Abstract
Recently it has become clear that the cost associated with the Warburg effect, which is inefficient production of ATP, is offset by selective advantages that are produced by resultant intracellular metabolic alterations. In fact tumors may be addicted to the Warburg effect. In addition these alterations result in changes in the extracellular tumor microenvironment that can also produce selective advantages for tumor cell growth and survival. One such extracellular alteration is increased adenosine concentrations that have been shown to impair T cell mediated rejection and support angiogenesis. The expression of the A2A receptor in non-small cell cancer (NSCLC) tissues, cell lines and cancer associated fibroblasts (CAF) was determined by performing immunohistrochemistry and immunoblot analysis. The efficacy of the A2A receptor antagonists in vivo was evaluated in a PC9 xenograft model. To determine the mode of cell death induced by A2A receptor antagonists flow cytometry, immunoblot, and cytotoxic analysis were performed. We found that a significant number of lung adenocarcinomas express adenosine A2A receptors. Antagonism of these receptors impaired CAF and tumor cell growth in vitro and inhibited human tumor xenograft growth in mice. These observations add to the rationale for testing adenosine A2A receptor antagonists as anticancer therapeutics. Not only could there be prevention of negative signaling in T cells within the tumor microenvironment and inhibition of angiogenesis, but also an inhibitory effect on tumor-promoting, immunosuppressive CAFs and a direct inhibitory effect on the tumor cells themselves.
Introduction
In addition to intrinsic properties of the tumor cell, various components of the tumor microenvironment contribute to cancer progression.Citation1-Citation3 One of these is extracellular adenosine, which is present in high concentrations in the tumor microenvironment, a consequence of anaerobic glycolysis in hypoxic regions; preferential utilization of aerobic glycolysis for energy metabolism in non-hypoxic regions (the Warburg effect); and tumor cell expression of the ectonucleotidase CD73 that catabolizes AMP to produce adenosine.Citation4,Citation5 Adenosine is a well recognized regulator of a variety of cellular processesCitation6 mediating its effects through its binding to four G-protein-coupled adenosine receptor subtypes, A1R, A2AR, A2BR, and A3R, expressed in a cell- and tissue-specific manner.Citation7 The differences between the receptors lie in their binding affinity to adenosine, the type of G proteins they recruit, and in the signaling pathways they activate.Citation8 A1R and A3R are Gi protein linked and inhibit adenylyl cyclase, while A2AR and A2BR are Gs linked and stimulate adenylyl cyclase.Citation9 A2AR signaling influences cancer progression in a variety of different ways including interference with immune mediated control of tumors by suppressing T cell activation,Citation10,Citation11 support of angiogenesis,Citation12 and promotion of tumor cell migration.Citation13,Citation14
The discovery of the involvement of adenosine in tumor protection from T cell-mediated destruction came from the knowledge that in non-malignant inflamed tissues, adenosine produced within the hypoxic microenvironment functions to limit the exuberance of the inflammatory response to reduce collateral damage of normal tissue by inflammatory cells. This is due to a direct inhibitory effect on T cells that express A2ARs.Citation11,Citation15 However, the inhibitory effect on T cells does not account for the full protection tumors have from immune mediated rejection. For example, it was shown in mice that fibroblast activation protein-α (FAPα)-expressing CAFs are immunosuppressive. Ablation of these cells in established tumors resulted in rejection mediated by TNF-α and interferon-γ.Citation16 CAFs also enhance tumor-promoting inflammationCitation17 and therefore contribute to tumor progression. Studies have shown that CAFs release TGF-β and VEGF, potential oncogenic signals involved in tumor progression.Citation18,Citation19 In addition, studies done with human prostatic CAFs show that co-culture of CAFs with prostatic epithelial cells dramatically stimulated the growth of the cancer cells ultimately changing their histology.Citation20 Furthermore, in non-small cell lung cancer (NSCLC), it has been shown that co-cultures of normal pulmonary fibroblasts and cancer cells modulate gene expression in fibroblasts, potentially affecting angiogenesis, invasion, cell growth, and survival.Citation21 Therefore, it is important to understand the growth pathways involved in CAFs in an effort to design effective strategies to inhibit their development.
Non-cancer associated fibroblasts are known to be responsive to adenosine in wound healing and inflammation-induced fibrosis with, for example, increased collagen production.Citation22 This, together with the fact that CAFs are exposed to high concentrations of extracellular adenosine led us to hypothesize that adenosine may be a paracrine or autocrine growth factor for CAFs. We also reasoned that adenosine might similarly function as a paracrine growth factor for the tumor cells themselves. We report here that CAFs express A2AR, and found that A2AR antagonists can decrease CAF and tumor cell growth in vitro, and human tumors transplanted into mice. These data supplement the previously described pro-tumorigenic mechanisms of adenosine through its inhibition of antitumor T cells and stimulation of angiogenesis, suggesting that A2A receptor antagonism may be a useful anticancer therapeutic modality.
