Abstract
The anti-tumor effects of arsenic trioxide (ATO) were well established in acute promyelocytic leukemia, but not in renal cell carcinoma (RCC). Recent evidences indicate that galectin-3 (Gal-3) plays an anti-apoptotic role in chemotherapy induced tumor cell death. This study was intended to clarify the exact roles of Gal-3 performed in ATO-induced apoptosis in RCC cells. Weak apoptosis was observed in Gal-3-positive RCC cells (Caki-1, Caki-2, 786-0, and ACHN) following ATO treatment. However, ATO treatment upregulated Gal-3 expression concurrently caused a Synexin-cooperated translocation of Gal-3 from the nucleus to the cytoplasm. Gal-3-knockdown cells were more sensitive to ATO treatment as indicated by a strong mitochondria-dependent apoptosis following ATO treatment. Meanwhile, Gal-3 was found to inhibit ATO-induced apoptosis through enhancing Bcl-2 expression and stabilizing mitochondria. To confirm the results obtained from genetic method, we employed a Gal-3 inhibitor, modified citrus prectin (MCP), and co-treated the RCC cells with ATO. The cells showed an increased apoptosis in the syngeneic application of Gal-3 inhibition and ATO compared with ATO application alone. Based on these results, we conclude that Gal-3 inhibition sensitizes human renal cell carcinoma cells to ATO treatment through increasing mitochondria-dependent apoptosis. Our studies implicate synergetic application of ATO and Gal-3 inhibition as a potential strategy for RCC treatment.
Introduction
Arsenic trioxide (ATO) is an FDA-approved drug, which has been used as a component of acute promyelocytic leukemia (APL) therapy. The clinical complete remission rate is about 65.6–84%, and more than 28.2% patients survive at least 10 y following ATO treatment.Citation1 Anti-tumor activity of ATO is mainly though apoptosis-eliciting, which has been verified in a series of malignant tumor cells, including in non-APL acute myeloid leukemia cells,Citation2 myeloma cells,Citation3 chronic myeloid leukemia cells,Citation4 and various solid tumor cells from head and neck,Citation5 gallbladder,Citation6 prostate/ovarian carcinomas,Citation7,Citation8 and glioma.Citation9 In agreement, ATO was reported to elicit renal carcinoma cells (RCCs) apoptosis.Citation10 However, these tumor cells show a low sensitivity to ATO, as high concentration (normally 10-fold) of ATO is required to induce apoptosis.Citation11 It is unlikely to apply ATO to treat these tumors in clinical practice because of the toxicity due to the increased concentration. In phase two clinical trials, ATO just partially inhibits RCC.Citation12 The limitation of ATO for RCCs treatment may be partially due to the enhanced tumor resistance to apoptosis. Hence, it is of great significance to find out the potential molecular mechanism underlying the tumor resistance to ATO.
Galectin-3 (Gal-3), a specific member of β-galectin family is well-known for its multi-function involved in tumor aggressiveness, including adhesion,Citation13 proliferation,Citation14 differentiation,Citation15 angiogenesis,Citation16 and metastasis.Citation17 Recently, it has been reported that Gal-3 performs an anti-apoptotic role in response to chemotherapeutic drugs.Citation17-Citation19 Gal-3 expression was enhanced in the survived cells after treatment of myeloid leukemia K562 cells with chemotherapeutic drugs.Citation20 Gal-3 translocated from the nucleus to the cytoplasm following chemotherapeutic treatment and enriched in mitochondrial membrane, which exempted mitochondria from damage through inhibiting the disruption of mitochondria membrane potential.Citation21 Importantly, the replacement of amino acid possessing phosphorylation site (Ser-6) with one without phosphorylation site, Gal-3 lost its ability to shuttle between the nucleus and the cytoplasm, contributing to increased apoptosis.Citation19 In the present study, we evaluated the role of Gal-3 in ATO-induced apoptosis in RCC cell lines using two approaches: Gal-3 knockdown by shRNA and blockage of Gal-3 functions using modified citrus pectin (MCP). Our results present that Gal-3 inhibition sensitizes human renal cell carcinoma cells to arsenic trioxide treatment through increasing mitochondria-dependent apoptosis.
Results
The effects of ATO on apoptosis and Gal-3 expression in RCCs
Initially, we tested whether ATO induced apoptosis in RCC cell lines. As evidenced by Park et al.,Citation10 ATO at the concentration range of 2.5–10 µM was tested in RCC cells. In this study, we treated four RCC cells (786-0, ACHN, Caki-1, and Caki-2) with a concentration of 2.5 µM ATO for 72 h and compared the apoptosis using PI and Annexin V-FITC double staining. As shown in , ATO application triggered apoptosis in ACHN and Caki-2 cells, but not in Caki-1 and 786-0 cells.
