that were either inter-chromosomal, or intra-chromosomal with the following characteristics
• The two ends of the breakpoint call were separated by >1000 bp
• The chromPETs comprising the breakpoint call had, on average, <1000 mapping combinations between the two ends (if first read in a chromPET maps m number of times and second read maps n number of times, then m × n < 1000),
• The breakpoint call was supported by >2 discordant chromPETs, and
• The average number of mismatches in the sequence of the two ends comprising the call (relative to the reference genome) was <2.
AbCNV algorithm for detecting copy number variants
The AbCNV algorithm uses the depth of coverage to estimate the copy number for a genomic segment.Citation17 The chromPETs that map within the expected distance are used to construct a coverage profile across the genome. We used non-overlapping windows across the chromosome and calculate coverage in each window. The binned data are then normalized to the total number of chromPETs by converting the average coverage per window to Tags per million (TPM) per window. We then calculate the log2 ratio of the TPM for each window in the WPE1-NB26 (C) vs. the RWPE1 (N) cell lines (Ratio_C_N).
The Ratio_C_N per window is then fed into a simple decision machine to determine the copy number of a window given all windows we have seen so far, as shown in Figure S1. Based on the parameters given, it converts the log2 ratio data into one of three states, normal, low or high connected together by edges. The different state transitions were governed by the following functions (C.N. = Log2 ratio copy number):
• Normal → High: C.N. > Median + Threshold
• Normal → Low: C.N. < Median − Threshold
• Low → Normal: C.N. > Median − Threshold + Tolerance * (Threshold)
• High → Normal: C.N. < Median + Threshold − Tolerance * (Threshold)
The threshold we used was twice the Median Absolute Deviations (MAD).
The tolerance we used was 75%.
The machine does not change state unless the criteria for a state transition (as shown in Fig. S1) are fulfilled for 3 consecutive probes. This allows the segmentation algorithm to overcome minor variances in the data that could arise from noise in the data. The genomic regions marked as low/high are regions where the Ratio_C_N was either in the High state or the Low state.
PCR validations
The copy numbers of target loci were measured by real time quantitative PCR using isolated genomic DNA from RWPE1, WPE1-NB26 and normal lung tissue cells. Genomic DNA from normal lung tissue is used as a control. For normalization we used the copy number of the Orc2 locus. qPCR values of target loci are normalized to the Orc2 locus and again normalized to qPCR value of the normal lung data.
Additional material
Download Zip (396.1 KB)Acknowledgments
AD, AM, and YS contributed to the conception of this project. YS prepared the chromPET libraries and validated predicted regions by PCR. AM performed the sequence alignments, designed a strategy of data analysis, wrote the algorithms and programs for AbCNV, and performed all the computational analysis for the project. AD devised and supervised the project. IH participated in discussions of analysis and coordination. All authors contributed to the drafting of the manuscript and the project was supported by P01CA104106. All authors read and approved the final manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cbt/article/25329
References
- Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med 2003; 138:819 - 30; http://dx.doi.org/10.7326/0003-4819-138-10-200305200-00010; PMID: 12755554
- Campbell PJ, Stephens PJ, Pleasance ED, O’Meara S, Li H, Santarius T, et al. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 2008; 40:722 - 9; http://dx.doi.org/10.1038/ng.128; PMID: 18438408
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009; 361:1058 - 66; http://dx.doi.org/10.1056/NEJMoa0903840; PMID: 19657110
- Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature 2010; 463:184 - 90; http://dx.doi.org/10.1038/nature08629; PMID: 20016488