Results
A2AR is expressed in NSCLC tumors and cell lines
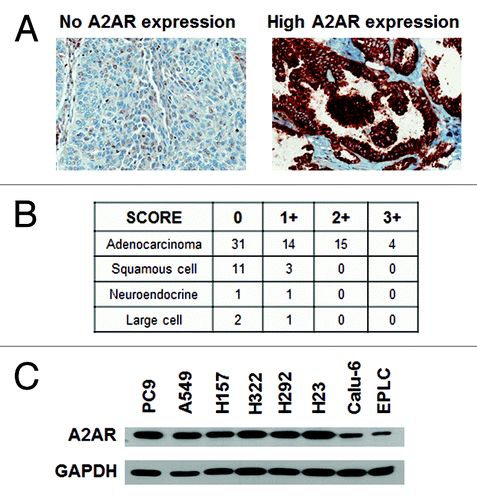
Expression of the A2AR has been reported on monocytes/macrophages, mast cells, granulocytes, lymphocytes, DCs, natural killer (NK) cells, endothelial cells, and airway epithelial cells.Citation12,Citation23 To determine the expression of A2AR in human lung cancers, a TMA was constructed that contained 83 tumors from Moffitt Cancer Center NSCLC patients. Immunohistochemical (IHC) analysis showed expression of the A2AR in 46% (38 out of 83) of the tumors, mainly in the membrane of malignant cells (). provides details on the expression intensity in the different histologic subtypes of NSCLC tumors. A2AR was expressed most commonly in the adenocarcinomas and no substantial correlation was observed between the staining of the A2AR and the stages of the tumor. Additionally, western blot analysis was performed on a panel of 8 NSCLC cell lines which included PC9, A549, H157, H322, H292, H23, Calu-6, and EPLC. shows that all of the NSCLC cell lines express the A2AR at varying levels.
Figure 1. NSCLC cells express A2AR. (A) IHC analysis of A2AR expression in a lung cancer TMA. Representative pictures of 0 and 3+ A2AR expressing tumors are shown. (B) Table showing the expression of A2AR in lung tumors from the TMA. 0, no expression; +1 to +3, increasing expression of A2AR. (C) Immunoblot analysis of 8 NSCLC cell lines show expression of the A2AR.
Cancer-associated fibroblasts (CAFs) express the A2AR
Interestingly, in some of the tumors examined for A2AR expression by IHC, we observed that non-malignant fibroblasts also were positive (). A2AR expression has been previously shown to be expressed by fibroblasts at sites of wound healing or pathologic fibrosis but not by CAFs.Citation22,Citation24,Citation25 To examine this further we established primary cell lines of CAFs from human lung cancer tumors. Portions of lung tumors resected from patients for clinically indicated reasons were mechanically and enzymatically digested, and cultured in DMEM. Within approximately one week, tumor and immune cells died out and fibroblasts survived. Five CAF cell lines were developed which proliferated vigorously for greater than 15 passages. CAFs are commonly identified by their expression of α-SMA and FAP-α 26. α-SMA expression was demonstrated by immunoblot analysis of all 5 CAF cell lines (). To further identify these cells as CAFs, the expression of the FAP-α protein was observed by flow cytometric analysis (). These results confirm that all 5 cell lines are indeed CAFs, and all of these expressed the A2AR ().
Figure 2. CAFs express A2AR. (A) IHC analysis of A2AR expression in a lung cancer TMA. Representative pictures of 0 and 2+ A2AR expressing fibroblasts are shown. Arrow shows the fibroblast in the picture. (B) Table showing the expression of A2AR in the fibroblasts of lung tumors from the TMA. 0, no expression; +1 to +3, increasing expression of A2AR. (C) Immunoblot analysis of A2AR and α-SMA in a panel of 5 CAF. Expression of (D) FAP-α and (E) CD73 were detected by flow cytometric analysis on lymphocytes (dotted line, negative control) and a panel of 5 CAF (all other lines).
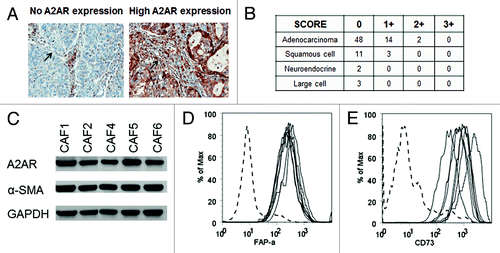
Furthermore, we found that the CAFs expressed CD73 as has been previously describedCitation27 (). Because CD73 is a 5′-ectonucleotidase that cleaves AMP to produce adenosine, it could be an important source of adenosine in the tumor microenvironment. This suggests that CAFs can both produce (Fig. S1) and respond to adenosine suggesting the possibility that adenosine could function as an autocrine growth factor.
A2AR antagonists cause a decrease in the tumor burden in an in vivo model
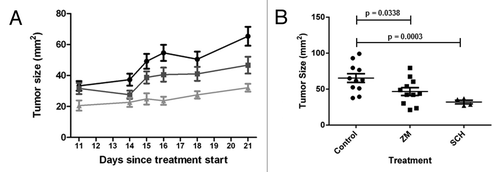
To determine whether A2AR signaling confers an advantage in tumor growth in vivo, PC9 cells were transplanted subcutaneously into nude mice. Mice were treated daily with A2AR antagonists ZM241385 (10 mg/kg) or SCH58261 (2 mg/kg). Animals receiving either antagonist showed a significant decreased in tumor growth (). Notably, when compared with the control group both ZM241385 (P < 0.05) and SCH58261 (P < 0.001) showed statistical significance. These findings strongly support the efficacy of using an A2AR antagonist in decreasing tumor growth in a NSCLC mouse model.
Figure 3. A2AR antagonists decrease tumor growth in a mouse xenograft model. (A) Nude mice (4‒6 wks old) were inoculated s.c. with 7.5 × 106 PC9 cells in the right flank. After 1 week the tumors were palpable and treatment with vehicle control (● 15% DMSO, 15% Cremophore EL, 70% H2O), SCH58261 2 mg/kg (▲), and ZM241385 10 mg/kg (■) started. Drugs were given via i.p. injections for 20 d. (B) A significant decrease in tumor burden was observed with both ZM241385 and SCH58261 treatment.