Figure 1. ATO has a weak apoptosis effect in renal cell carcinoma cells and upregulates galectin-3 (Gla-3) protein expression. (A) ATO treatment (2.5 μM) did not significantly induce apoptosis in RCC cells, especially in Caki-1 and 786-0 cells. Cells were treated with ATO followed by staining with PI and Annexin V as described in Materials and Methods. Apoptotic analysis was applied to cells before (top panel) and after (bottom panel) ATO treatment using flow cytometer, and the percentage of each population is labeled in the figure. (B) ATO treatment (2.5 μM) increased endogenous galectin-3 expression in 786-0, ACHN, Caki-1 and Caki-2 cells. Cells were treated with or without ATO as described in Materials and Methods. Anti-Gal-3 antibody (1:1000) was used to detect endogenous Gal-3 proteins. GAPDH was used as loading control. Quantifications of respective Gal-3 levels are presented based on at least three-time repeats. *P < 0.05, **P < 0.01.
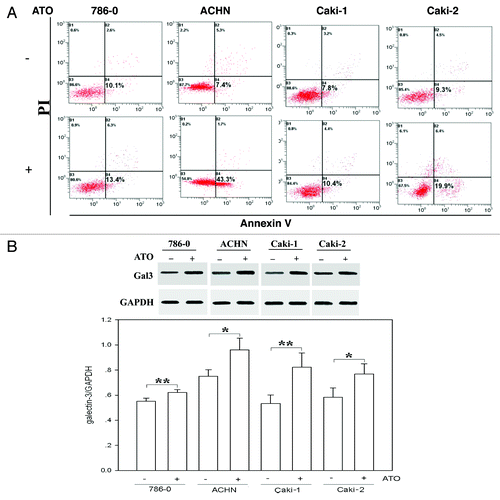
In order to determine whether Gal-3 expression was affected in ATO-induced apoptosis, we compared the protein level of Gal-3 before and after ATO treatment in four RCC cell lines under hypoxia conditions. The results showed that Gal-3 protein was originally expressed in all four cell lines. Moreover, in comparison with pretreatment, Gal-3 level was significantly upregulated following ATO treatment (P < 0.01) ().
ATO affects subcellular distribution of Gal-3
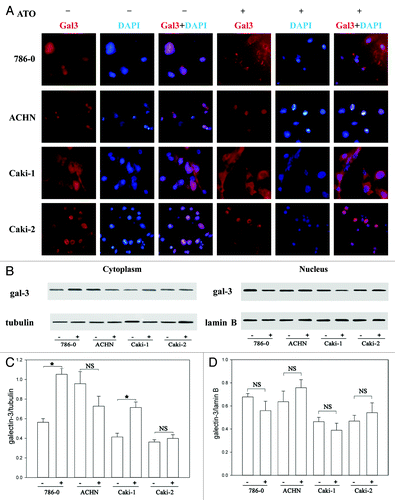
Previous study showed that the translocation of Gal-3 from the nucleus to the cytoplasm contributed to anti-apoptotic activity of Gal-3.Citation19 Accordingly, we detected the subcellular distribution of Gal-3 before and after ATO treatment using immunofluorescence. Gal-3 was uniformly distributed in the nucleus as well as in the cytoplasm in all cell lines studied (). However, after treatment with 5 μM ATO for 72 h, the nucleus Gal-3 was decreased in both Caki-1 and 786-0 cells, while cytoplasmic Gal-3 was obviously increased (). Statistical analysis showed that the increase of cytoplasmic Gal-3 following ATO treatment was significant in both of 786-0 and Caki-1 cells (P < 0.05). By contrast, Gal-3 distribution was not obviously affected in Caki-2 cells and ACHN cells. The immunochemistry results were further confirmed by western blotting ().
Figure 2. ATO induces the translocation of Gal-3 from the nucleus to the cytoplasm. (A) Gal-3 distribution before and after ATO treatment. The red signal shows Gal-3, and the blue one is nuclei. The staining results in 786-0, ACHN, Caki-1, and Caki-2 cells are presented from top to bottom, respectively. Gal-3 transferred from the nucleus to the cytoplasm in Caki-1 and 786-0 cells. (B and C) Western blotting was used to confirm the translocation of Gal-3. The anti-Gal-3 antibodies were used to detect Gal-3 in the nucleus and cytoplasm. Quantifications of respective Gal-3 levels are presented based on at least three-time repeats (*P < 0.05).
Synexin is co-translocated with Gal-3 in RCC cells following ATO treatment
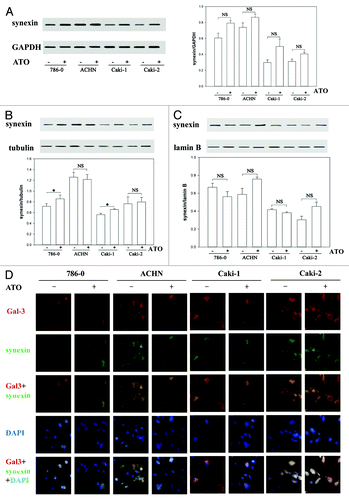
Synexin was reported to regulate Gal-3 translocation from the nucleus to the cytoplasm.Citation21 Hence, we intended to determine whether synexin was co-translocated with Gal-3 in RCCs in response to ATO treatment. Western blotting results showed that the total amount of synexin protein was not obviously changed in four RCC cell lines after ATO treatment (). However, consistent with Gal-3 translocation, the translocation of synexin from nucleus to cytoplasm was also found in Caki-1 and 786-0 cells (). Consistent with the results from western blotting measurements, immunochemistry data further verified the translocation of Synexin from nucleus to cytoplasm following ATO treatment ().