- Stephens PJ, McBride DJ, Lin ML, Varela I, Pleasance ED, Simpson JT, et al. Complex landscapes of somatic rearrangement in human breast cancer genomes. Nature 2009; 462:1005 - 10; http://dx.doi.org/10.1038/nature08645; PMID: 20033038
- Hampton OA, Den Hollander P, Miller CA, Delgado DA, Li J, Coarfa C, et al. A sequence-level map of chromosomal breakpoints in the MCF-7 breast cancer cell line yields insights into the evolution of a cancer genome. Genome Res 2009; 19:167 - 77; http://dx.doi.org/10.1101/gr.080259.108; PMID: 19056696
- Pflueger D, Rickman DS, Sboner A, Perner S, LaFargue CJ, Svensson MA, et al. N-myc downstream regulated gene 1 (NDRG1) is fused to ERG in prostate cancer. Neoplasia 2009; 11:804 - 11; PMID: 19649210
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010; 463:191 - 6; http://dx.doi.org/10.1038/nature08658; PMID: 20016485
- Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. J Clin Oncol 2011; 29:3659 - 68; http://dx.doi.org/10.1200/JCO.2011.35.1916; PMID: 21859993
- Wang XS, Shankar S, Dhanasekaran SM, Ateeq B, Sasaki AT, Jing X, Robinson D, Cao Q, Prensner JR, Yocum AK, et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov 2011; 1:35 - 43; http://dx.doi.org/10.1158/2159-8274.CD-10-0022; PMID: 22140652
- Wu C, Wyatt AW, Lapuk AV, McPherson A, McConeghy BJ, Bell RH, et al. Integrated genome and transcriptome sequencing identifies a novel form of hybrid and aggressive prostate cancer. J Pathol 2012; 227:53 - 61; http://dx.doi.org/10.1002/path.3987; PMID: 22294438
- Webber MM, Quader ST, Kleinman HK, Bello-DeOcampo D, Storto PD, Bice G, et al. Human cell lines as an in vitro/in vivo model for prostate carcinogenesis and progression. Prostate 2001; 47:1 - 13; http://dx.doi.org/10.1002/pros.1041; PMID: 11304724
- Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res 2010; 20:623 - 35; http://dx.doi.org/10.1101/gr.102970.109; PMID: 20308636
- Novocraft.com: Novoalign an aligner for single-ended and paired-end reads from the Illumina Genome Analyser [Internet]. Selangor (Malaysia); 2008- [Updated 2013 Sep 9; cited 2013 Sep 24]. Available from: http://www.novocraft.com/
- Chiang DY, Getz G, Jaffe DB, O’Kelly MJ, Zhao X, Carter SL, et al. High-resolution mapping of copy-number alterations with massively parallel sequencing. Nat Methods 2009; 6:99 - 103; http://dx.doi.org/10.1038/nmeth.1276; PMID: 19043412
- Hall IM, Quinlan AR. Detection and interpretation of genomic structural variation in mammals. Methods Mol Biol 2012; 838:225 - 48; http://dx.doi.org/10.1007/978-1-61779-507-7_11; PMID: 22228015
- Yoon S, Xuan Z, Makarov V, Ye K, Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res 2009; 19:1586 - 92; http://dx.doi.org/10.1101/gr.092981.109; PMID: 19657104
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, et al, 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010; 467:1061 - 73; http://dx.doi.org/10.1038/nature09534; PMID: 20981092
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, et al. Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19:1639 - 45; http://dx.doi.org/10.1101/gr.092759.109; PMID: 19541911
- Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res 2007; 35:Web Server issue W193-200; http://dx.doi.org/10.1093/nar/gkm226; PMID: 17478515
- Reimand J, Arak T, Vilo J. g:Profiler--a web server for functional interpretation of gene lists (2011 update). Nucleic Acids Res 2011; 39:Web Server issue W307 - 15; http://dx.doi.org/10.1093/nar/gkr378; PMID: 21646343
- Xie C, Tammi MT. CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinformatics 2009; 10:80; http://dx.doi.org/10.1186/1471-2105-10-80; PMID: 19267900
- Daines B, Wang H, Li Y, Han Y, Gibbs R, Chen R. High-throughput multiplex sequencing to discover copy number variants in Drosophila. Genetics 2009; 182:935 - 41; http://dx.doi.org/10.1534/genetics.109.103218; PMID: 19528327
- Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, et al. TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res 2006; 66:3396 - 400; http://dx.doi.org/10.1158/0008-5472.CAN-06-0168; PMID: 16585160
- Perner S, Demichelis F, Beroukhim R, Schmidt FH, Mosquera JM, Setlur S, et al. TMPRSS2:ERG fusion-associated deletions provide insight into the heterogeneity of prostate cancer. Cancer Res 2006; 66:8337 - 41; http://dx.doi.org/10.1158/0008-5472.CAN-06-1482; PMID: 16951139