A2AR antagonists induce apoptotic cell death in NSCLC cells
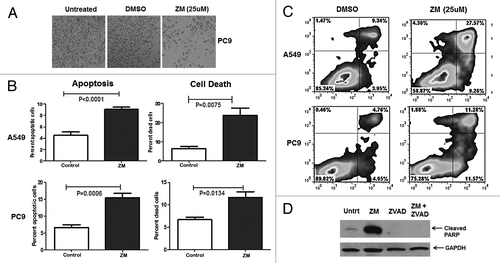
A2AR antagonists have mainly been studied as a means of preventing inhibition of T cells and enhancement of cancer immunotherapy. Our observation that tumor cells express the A2AR together with the knowledge that the adenosine level in the tumor microenvironment is high suggested that adenosine may be a paracrine growth or survival factor for tumor cells. Recently, a study showed that the use of the A2AR antagonist SCH58261 as well as the knockdown of the A2AR decreased cell viability in the NSCLC cell line H1975.Citation28 Even though it has been shown that A2AR antagonists decrease cell viability in NSCLC, the exact mechanism by which this occurs is yet to be elucidated. We found, using HPLC, that the two NSCLC cell lines PC9 and A549 produced extracellular adenosine (3.73 ng/ml and 0.45 ng/ml, respectively) (Fig. S2). Visual analysis of these two cell lines, PC9 () and A549 (Fig. S3), demonstrated a decrease in the number of adherent cells in culture after a 48 h treatment with the A2AR antagonist ZM241385 (25 μM) when compared with untreated and vehicle control (DMSO). Given the high concentration of A2AR antagonist, which was determined by our laboratory, we do not dismiss the possibility that we might non-selectively antagonize other receptors, in fact an even a higher concentration than the one reported in our study was previously used by Escudero et at.Citation29 To determine if A2AR antagonists induce cell death in these cell lines, flow cytometric analysis was performed after staining with APC-annexin V and propidium iodide. A549 and PC9 cells were treated with ZM241385 (25 μM) or vehicle control (DMSO) for 48 h (). In A549 and PC9 cells the apoptotic cells (9% and 15% annexin V-postive cells respectively, P ≤ 0.001) were significantly increased after ZM241385 treatment. The total proportion of dead cells was also increased (23% and 12% annexin V/PI-positve cells respectively, P ≤ 0.05) (). The induction of apoptosis by ZM241385 was further confirmed by immunoblot analysis of PARP cleavage (). In the presence of an apoptotic inducer, full length PARP (116 kDa) is cleaved into an 89 kDa fragment as a result of caspase cleavage. We found that PC9 () and A549 (Fig. S4) cells, in the presence of ZM241385 (25 μM), had an increase in the 89 kDa fragment, when compared with vehicle control (DMSO). The cleavage of PARP induced by ZM241385 was abrogated when the cells were pre-treated for 1 h with the pan-caspase inhibitor Z-VAD.fmk (50 μM). In addition, a caspase 3/7 assay was performed in A549 cells treated with vehicle control (DMSO), ZM241385, and ZM241385 plus Z-VAD.fmk (50μM). Caspase 3/7 activity was reduced by 16-fold in the ZM241385 plus Z-VAD.fmk treatment when compared with ZM241385 alone (Fig. S5). Furthermore, a flow cytometric analysis of the cell cycle was performed in PC9 cells and no apparent difference was observed between vehicle control (DMSO) treated cells and ZM241385 (25 μM) treated cells (data not shown). Moreover, in order to show specificity of ZM241385 at 25 μM, we silenced the A2AR in A549 cells and examined whether the cells showed a similar phenotype as to the one observed when the cells were in the presence of the A2AR antagonist. The data demonstrates (Fig. S6) that when the A2AR is silenced there is an increase in apoptotic cells analogous to that induced by the A2AR antagonist. Thus, we can conclude that A2AR antagonists reduce tumor growth at least in part due to the induction of apoptosis in NSCLC tumor cells. Conversely this is consistent with adenosine serving as a paracrine pro-survival factor.
Figure 4. A2AR antagonists induce apoptotic cell death. (A) Morphological analysis PC9 cells untreated, vehicle control (DMSO), and treated with ZM241385 (25 μM; 48 h). Notice the marked decrease in adhering cells in ZM241385 treated cells. (B) A549 and PC9 cells were treated with vehicle control (DMSO) and ZM241385 (25 μM; 48 h) and the percentage of apoptotic and dead cells determined as described in Materials and Methods. ZM241385 causes significant apoptosis and cell death as compared with vehicle control (P < 0.05). Means ± SD from 6 experiments are presented. (C) Representative of an Annexin V/PI histogram. (D) PC9 cells were treated with vehicle control, ZM241385 (25 μM; 48 h), the pan-caspase inhibitor Z-VAD.fmk (50 μM; 1 h pre-treatment) and ZM241385 in the presence of Z-VAD.fmk and immunoblotting analysis of PARP cleavage was performed. ZM241385 treatment causes significant PARP cleavage, while pre-treatment with Z-VAD.fmk prevented cleavage of PARP.
A2AR antagonists decrease the proliferation of CAFs
Because CAFs contribute to accelerated tumor growth, and they express A2A receptors we hypothesized that the A2AR antagonist-mediated tumor growth inhibition () could be due to CAF growth inhibition in addition to a direct effect on the tumor cells. As we observed with tumor cells, both A2AR antagonists, ZM241385 (25 μM) and SCH58261 (25 μM), could inhibit the growth of CAF cells in vitro. Adenosine was produced by CAFs (1‒1.5 ng/ml by HPLC analysis; Fig. S1), and significant cell growth inhibition (30‒50%) was observed in all 5 CAF cell lines in the presence of ZM241385 (). In the presence of SCH58261 there was some cell growth inhibition (10‒20%) but this was not significant and it was not observed in all 5 CAFs (Fig. S7). Furthermore, treatment of CAF cells with the A2AR agonist CGS21680 (25 μM) increased cell growth in 3 out of 5 CAF cell lines (Fig. S8).