Figure 3. Synexin is co-translocated with Gal-3 in RCC cells. (A) The total protein levels of synexin were the same before and after ATO treatment in all RCC cells tested. (B) ATO triggered the translocation of synexin from the nucleus to the cytoplasm in Caki-1 and 786-0 cells. The protein levels of synexin in the nucleus and cytoplasm were determined using western blotting. (C) Immunofluerescence experiment confirmed that Gal-3 (red) was co-translocated with synexin (green) in RCC cells.
Knockdown of Gal-3 increases the sensitivity of Caki-1 cells to ATO-induced apoptosis
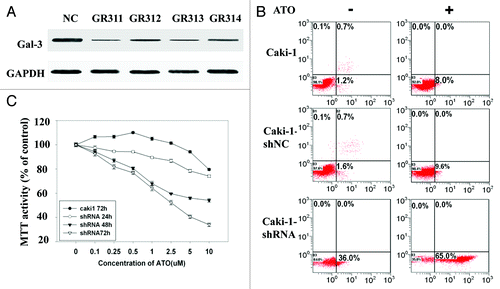
To study whether Gal-3 is a key factor preventing cells from ATO-induced apoptosis in RCCs, we used shRNA to knockdown Gal-3 expression in Caki-1 cell line. Four independent shRNA constructs (GR311, GR312, GR313, and GR314) were used to knock down endogenous Gal-3. The Gal-3 protein level was significantly reduced by all shRNAs (). GR311 possessed the optimum effect in reducing Gal-3 expression (about 10% of the control level) and hence was chosen for the following experiments. Control shRNA alone did not induce apoptosis in Caki-1 cells or affect ATO’s effects on apoptosis (). However, Gal-3-knockdown Caki-1 cells showed dramatically increased apoptosis, about 20-fold more than control group (P < 0.05). Interestingly, when galectin-3-knockdown Caki-1 cells were treated with ATO, apoptosis was further aggravated (about 2-fold) (). An additive effect was observed following synergic application of galectin-3 inhibition and ATO treatment, indicating that the reduction of Gal-3 likely increased the sensitivity of Caki-1 cells to ATO-induced apoptosis. To confirm this point, Gal-3-knockdown Caki-1 cells were treated with different concentrations of ATO for 24, 48, and 72 h, respectively. MTT assay was employed to monitor cell proliferation of Caki-1 cells. The results showed that ATO-induced proliferation inhibition was obviously facilitated by Gal-3-knockdown. The proliferation inhibition in Gal-3-knockdown Caki-1 cells treated with 0.5 μM ATO for 48 h was equal to that in wild-type Caki-1 cells treated with 10 μM ATO for 72 h (). IC50 of Gal-3- knockdown Caki-1 cells after 72 h ATO treatment was reduced to 1 μM, indicating that the knockdown of Gal-3 enhances the sensitivity of Caki-1 cells to ATO treatment.
Figure 4. The sensitivity of Caki-1 cells to ATO-induced apoptosis is increased by Gal-3 knockdown. (A) The protein level of Gal-3 was reduced after shRNA treatment. Four independent shRNAs against Gal-3 were used to construct stable cell lines. (B) The apoptosis of Caki-1 cells was enhanced by Gal-3 knockdown, and ATO treatment further increased apoptotic effect. Cells were labeled with Annexin V (x-axis) and PI (y-axis), and apoptosis was analyzed using flow cytometer. (C) Cell viability was reduced by Gal-3 knockdown combined with ATO treatment.
Gal-3 knockdown reduces mitochondrial membrane potential and increases mitochondria-related apoptosis following ATO treatment
Following ATO treatment, mitochondrial membrane permeability and mitochondrial membrane potential of the tumor cells are disrupted. As reported, Gal-3 is proven to mediate mitochondrial-dependent apoptosis, including abolishment of mitochondrial membrane potential, the release of cytochrome c, and the activation of caspase-3.Citation22 To this end, we detected mitochondrial features in Gal-3-knockdown Caki-1 cells to determine the effect of Gal-3 on mitochondria function following ATO treatment. JC-1 was used to label healthy mitochondria, JC-1 forms complex inside mitochondria and give red fluorescence signals when mitochondrial potential is high. As shown in , compared with the control group, treatment with ATO for 72 h did not obviously alter mitochondrial membrane potential in wild-type Caki-1 cells. That implicated that mitochondrial membrane potential was not affected by ATO in wild-type Caki-1 cells, which was consistent with the weak apoptotic effect of ATO on Caki-1 cells. Gal-3-knockdown significantly reduced the fluorescence signals (from 18.6 to 8.15) compared with control group (). Moreover, ATO treatment further increased the reduction of membrane potential (about 1.5-fold) in Gal-3-knockdown Caki-1 cells. Following the disruption of mitochondria, cytochrome c would be released into the cytoplasm, leading to caspase-dependent apoptosis. Hence, we further detected cytochrome c release in response to Gal-3 knockdown. As expected, compared with wild-type Caki-1 cells, the knockdown of Gal-3 increased the release of cytochrome c. Additionally, ATO (2.5 μM) treatment further enhanced cytochrome c release in Gal-3 knockdown cells (). We next examined the effects of Gal-3 knockdown and ATO on caspase-3 activity. In consistent with cytochrome c release, the activity of caspase-3 was enhanced in Gal-3 knockdown cells, which were further increased by ATO treatment (). In consistency, cell viability was significantly decreased in Gal-3 knockdown cells and further decreased by ATO treatment (). These results implicated that the knockdown of Gal-3 increased mitochondria-dependent apoptosis following ATO treatment.