- Rubin MA, Chinnaiyan AM. Bioinformatics approach leads to the discovery of the TMPRSS2:ETS gene fusion in prostate cancer. Lab Invest 2006; 86:1099 - 102; PMID: 16983328
- Hanauer DA, Rhodes DR, Sinha-Kumar C, Chinnaiyan AM. Bioinformatics approaches in the study of cancer. Curr Mol Med 2007; 7:133 - 41; http://dx.doi.org/10.2174/156652407779940431; PMID: 17311538
- Lapointe J, Kim YH, Miller MA, Li C, Kaygusuz G, van de Rijn M, et al. A variant TMPRSS2 isoform and ERG fusion product in prostate cancer with implications for molecular diagnosis. Modern pathology: an official journal of the United States and Canadian Academy of Pathology. Inc 2007; 20:467 - 73
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009; 458:97 - 101; http://dx.doi.org/10.1038/nature07638; PMID: 19136943
- Palanisamy N, Ateeq B, Kalyana-Sundaram S, Pflueger D, Ramnarayanan K, Shankar S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010; 16:793 - 8; http://dx.doi.org/10.1038/nm.2166; PMID: 20526349
- Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, et al. The genomic complexity of primary human prostate cancer. Nature 2011; 470:214 - 20; http://dx.doi.org/10.1038/nature09744; PMID: 21307934
- Zhang Y, Gong M, Yuan H, Park HG, Frierson HF, Li H. Chimeric transcript generated by cis-splicing of adjacent genes regulates prostate cancer cell proliferation. Cancer Discov 2012; 2:598 - 607; http://dx.doi.org/10.1158/2159-8290.CD-12-0042; PMID: 22719019
- Cui Y, Ying Y, van Hasselt A, Ng KM, Yu J, Zhang Q, et al. OPCML is a broad tumor suppressor for multiple carcinomas and lymphomas with frequently epigenetic inactivation. PLoS One 2008; 3:e2990; http://dx.doi.org/10.1371/journal.pone.0002990; PMID: 18714356
- Kawana Y, Ichikawa T, Suzuki H, Ueda T, Komiya A, Ichikawa Y, et al. Loss of heterozygosity at 7q31.1 and 12p13-12 in advanced prostate cancer. Prostate 2002; 53:60 - 4; http://dx.doi.org/10.1002/pros.10131; PMID: 12210480
- Chêne L, Giroud C, Desgrandchamps F, Boccon-Gibod L, Cussenot O, Berthon P, et al. Extensive analysis of the 7q31 region in human prostate tumors supports TES as the best candidate tumor suppressor gene. Int J Cancer 2004; 111:798 - 804; http://dx.doi.org/10.1002/ijc.20337; PMID: 15252854
- Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol 2003; 162:2029 - 39; http://dx.doi.org/10.1016/S0002-9440(10)64335-0; PMID: 12759258
- Fiucci G, Ravid D, Reich R, Liscovitch M. Caveolin-1 inhibits anchorage-independent growth, anoikis and invasiveness in MCF-7 human breast cancer cells. Oncogene 2002; 21:2365 - 75; http://dx.doi.org/10.1038/sj.onc.1205300; PMID: 11948420
- Horiguchi A, Zheng R, Shen R, Nanus DM. Inactivation of the NF2 tumor suppressor protein merlin in DU145 prostate cancer cells. Prostate 2008; 68:975 - 84; http://dx.doi.org/10.1002/pros.20760; PMID: 18361411
- Hanemann CO. Magic but treatable? Tumours due to loss of merlin. Brain 2008; 131:606 - 15; http://dx.doi.org/10.1093/brain/awm249; PMID: 17940085
- Schoeber JPH, Topala CN, Lee KP, Lambers TT, Ricard G, van der Kemp AWCM, et al. Identification of Nipsnap1 as a novel auxiliary protein inhibiting TRPV6 activity. Pflugers Arch 2008; 457:91 - 101; http://dx.doi.org/10.1007/s00424-008-0494-5; PMID: 18392847
- Lehen’kyi V, Flourakis M, Skryma R, Prevarskaya N. TRPV6 channel controls prostate cancer cell proliferation via Ca(2+)/NFAT-dependent pathways. Oncogene 2007; 26:7380 - 5; http://dx.doi.org/10.1038/sj.onc.1210545; PMID: 17533368
- Barbier O, Bélanger A. Inactivation of androgens by UDP-glucuronosyltransferases in the human prostate. Best Pract Res Clin Endocrinol Metab 2008; 22:259 - 70; http://dx.doi.org/10.1016/j.beem.2008.01.001; PMID: 18471784
- Riento K, Ridley AJ. Rocks: multifunctional kinases in cell behaviour. Nat Rev Mol Cell Biol 2003; 4:446 - 56; http://dx.doi.org/10.1038/nrm1128; PMID: 12778124
- Itoh K, Yoshioka K, Akedo H, Uehata M, Ishizaki T, Narumiya S. An essential part for Rho-associated kinase in the transcellular invasion of tumor cells. Nat Med 1999; 5:221 - 5; http://dx.doi.org/10.1038/5587; PMID: 9930872