Figure 5. A2AR antagonists induce inhibition of cell proliferation. (A) CAFs were treated with vehicle control (DMSO; D) or ZM241385 (25 μM; Z). After 72 h an MTS assay was performed. ZM241385 significantly inhibited the growth in all 5 CAFs (*P < 0.05). Means +/− SEM from 3 experiments are presented. (B) CAF5 cells were treated with vehicle control (DMSO) and ZM241385 (25 μM; 96 h). ZM241385 does not cause apoptosis as compared with vehicle control as shown in the representative histogram. (C) CAF5 cells were treated with vehicle control (DMSO) and ZM241385 (25 μM; 4 h) and immunoblotting analysis of PARP cleavage was performed. ZM241385 treatment did not cause PARP cleavage. (D) Decrease in cell proliferation (3HdT assay) on CAF5 in the presence of ZM241385 (25 μM; 48 h) is significant when compared with vehicle control (DMSO). Means ± SEM from 3 experiments are presented.
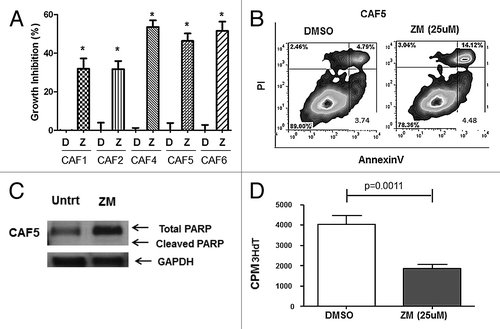
The mechanism whereby A2AR signaling favors cell growth in CAFs differed from what we observed with the tumor cells. Flow cytometric analysis after annexin V/PI staining was performed in CAFs treated with ZM241385 (25 μM) and vehicle control (DMSO) for 96 h. The A2AR antagonist did not induce apoptosis in CAF5 cells, which had no increase in annexin V positive cells, when compared with vehicle control (representative histogram in ). To further confirm that ZM241385 was not inducing apoptotic cell death in CAFs, an immunobloting analysis of PARP cleavage was performed. We were able to observe no cleaved PARP (89 kDa fragment) in CAFs treated with ZM241385 for 4 h (). Immunoblotting analysis of PARP cleavage was also performed at 24 and 48 h (data not shown) but no total or cleaved PARP was observed at these time points. Since no apoptotic cell death was observed, but there was a decrease in CAF growth we hypothesized that A2AR antagonists decrease cell proliferation in the CAFs. Tritiated thymidine incorporation assays showed a decrease in CAF proliferation (P ≤ 0.05) when CAFs were treated with ZM241385 (25 μM for 48 h) when compared with vehicle control (, only CAF5 is shown).
Discussion
The metabolic alterations responsible for the Warburg effect and other metabolic alterations produce a selective advantage for tumor growth.Citation30 So despite there being a relative cost (inefficient production of ATP), tumor cells can be “addicted” to aerobic glycolysis. In addition to influencing intracellular processes, these metabolic alterations also result in alteration of the extracellular tumor microenvironment. For example elevated levels of lactate that can provide a selective advantage for tumor cell growth.Citation31 Another such extracellular alteration is an increase in adenosine concentration due to excess AMP and the expression of CD73 by tumor cells and CAFs. Adenosine modulates the function of a variety of different cell types through its binding to several cell surface receptors.Citation9 In tumors, extracellular adenosine is pro-tumorigenic through its known ability to inhibit T cell function and support angiogenesis.Citation6 The latter is due to a direct effect on blood vessels,Citation32 but may also be due to the fact that A2AR signaling in macrophages (another prominent component of the tumor microenvironment) increases VEGF production.Citation33
It was previously reported that A2A receptors can be expressed in human lung cancers, with expression on endothelial cells and tumor macrophages in the stroma.Citation12 We discovered through an immunohistochemical analysis of 83 tumors that CAFs and tumor cells also express the A2A receptor, most notably in adenocarcinomas. Fibroblasts at sites of wound healingCitation25 and pathologically fibrosing conditionsCitation22,Citation24 share many characteristics with CAFs, for example they express FAP-α whereas fibroblasts in normal tissue do not.Citation26 Because it is known that adenosine signaling through the A2AR on these fibroblasts promotes wound healing,Citation22 we hypothesized that adenosine signaling may similarly produce a selective advantage to CAFs which promote tumor growth. We found that adenosine was produced by tumor cells and CAFs in vitro, and antagonism of the A2AR inhibited the growth of both of these cell types in vitro. Interestingly, the CAFs express CD73Citation27 () suggesting that CAFs both produce and respond to adenosine, and therefore can be considered an autocrine growth factor as well as a paracrine growth factor for tumor cells.
Clearly A2AR signaling is only partly responsible for tumor growth as induction of death in tumor cells and inhibition of CAF proliferation was only partial, and in the xenograft model tumor progression was only slowed, not stopped. Combinations of A2AR antagonism with chemotherapy, radiation or other apoptosis-inducing targeted therapies may be additive or synergistic. Although not tested in our xenograft model, we would predict that there would be a greater magnitude of the A2AR antagonist effect in a syngeneic immunocompetent model, due to the known ability of A2AR antagonists to prevent the negative impact of adenosine on T cells. Furthermore, our data suggest that A2AR antagonist inhibition of CAFs, which are themselves known to be immunoinhibitoryCitation5 would result in improved immune-mediated rejection of tumors.
We have not yet determined the relevant downstream signaling pathways linked to the A2AR in CAFs and tumor cells. They will likely differ, as the apparent mechanism of growth inhibition produced by A2AR antagonists is through apoptosis in tumor cells and inhibition of proliferation in the CAFs. An understanding of the signaling pathways involved could guide more rational combinations of targeted agents with A2AR antagonism to enhance tumor cell and CAF growth inhibition.