Figure 5. Gal-3 knockdown decreases mitochondrial membrane potential and increases mitochondria-related apoptosis after ATO treatment. (A) Gal-3-knockdown Caki-1 cells showed reduced mitochondrial membrane potential, and the reduction was further enhanced by ATO treatment. JC-1 mitochondrial membrane potential assay was performed as described in methods. (B) Cytochrome c was released in response to Gal-3 knockdown and ATO treatment. Anti-cytochrome c antibodies were used to detect cytochrome c in the cytoplasm. (C) Caspase-3 activity was increased and (D) cell viability was reduced after Gal-3 knockdown and ATO treatment (P < 0.05).
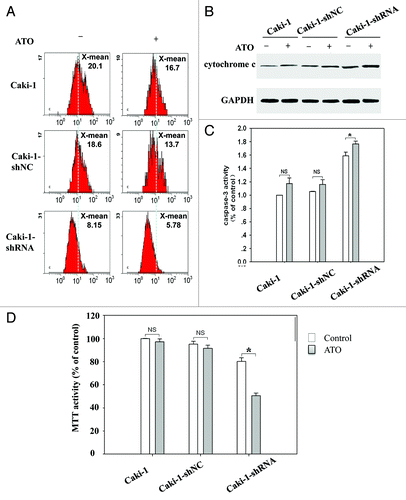
The Bcl-2 family plays a critical role in mitochondria-dependent apoptosis. The balance between anti-apoptotic protein, such as Bcl-2 and Bcl-xl, and pro-apoptotic protein like Bax and Bad, is critical for mitochondria-related apoptosis. It has been known that ATO induces apoptosis via inhibiting Bcl-2.Citation23 We found that ATO did not inhibit the protein level of Bcl-2 in either wild-type Caki-1 cells or control shRNA treated Caki-1 cells. Interestingly, when Gal-3 expression was knocked down, Bcl-2 level was sharply decreased in response to ATO treatment (). In contrast, an equal decrease of Bcl-xl level was observed in wild-type, control shRNA-treated, and Gal-3 shRNA-treated Caki-1 cells following ATO treatment ().
Figure 6. Gal-3 knockdown increases the apoptotic effect of ATO in RCC cells. (A) Gal-3 protected Bcl-2 from ATO induced reduction. (B) Gal-3 did not affect Bcl-xl levels before and after ATO treatment. (C) The Galecin-3 inhibitor, MCP, facilitated ATO to induce aggressive apoptosis in RCC cells, including 786-0, ACHN, Caki-1, and Caki-2.
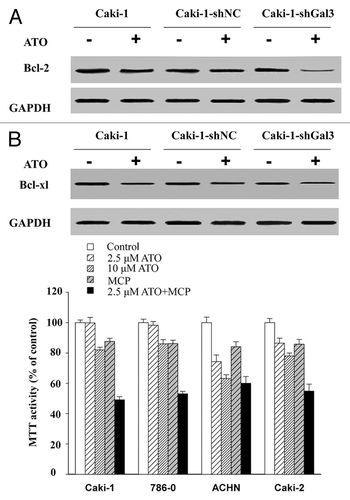
The synergetic effect of MCP and ATO in RCC cell lines
To confirm the results obtained from the Gal-3 knockdown, a Gal-3 functional inhibitor, MCP was employed. As shown in , MCP acted together with ATO to increase the apoptosis effect of ATO in all four cell lines. The application of MCP largely reduced the concentration of ATO required to attain the same apoptotic effect as ATO alone did.
Discussion
We hypothesized that the ineffectiveness of ATO in treating RCC was due to the apoptosis resistance in this study. As Gal-3 shows anti-apoptosis function in multiple cells following chemotherapeutic agents, it is possible that Gal-3 prohibits apoptosis pathway activated by ATO in RCCs, resulting in tumor resistance to ATO.
Initially, we detected apoptosis in four RCCs following ATO treatment for 72 h. Consistent with a previous study,Citation10 we showed that ATO treatment only induced apoptosis in ACHN and Caki-2 cells, but not in Caki-1 and 786-0 cells. Gal-3 was expressed in all RCC cell lines studied. Concurrently, ATO treatment significantly upregulated the expression of Gal-3 in RCC cell lines. Since Gal-3 was known for its anti-apoptotic role in chemotherapy-induced apoptosis, the dramatically increased Gal-3 level in RCC cells might contribute to the inhibition of ATO-induced apoptosis. In support of our presumption, our preliminary studies indicated that ATO weakly inhibited cell proliferation in these RCC cells (data not shown).
Previous studies showed that the translocation of Gal-3 from nucleus to cytoplasm contributed to anti-apoptotic activity of Gal-3.Citation17 Because the apoptotic function of Gal-3 was related to its subcellular location. We further studied the effect of ATO on Gal-3 translocation, and discovered that the localization of Gal-3 was affected by ATO treatment, transferring from the nucleus to the cytoplasm in Caki-1 and 786-0 cells. By contrast, the translocation of Gal-3 was not obviously affected in Caki-2 cells and ACHN cells. This discrepancy might be due to different cell types.
As a Ca2+ and phospholipid binding protein, synexin facilitates Gal-3 to prevent mitochondrial damage and cytochrome c release in cisplatin-induced BT549 cells (24). To date, it has been discovered that ATO-induced apoptosis is related to mitochondrial apoptosis.Citation24 It is worth to mention that when the expression of synexin is blocked by antisense oligonucleotides, Gal-3 cannot be transferred to perinuclear membrane and therefore loses its anti-apoptotic activity.Citation21 In agreement, our data also suggested that ATO did not influence the expression of total synexin, but synexin did move together with Gal-3 in response to ATO treatment.
To directly confirm that Gal-3 is the key factor in RCC cells against ATO-induced apoptosis, we used shRNA technique to knockdown Gal-3 expression. According to Park et al.,Citation10 Caki-1 cells were the primary cells, which have immunity to ATO treatment. To this end, we transferred the shRNA to knock down Gal-3 expression in Caki-3 cells. We found that Gal-3 inhibition by shRNA significantly induced Caki-1 cells apoptosis and proliferation inhibition. These effects have been demonstrated in prostate cancer cells.Citation25 Most importantly, Gal-3 inhibition sensitized the cells to ATO treatment. The effect of ATO treatment on apoptosis and proliferation inhibition were amplified in the knockdown cells. In wild-type Caki-3 cells, the cell viability was even increased following low doses of ATO (0–1 μM) treatment. Although we did not figure out the detailed mechanisms, ATO at low concentration did enhance tumor cell proliferation in prostate cancer cell lines.Citation26 In addition, the reduced expression of Gal-3 facilitated the damage of mitochondrial membrane caused by ATO, resulting in the increase of cytochrome c release and caspase-3 activation. Based on our results, the knockdown of Gal-3 enhanced the sensitivity of Caki-1 cells to ATO-induced apoptosis. This conclusion was further confirmed using a functional inhibitor of Gal-3, MCP.
Furthermore, Gal-3-knockdown Caki-1 cells had an obvious reduction of Bcl-2 protein level following ATO stimulation compared with wild-type cells, which indicated that Gal-3 maintained the level of Bcl-2. Although Gal-3 does not belong to Bcl-2 family, it shares a few structural features with Bcl-2, including containing proline, glycine and alanine enriched N-terminus. In addition, its C-terminal also carries an anti-death sequence, Asp-Trp-Gly-Arg (NWGR),Citation27,Citation28 which exists in the BH1 domain of Bcl-2 proteins and plays a key role in their anti-apoptotic activities.Citation29 Gal-3 may mimic members of the Bcl-2 family and bind to Bcl-2 protein through the similar domain. The Gal-3-Bcl-2 heterodimer may inhibit cell apoptosis through the Bcl-2 related cell death pathway. Other studies showed that cis-diammine-dichloroplatinum (CDDP) induced the translocation of Gal-3 from the nucleus to the cytoplasm, reduced the expression of apoptotic protein Bad, enhanced the phosphorylation of Bad, which stabilized mitochondrial membrane and inhibited apoptosis.Citation18
Additionally, we evaluated the syngeneic application of Gal-3 inhibitor, MCP, with ATO in RCC cells. An additive effect was observed, which was similar to what we observed in Gal-3-knockdown Caki-1 cells. However, it is still unclear how Gal-3 is regulated and how the complex is formed in the cytoplasm.
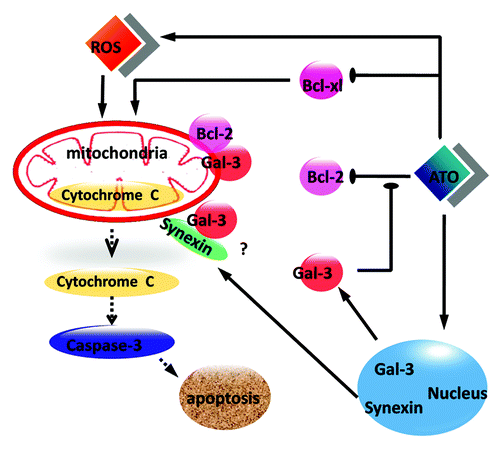
We illustrated in a mechanism which elucidated the role of Gal-3 in protecting RCC cells against ATO-medicated apoptosis. In response to ATO treatment, DNA is damaged and Gal-3 is translocated from the nucleus to the cytoplasm together with synexin. However, Gal-3 in the cytoplasm might bind to Bcl-2 to produce a dimer and prevent Bcl-2 reduction following ATO treatment. Finally, the damages of mitochondria following ATO treatment, including disruption of mitochondrial membrane potential, cytochrome c release was blocked by Gal-3/Bcl-2 dimer.