Our work contributes to the growing body of evidence that targeting signaling through the adenosine A2A receptor may be a useful, novel anti-cancer therapeutic modality. Multiple mechanisms could contribute to A2AR antagonism-induced tumor regression including: (1) enhanced T cell mediated killing by lessening the immunosuppressive microenvironment by both removing the direct inhibitory signal in T cells, and inhibiting the growth of immunosuppressive CAFs; (2) inhibition of angiogenesis; (3) reduced VEGF production by tumor associated macrophages; (4) inhibition of growth-promoting CAFs; and (5) direct tumor cell growth inhibition. A reduction in A2AR signaling in tumors could be accomplished by either reducing the extracellular microenvironmental adenosine concentration, or by inhibiting signaling by the A2AR. The former could be accomplished by treating patients with, for example an inhibitory monoclonal antibody directed at the AMP-degrading ectonucleotidase CD73.Citation34,Citation35 Inhibition of A2AR signaling could be accomplished with the use A2AR antagonists. These are currently being developed for the treatment of Parkinson disease.Citation36
Materials and Methods
Cell culture and reagents
Primary human fibroblasts were isolated from portions of lung tumors resected from patients for clinically indicated reasons. The tumors were mechanically and enzymatically (CPD; collagenase, protease and DNase) digested and the cells were cultured in DMEM 10% FBS, PenStrep, and l-glutamine at 37 °C. After one week of culture, tumor and immune cells died; however the cancer-associated fibroblasts (CAFs) proliferated vigorously and survived for greater than 15 passages. A549 and PC9 cells were purchased from ATCC and cultured in RPMI 10% FBS, PenStrep, and l-glutamine at 37°C.
Adenosine agonists and antagonists
The following adenosine agonists and antagonists were used: A2AR agonist 2-p-(2-Carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride hydrate (CGS21680 hydrochloride hydrate, Sigma-Aldrich); A2AR antagonist 4-(2-[7-Amino-2-(2-furyl)[1,2,4]tri azolo[2,3-a][1,3,5]triazin-5-ylamino]ethyl)phenol (ZM241385, Tocris); and A2AR antagonist 2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5c] pyrimidin-5-amine (SCH58261, Tocris).
Western blots
Whole cell lystates were collected in 1× CHAPS buffer (Cell Signaling), from a panel of NSCLC cell lines as well as human CAF lines. Protein concentrations were quantified using the Bio-Rad Protein Assay dye. Equal amounts of protein (10 μg) were loaded into the wells of a 10% SDS-PAGE gel and resolved at 100 V for 90 min. Proteins were then transferred to a PVDF membrane, blocked and then probed for A2AR at 1:500, overnight incubation at 4 °C (Enzo Life Sciences; SA-654), α-SMA at 1:25 000, 1 h at room temperature (Abcam; ab32575), PARP at 1:2000, overnight incubation at 4 °C (Cell Signaling; 9542), or GAPDH at 1:2000, 30 min at room temperature (Cell Signaling; 2118S).
Immunohistochemistry (IHC)
A human NSCLC tissue microarray (TMA), was constructed from 83 tissue cores from NSCLC patients resected at the H. Lee Moffitt Cancer Center. The TMA was stained using a Ventana Discovery XT automated system (Ventana Medical Systems) as per manufacturer's protocol with proprietary reagents. Briefly, slides were deparaffinized on the automated system with EZ Prep solution (Ventana). Heat-induced antigen retrieval method was used in Cell Conditioning 1 (Ventana). TMA slides were incubated with a rabbit primary antibody for A2AR (Enzo Life Sciences; SA-654) at a concentration of 1:50 in Dako antibody diluent (Dako) and incubated for 1 h. The Ventana OmniMap Anti-Rabbit Secondary Antibody was used for 20 min. The detection system used was the Ventana ChromoMap kit and slides were then counterstained with Hematoxylin. Slides were then dehydrated and coverslipped as per normal laboratory protocol. The immunostained TMA was scored for A2AR immunoreactivity using a 4-tier scoring system (0 = negative, 1 = weak, 2 = moderate, 3 = strong) was used to evaluate staining intensity.
Morphologic analysis
To examine the morphology of cultured cells after treatment with an A2AR antagonist 3 × 105 cells/well PC9 or A549 cells were seeded in a 6-well culture plate in RPMI. After 24 h the cells were treated with 25 μM of ZM241385 or vehicle control for 48 h. Pictures were taken under a brightfield light with an automated Zeiss Observer Z.1 inverted microscope through a 10×/0.3NA objective. Images were produced using the AxioCam MRm CCD camera and Axiovision version 4.7 softer suite (Carl Zeiss Inc.).
AnnexinV/PI analysis
To examine apoptotic cell death, 3 × 105 cells/well CAFs or NSCLC cells were seeded onto a 6-well culture plate in DMEM or RPMI. After 24 h the cells were treated with 25 μM ZM241385 or vehicle control (DMSO). Supernatant and cells were collected 24, 48, 72, and 96 h later. The adherent cells were removed from the plate using 500 μl Accutase (Sigma) and allowed to rest in complete media for 15 min. Cells were suspended in 100 μl Annexin V staining buffer (Invitrogen) with 5 μl Annexin V–APC (BD Bioscience) at room temperature for 20 min. After staining, cells were diluted in an additional 300 μl binding buffer and PI was added at 1.25 μg/ml (Sigma). Fluorescence was measured using a FACSCalibur (BD Bioscience) and data was analyzed using FlowJo software (Treestar). Annexin V positive, PI negative cells were identified as early apoptotic.