Figure 7. A scheme illustrates the anti-apoptotic role of Gal-3 in ATO-treated RCC cells. ATO increases the protein level of Gal-3 and triggers the co-translocation of Gal-3 from the nucleus to the cytoplasm with synexin. Cytoplasmic Gal-3 blocks the Bcl-2 reduction medicated by ATO and further protects mitochondrial membrane potential so that the cytochrome c release is decreased.
Based on the present study, the application of Gal-3 inhibition with ATO may be a potential strategy for renal cell carcinoma treatment. Our findings also implicate new ways to increase chemosensitivity and reduce the side effects of chemotherapy.
Materials and Methods
Cell culture
The human clear cell renal cell carcinoma (ccRCC) cell lines Caki-1 (Cell Bank of Chinese Academy of Sciences) and Caki-2 (ATCC) were maintained in McCoy 5A (Sigma) supplemented with 10% fetal bovine serum. The human renal cell adenocarcinoma cell lines 786-0 and ACHN (Cell Bank of Chinese Academy of Sciences) were grown in RPMI-1640 (NaHCO3 1.5 g/L, glucose 2.5 g/L, sodium pyruvate 0.11 g/L) and MEM (NaHCO3 1.5 g/L, Sodium Pyruvate 0.11 g/L [Gibco]), respectively, with 10% fetal bovine serum. Cells were cultured in a humidified incubator at 37 °C with 5% CO2.
Measurement of cell growth
In vitro growth inhibition effects of arsenic trioxide on renal cell carcinoma cells were determined using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assay.Citation21 Briefly, 3 × 103 Caki-1, Caki-2, 786-0, or ACHN cells were plated in 96-well plates and allowed to attach overnight. Cells were treated with 0, 0.25, 0.5, 1, 2.5, 5, or 10 µM of ATO (Harbin Yida Pharmaceutical Co., Ltd.) respectively for 72 h at 37 °C in 5% CO2. Five independent wells were repeated for each treatment. Twenty microliters of MTT (Sigma) solution (5 mg/mL in PBS) was added to each well and the plates were incubated for additional 4 h at 37 °C. To achieve solubilization of the formazan crystal formed in viable cells, 150 μl DMSO was added to each well before measurement at absorbance of 490 nm.
Apoptosis assay
Apoptosis was determined using Annexin V-FITC and PI double staining. Two hundred microliters of desired cells (1 × 106 cells per mL) were centrifuged at 1000 rpm for 5 min (4 °C). Cells were washed with pre-cooled PBS and resuspended in 100 µl binding buffer with 2 µl Annexin-V-FITC (20 µg/mL). The mixed samples were placed on ice in dark for 15 min. Four hundred microliters of PBS and 1 µl PI (50 µg/mL) were added to each sample before measurement. The value was read 2 min later using flow cytometer (Beckman Coulter EPICS XL). Cells without Annexin V-FITC and PI staining were used as negative controls.
Measurement of mitochondrial membrane potential
JC-1 mitochondrial membrane potential assay kit (Ocbio Science and Technologies Co., Ltd.) was used to detect mitochondrial membrane potential according to manufacturer’s instruction. In healthy cells, JC-1 forms complexes with intense red fluorescence. In contrast, in apoptotic or unhealthy cells with low mitochondrial potential, JC-1 remains monomeric structure and gives green fluorescence. Briefly, 5 × 105 cells were suspended in 0.5 mL PBS. For the control tube, CCCP (10 μM final concentration) was added to cells followed by incubation at 37 °C for 20 min. For each sample, 0.5 mL prepared JC-1 solution was added to cells and the final concentration of JC-1 was 5 μM. Cells was incubated at 37 °C for 20 min, and then collected by centrifugation at 600 g for 4 min at 4 °C. Pellet was resuspended and washed with JC-1 buffer. In the end, flow cytometer was used to collect fluorescence signals.
Immunoblot analysis
Proteins in cytoplasm and nucleus were extracted as described previously.Citation30 Cells were grown to subconfluency on 6-well plates and exposed to ATO (10 μM) for 72 h. Cells were collected, washed with PBS, and resuspended in hypotonic buffer (20 mM HEPES, 10 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.1% Triton X-100, 20% glycerol, 5 μg leupeptin per mL, 10 μg aprotinin per mL, and 500 μM phenyl-methylsulfonyl fluoride). The cells were centrifuged at 850 g for 5 min. The supernatant was used as the cytoplasmic fraction. The pellet was suspended in extraction buffer (20 mM HEPES, 10 mM KCl, 1 mM MgCl2, 0.5 mM DTT, 0.1% Triton X-100, 20% glycerol, 420 mM NaCl, 5 μg leupeptin per mL, 10 μg aprotinin per mL, and 500 μM phenylmethylsulfonyl fluoride). This suspension was centrifuged at 15 000 g for 30 min. The supernatant contained the nuclear fraction. Protein concentrations were measured using BCA Protein Assay Kit (Beyotime Biotechnology). Cytoplasmic proteins (10 μg), nuclear proteins (20 μg), and total proteins were separated by SDS-PAGE (12.5% polyacrylamide) by the method of Laemmli and transferred to a PVDF membrane. The presence of proteins was detected using human anti-Gal-3 antibody H-160 (Santa Cruz), anti-GAPDH antibody (Sigma), anti-BCL-xl antibody and anti-Bcl-2 antibody (Santa Cruz). Expression levels of proteins were detected using the enhanced chemiluminescence system. For densitometric analysis of western blots, Image J software was used.
Immunofluorescence
Immunofluorescence was performed as described previously.Citation19 In brief, cells were fixed in 4% paraformaldehyde for 15 min and blocked in PBS containing 3% BSA for 30 min. The cells were incubated in Gal-3 rabbit polyclonal antibody (1:50) at room temperature (RT) for 1 h followed by incubation with anti-rabbit secondary antibody Alexa Fluor 488 or FITC (1:200) at RT for 1 h. DAPI was used to stain nuclei. Immunofluorescence was visualized with a laser confocal fluorescence microscope (Zeiss, Imager Z1).
Quantification of Caspase-3 activity
The activity of caspase-3 was determined using Caspase-3 Assay Kit (Beyotime Institute of Biotechnology) by detecting the color reporter molecule, p-nitroanilide (pNA) after cleavage from the substrate of Ac-DEVD-pNA. Cells were seeded at 4 × 105 cells per well in a 6-well plate and incubated overnight. Cells were treated with indicated drug for 24 h, and then cell lysates were assayed for caspase-3 activity. Ten microliters of cell lysate was mixed with pre-cooled substrate solution Ac-DEVD-pNA (0.2 mM) and incubated at 37 °C for 60‒120 min. The mixture without cell lysate was used as blank control. Relative fluorescent value was acquired at 405 nm wavelength. The activity of caspase-3 in cell lysates was obtained by comparing to blank control and normalizing with the standard curve.
Generation of stable Gal-3 knockdown cell lines
The shRNA retrovirus used to establish Gal-3-knockdown stable cell lines was made from 293T cells and used to transduce Caki-1 cells. Stable transductants were selected after 14 d of puromycin (2 μM, Sigma). Four candidate shRNAs were obtained from NeuronBiotech and transduced into Caki-1 cells. The target sequences were GR311: CCCACGCTTC AATGAGAACA A, GR312: GCAAACAGAA TTGCTTTAG AT, GR313: GCCACTGATT GTGCCTTATA A and GR314: GCTCCATGAT GCGTTATCTG G.
MCP preparation and cell treatment
A 0.1% aqueous pectin dispersion was prepared by dissolving 200 mg of unmodified CP (citrus pectin, Sigma) in 200 mL of deionized and filtered water as demonstrated previously.Citation31 Briefly, the solution was autoclaved at 123.2 °C for 30 min. At the end of the heat treatment, the solution was allowed to cool to RT and stored overnight at 4 °C, allowing formation of a gel-like precipitate. The next day, the aqueous phase was collected, frozen at −80 °C and lyophilized. Cells were treated with 3.0 mg/mL MCP for 48 h.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood 1997; 89:3354 - 60; PMID: 9129042
- Rojewski MT, Baldus C, Knauf W, Thiel E, Schrezenmeier H. Dual effects of arsenic trioxide (As2O3) on non-acute promyelocytic leukaemia myeloid cell lines: induction of apoptosis and inhibition of proliferation. Br J Haematol 2002; 116:555 - 63; http://dx.doi.org/10.1046/j.0007-1048.2001.03298.x; PMID: 11849211
- Liu Q, Hilsenbeck S, Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 2003; 101:4078 - 87; http://dx.doi.org/10.1182/blood-2002-10-3231; PMID: 12531793