Flow cytometry
The fibroblasts’ identity as CAFs was confirmed by expression of fibroblast activation protein-α (FAP-α). Briefly, the cells were stained for 30 min at room temperature with anti-FAP-α (R&D Systems; MAB3715), washed and stained with a rabbit anti-mouse Alexa Fluor 488 (Molecular Probes; A11059). In addition, CAFs were stained with anti-CD73 (BD Pharmigen; 550257) to observe if they expressed this 5′ ectonucleotidase. Fluorescence was measured using a FACSCalibur (BD Bioscience) and data were analyzed using FlowJo software (Treestar). Lymphocytes were used as a negative control since they do not express FAP-α or CD73.
Cell viability assay
The CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega) was used to examine cell viability and was performed according to the manufacturer’s protocol. Briefly, cells were seeded into a 96-well plate at 5 × 103 cells/well. They were treated with escalating doses of SCH58261, ZM241385, or CGS21680 for 72 h. After the treatment period, 20 μl of the MTS solution was added and incubated at 37 °C for 1 h. Plates were read at 490 nm in a BioTek EL808 microplate reader. Treatments were compared with their vehicle control.
Proliferation analysis
Cell proliferation was assessed after 48 h of ZM241385 (25 μM) treatment by incubating overnight with 1 μCi of [3H]TTP (diluted in 20 ul of complete DMEM medium). Cells were then harvested onto glass fiber filters using a cell harvester (Filtermate; Packard Bioscience Co.) and radioactivity was measured with MicroScint™ PS solution (Packard Bioscience Co.) using a Top Count® NXT™ (Packard Bioscience Co.) microplate scintillation counter.
Caspase 3/7 activity assay
The CellPlayer 96-Well Kinetic Caspase 3/7 Reagent (Essen Bioscience) was used to assess caspase 3/7 activity and was performed according to the manufacture’s protocol. Briefly, A549 cells were seeded in a 96-well plate at 5 × 103 cells/well. They were pre-treated with Z-VAD.fmk (50 μM) and then treated with ZM241385 (25μM) for 48 h. After treatment, the CellPlayer 96-Well Kinetic Caspase 3/7 Reagent was added to the cells at a final concentration of 5μM. The plate was placed on the IncuCyte™ FLR in which the caspase 3/7 activity was monitored in a non-invasive form. The first and last image of each image set was extracted for analysis with Definiens Developer version 1.5 (Definiens Inc.). Caspase 3/7 positive cells were identified and segmented with an auto-threshold segmentation algorithm. This segmentation was further refined by object size and finally the number of Caspase 3/7 cells was enumerated.
Mouse model
PC9 cells (7.5 × 106) were injected s.c. (subcutaneous) into 4‒6 week old athymic nude mice (NCI). When tumors were palpable, mice were randomly allocated into three groups and treated by daily i.p. (intraperitoneal) injections of ZM241385 (10 mg/kg), SCH58261 (2 mg/kg) both in carrier solution 15% DMSO, 15% Cremophore EL, 70% H2O to a total injection volume of 0.1 ml or vehicle (carrier alone) for 20 d. The experiment was terminated when tumors became ulcerated. Animal experiments were performed according to a protocol approved by the Institutional Animal Care and Use Committee of the University of South Florida.
LC/MS/MS for adenosine concentration determination
Calibration and quality control (QC) samples were made by adding known amounts of adenosine to blank matrix. All calibration, QC and unknown cell line samples were prepared in the following manner. Wells of a 96-well plate were filled with 50 μl of media followed by 10 μl of the internal standard (adenosine-13C5). Next, 250 μl of 0.1% acetic acid was added to each. The plate was sealed and vortexed for 1 min. Ten microliters of this was injected into an Accela UHPLC (Thermo Electron) coupled to a Thermo TSQ Quantum tandem mass spectrometer. Gradient elution was achieved with mobile phases of water and methanol, both containing 0.1% acetic acid. The flow rate was 0.4 ml/min with a run time of 6.5 min. A Zorbax SB-C18 reverse phase column 2.1 × 50 mm, 3.5 μm (Agilent Technologies) was used to separate compounds and the column eluate entered the MS system by way of a heated electrospray ionization source (H-ESI). Selected reaction monitoring (SRM) of the target compound and internal standard was performed. The following SRM transitions were monitored for quantitation: 268.0 → 119.0 for adenosine and 273.0 → 119.0 for adenosine 13C5. The resulting chromatographic peaks were integrated by Thermo Xcaliber software. Linear regression was used to form the calibration curve from standards; QCs were checked against the regression line and unknowns were plotted for back calculation of the raw concentrations. The assay has a linear range from 1–2500 ng/ml. Inter- and intra-assay variability was less than 8% with a relative mean error of less than 13%. There was no significant ion suppression or enhancement to report based on the retention times and the dilutions utilized.
Silencing of A2AR
To silence the A2AR, A549 cells (1.75 × 105) were plated in a 6-well plate. After 24 h, cells were transfected using Lipofectamine® RNAiMAX transfection reagent (Invitrogen). Briefly, 4 μl of the transfection reagent was added to 250μl of Opti-MEM (Invitrogen) as well as 250 pmol of A2AR siRNA (Silencer® Select Validated siRNA, Invitrogen) to 250 μl of Opti-MEM. Solutions were incubated for 5 min at room temperature and then mixed together and incubated for 20 min at room temperature. The final solution was added dropwise to the well and incubated at 37 °C for 4 h. The media was changed and incubated for another 48 h before the RNA was extracted.
Quantitative real time (qRT)-PCR analysis
Total RNA was extracted using TriZol reagent (Invitrogen) and cDNA obtained with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Target mRNA was quantified using the A2AR TaqMan® Gene Expression Assays (Applied Biosystems) and the 7900HT Fast Real-Time PCR System (Applied Biosystems). PCR amplification cycling parameters were 3 min 95 °C, 15 sec 95 °C, 30 sec 60 °C 40 reps, 1 min 95 °C. Single product amplification was confirmed by melting curve analysis. Quantification is expressed in arbitrary units and target mRNA levels were normalized to GAPDH expression using the method of Pfaffl.Citation37
Statistical analysis
Data represent mean ± SEM. Statistical calculations were performed using the Student t-test. Statistical significance was accepted for P values less than 0.05.
| Abbreviations: | ||
| A1R | = | adenosine A1 receptor |
| A2AR | = | adenosine A2A receptor |
| A2BR | = | adenosine A2B receptor |
| A3R | = | adenosine A3 receptor |
| CAF | = | cancer associated fibroblast |
| CGS21680 | = | 2-p-(2-Carboxyethyl)phenethylamino-5′-N-ethylcarboxamidoadenosine hydrochloride hydrate |
| CPD | = | collagenase protease DNase |
| FAPα | = | fibroblast activation protein alpha |
| IHC | = | immunohistochemical |
| i.p. | = | intra-peritoneal |
| NK | = | natural killer |
| NSCLC | = | non small cell lung cancer |
| s.c. | = | subcutaneous |
| SCH58261 | = | 2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5c] pyrimidin-5-amine |
| TMA | = | tissue microarray |
Additional material
Download Zip (1.7 MB)Acknowledgments
This work has been supported in part by the Flow Cytometry Core Facility, the Translational Research Core’s Clinical Pharmacology Laboratory and the Analytic Microscopy Facility at the H. Lee Moffitt Cancer Center and Research Institute, a comprehensive cancer center designated by the National Cancer Institute.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Bremnes RM, Dønnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, et al. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol 2011; 6:209 - 17; http://dx.doi.org/10.1097/JTO.0b013e3181f8a1bd; PMID: 21107292
- Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S, et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med 2012; 12:24; http://dx.doi.org/10.1186/1471-2466-12-24; PMID: 22694981
- Zhao XY, Zeng X, Li XM, Wang TL, Wang BE. Pirfenidone inhibits carbon tetrachloride- and albumin complex-induced liver fibrosis in rodents by preventing activation of hepatic stellate cells. Clin Exp Pharmacol Physiol 2009; 36:963 - 8; http://dx.doi.org/10.1111/j.1440-1681.2009.05194.x; PMID: 19413596
- Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res 2010; 70:2245 - 55; http://dx.doi.org/10.1158/0008-5472.CAN-09-3109; PMID: 20179192
- Stagg J, Smyth MJ. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010; 29:5346 - 58; http://dx.doi.org/10.1038/onc.2010.292; PMID: 20661219
- Choi K, Lee K, Ryu SW, Im M, Kook KH, Choi C. Pirfenidone inhibits transforming growth factor-β1-induced fibrogenesis by blocking nuclear translocation of Smads in human retinal pigment epithelial cell line ARPE-19. Mol Vis 2012; 18:1010 - 20; PMID: 22550395
- Furuta S, Watanabe L, Doi S, Horiuchi H, Matsumoto K, Kuzumaki N, et al. Subdiaphragmatic vagotomy increases the sensitivity of lumbar Aδ primary afferent neurons along with voltage-dependent potassium channels in rats. Synapse 2012; 66:95 - 105; http://dx.doi.org/10.1002/syn.20982; PMID: 21905127
- Gessi S, Merighi S, Sacchetto V, Simioni C, Borea PA. Adenosine receptors and cancer. Biochim Biophys Acta 2011; 1808:1400 - 12; http://dx.doi.org/10.1016/j.bbamem.2010.09.020; PMID: 20888788
- Fishman P, Bar-Yehuda S, Synowitz M, Powell JD, Klotz KN, Gessi S, et al. Adenosine receptors and cancer. Handb Exp Pharmacol 2009; 193:399 - 441; http://dx.doi.org/10.1007/978-3-540-89615-9_14; PMID: 19639290
- Visner GA, Liu F, Bizargity P, Liu H, Liu K, Yang J, et al. Pirfenidone inhibits T-cell activation, proliferation, cytokine and chemokine production, and host alloresponses. Transplantation 2009; 88:330 - 8; http://dx.doi.org/10.1097/TP.0b013e3181ae3392; PMID: 19667934
- Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 2006; 103:13132 - 7; http://dx.doi.org/10.1073/pnas.0605251103; PMID: 16916931
- Ahmad A, Ahmad S, Glover L, Miller SM, Shannon JM, Guo X, et al. Adenosine A2A receptor is a unique angiogenic target of HIF-2alpha in pulmonary endothelial cells. Proc Natl Acad Sci U S A 2009; 106:10684 - 9; http://dx.doi.org/10.1073/pnas.0901326106; PMID: 19541651
- Waller JR, Murphy GJ, Metcalfe MS, Bicknell GR, Saunders RN, Margolin SB, et al. Effects of pirfenidone on vascular smooth muscle cell proliferation and intimal hyperplasia following arterial balloon injury. Transplant Proc 2001; 33:3816 - 8; http://dx.doi.org/10.1016/S0041-1345(01)02615-X; PMID: 11750625
- Torigoe K, Nakahara K, Rahmadi M, Yoshizawa K, Horiuchi H, Hirayama S, et al. Usefulness of olanzapine as an adjunct to opioid treatment and for the treatment of neuropathic pain. Anesthesiology 2012; 116:159 - 69; http://dx.doi.org/10.1097/ALN.0b013e31823c7e56; PMID: 22126917
- Scheibner KA, Boodoo S, Collins S, Black KE, Chan-Li Y, Zarek P, et al. The adenosine a2a receptor inhibits matrix-induced inflammation in a novel fashion. Am J Respir Cell Mol Biol 2009; 40:251 - 9; http://dx.doi.org/10.1165/rcmb.2008-0168OC; PMID: 18703794
- Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010; 330:827 - 30; http://dx.doi.org/10.1126/science.1195300; PMID: 21051638
- Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010; 17:135 - 47; http://dx.doi.org/10.1016/j.ccr.2009.12.041; PMID: 20138012
- Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer 2008; 99:1375 - 9; http://dx.doi.org/10.1038/sj.bjc.6604662; PMID: 18797460
- Saigusa S, Toiyama Y, Tanaka K, Yokoe T, Okugawa Y, Fujikawa H, et al. Cancer-associated fibroblasts correlate with poor prognosis in rectal cancer after chemoradiotherapy. Int J Oncol 2011; 38:655 - 63; http://dx.doi.org/10.3892/ijo.2011.906; PMID: 21240461
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59:5002 - 11; PMID: 10519415
- Fromigué O, Louis K, Dayem M, Milanini J, Pages G, Tartare-Deckert S, et al. Gene expression profiling of normal human pulmonary fibroblasts following coculture with non-small-cell lung cancer cells reveals alterations related to matrix degradation, angiogenesis, cell growth and survival. Oncogene 2003; 22:8487 - 97; http://dx.doi.org/10.1038/sj.onc.1206918; PMID: 14627989
- Cronstein BN. Adenosine receptors and fibrosis: a translational review. F1000 Biol Rep 2011; 3:21; http://dx.doi.org/10.3410/B3-21; PMID: 22003368
- Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ 2007; 14:1315 - 23; http://dx.doi.org/10.1038/sj.cdd.4402132; PMID: 17396131
- Fernández P, Trzaska S, Wilder T, Chiriboga L, Blackburn MR, Cronstein BN, et al. Pharmacological blockade of A2A receptors prevents dermal fibrosis in a model of elevated tissue adenosine. Am J Pathol 2008; 172:1675 - 82; http://dx.doi.org/10.2353/ajpath.2008.070952; PMID: 18467695
- Montesinos MC, Shaw JP, Yee H, Shamamian P, Cronstein BN. Adenosine A(2A) receptor activation promotes wound neovascularization by stimulating angiogenesis and vasculogenesis. Am J Pathol 2004; 164:1887 - 92; http://dx.doi.org/10.1016/S0002-9440(10)63749-2; PMID: 15161625
- Ostman A, Augsten M. Cancer-associated fibroblasts and tumor growth--bystanders turning into key players. Curr Opin Genet Dev 2009; 19:67 - 73; http://dx.doi.org/10.1016/j.gde.2009.01.003; PMID: 19211240
- Paunescu V, Bojin FM, Tatu CA, Gavriliuc OI, Rosca A, Gruia AT, et al. Tumour-associated fibroblasts and mesenchymal stem cells: more similarities than differences. J Cell Mol Med 2011; 15:635 - 46; http://dx.doi.org/10.1111/j.1582-4934.2010.01044.x; PMID: 20184663
- Kuzumaki N, Suzuki A, Narita M, Hosoya T, Nagasawa A, Imai S, et al. Multiple analyses of G-protein coupled receptor (GPCR) expression in the development of gefitinib-resistance in transforming non-small-cell lung cancer. PLoS One 2012; 7:e44368; http://dx.doi.org/10.1371/journal.pone.0044368; PMID: 23144692
- Escudero C, Bertoglia P, Hernadez M, Celis C, Gonzalez M, Aguayo C, et al. Impaired A2A adenosine receptor/nitric oxide/VEGF signaling pathway in fetal endothelium during late- and early-onset preeclampsia. Purinergic Signal 2013; 9:215 - 26; http://dx.doi.org/10.1007/s11302-012-9341-4; PMID: 23179048
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85 - 95; http://dx.doi.org/10.1038/nrc2981; PMID: 21258394
- Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol 2010; 8:88; http://dx.doi.org/10.1186/1741-7007-8-88; PMID: 20598111
- Desai A, Victor-Vega C, Gadangi S, Montesinos MC, Chu CC, Cronstein BN. Adenosine A2A receptor stimulation increases angiogenesis by down-regulating production of the antiangiogenic matrix protein thrombospondin 1. Mol Pharmacol 2005; 67:1406 - 13; http://dx.doi.org/10.1124/mol.104.007807; PMID: 15673602
- Ramanathan M, Pinhal-Enfield G, Hao I, Leibovich SJ. Synergistic up-regulation of vascular endothelial growth factor (VEGF) expression in macrophages by adenosine A2A receptor agonists and endotoxin involves transcriptional regulation via the hypoxia response element in the VEGF promoter. Mol Biol Cell 2007; 18:14 - 23; http://dx.doi.org/10.1091/mbc.E06-07-0596; PMID: 17065555
- Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A 2010; 107:1547 - 52; http://dx.doi.org/10.1073/pnas.0908801107; PMID: 20080644
- Stagg J, Beavis PA, Divisekera U, Liu MC, Möller A, Darcy PK, et al. CD73-deficient mice are resistant to carcinogenesis. Cancer Res 2012; 72:2190 - 6; http://dx.doi.org/10.1158/0008-5472.CAN-12-0420; PMID: 22396496
- Park A, Stacy M. Istradefylline for the treatment of Parkinson’s disease. Expert Opin Pharmacother 2012; 13:111 - 4; http://dx.doi.org/10.1517/14656566.2012.643869; PMID: 22149371
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29:e45; http://dx.doi.org/10.1093/nar/29.9.e45; PMID: 11328886