- Nimmanapalli R, Bali P, O’Bryan E, Fuino L, Guo F, Wu J, Houghton P, Bhalla K. Arsenic trioxide inhibits translation of mRNA of bcr-abl, resulting in attenuation of Bcr-Abl levels and apoptosis of human leukemia cells. Cancer Res 2003; 63:7950 - 8; PMID: 14633726
- Seol JG, Park WH, Kim ES, Jung CW, Hyun JM, Kim BK, Lee YY. Effect of arsenic trioxide on cell cycle arrest in head and neck cancer cell line PCI-1. Biochem Biophys Res Commun 1999; 265:400 - 4; http://dx.doi.org/10.1006/bbrc.1999.1697; PMID: 10558879
- Ai Z, Lu W, Qin X. Arsenic trioxide induces gallbladder carcinoma cell apoptosis via downregulation of Bcl-2. Biochem Biophys Res Commun 2006; 348:1075 - 81; http://dx.doi.org/10.1016/j.bbrc.2006.07.181; PMID: 16904648
- Uslu R, Sanli UA, Sezgin C, Karabulut B, Terzioglu E, Omay SB, Goker E. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin Cancer Res 2000; 6:4957 - 64; PMID: 11156257
- Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res 2001; 61:5432 - 40; PMID: 11454688
- Chiu HW, Ho YS, Wang YJ. Arsenic trioxide induces autophagy and apoptosis in human glioma cells in vitro and in vivo through downregulation of survivin. J Mol Med (Berl) 2011; 89:927 - 41; http://dx.doi.org/10.1007/s00109-011-0763-1; PMID: 21594580
- Hyun Park W, Hee Cho Y, Won Jung C, Oh Park J, Kim K, Hyuck Im Y, Lee MH, Ki Kang W, Park K. Arsenic trioxide inhibits the growth of A498 renal cell carcinoma cells via cell cycle arrest or apoptosis. Biochem Biophys Res Commun 2003; 300:230 - 5; http://dx.doi.org/10.1016/S0006-291X(02)02831-0; PMID: 12480548
- Grad JM, Bahlis NJ, Reis I, Oshiro MM, Dalton WS, Boise LH. Ascorbic acid enhances arsenic trioxide-induced cytotoxicity in multiple myeloma cells. Blood 2001; 98:805 - 13; http://dx.doi.org/10.1182/blood.V98.3.805; PMID: 11468182
- Vuky J, Yu R, Schwartz L, Motzer RJ. Phase II trial of arsenic trioxide in patients with metastatic renal cell carcinoma. Invest New Drugs 2002; 20:327 - 30; http://dx.doi.org/10.1023/A:1016270206374; PMID: 12201495
- Kuwabara I, Liu FT. Galectin-3 promotes adhesion of human neutrophils to laminin. J Immunol 1996; 156:3939 - 44; PMID: 8621934
- Inohara H, Akahani S, Raz A. Galectin-3 stimulates cell proliferation. Exp Cell Res 1998; 245:294 - 302; http://dx.doi.org/10.1006/excr.1998.4253; PMID: 9851870
- Yoshii T, Fukumori T, Honjo Y, Inohara H, Kim HR, Raz A. Galectin-3 phosphorylation is required for its anti-apoptotic function and cell cycle arrest. J Biol Chem 2002; 277:6852 - 7; http://dx.doi.org/10.1074/jbc.M107668200; PMID: 11724777
- Nangia-Makker P, Honjo Y, Sarvis R, Akahani S, Hogan V, Pienta KJ, Raz A. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am J Pathol 2000; 156:899 - 909; http://dx.doi.org/10.1016/S0002-9440(10)64959-0; PMID: 10702407
- Takenaka Y, Fukumori T, Raz A. Galectin-3 and metastasis. Glycoconj J 2004; 19:543 - 9; http://dx.doi.org/10.1023/B:GLYC.0000014084.01324.15; PMID: 14758078
- Fukumori T, Kanayama HO, Raz A. The role of galectin-3 in cancer drug resistance. Drug Resist Updat 2007; 10:101 - 8; http://dx.doi.org/10.1016/j.drup.2007.04.001; PMID: 17544840
- Fukumori T, Oka N, Takenaka Y, Nangia-Makker P, Elsamman E, Kasai T, Shono M, Kanayama HO, Ellerhorst J, Lotan R, et al. Galectin-3 regulates mitochondrial stability and antiapoptotic function in response to anticancer drug in prostate cancer. Cancer Res 2006; 66:3114 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-05-3750; PMID: 16540661
- Cheng YL, Huang WC, Chen CL, Tsai CC, Wang CY, Chiu WH, Chen YL, Lin YS, Chang CF, Lin CF. Increased galectin-3 facilitates leukemia cell survival from apoptotic stimuli. Biochem Biophys Res Commun 2011; 412:334 - 40; http://dx.doi.org/10.1016/j.bbrc.2011.07.099; PMID: 21821001
- Yu F, Finley RL Jr., Raz A, Kim HR. Galectin-3 translocates to the perinuclear membranes and inhibits cytochrome c release from the mitochondria. A role for synexin in galectin-3 translocation. J Biol Chem 2002; 277:15819 - 27; http://dx.doi.org/10.1074/jbc.M200154200; PMID: 11839755
- Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, Chen SJ, et al. Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemia 2000; 14:262 - 70; http://dx.doi.org/10.1038/sj.leu.2401650; PMID: 10673743
- Han B, Zhou G, Zhang Q, Zhang J, Wang X, Tang W, Kakudo K. Effect of arsenic trioxide (ATO) on human lung carcinoma PG cell line: ATO induced apoptosis of PG cells and decreased expression of Bcl-2, Pgp. J Exp Ther Oncol 2004; 4:335 - 42; PMID: 15844663
- Akay C, Thomas C 3rd, Gazitt Y. Arsenic trioxide and paclitaxel induce apoptosis by different mechanisms. Cell Cycle 2004; 3:324 - 34; http://dx.doi.org/10.4161/cc.3.3.657; PMID: 14726646
- Wang Y, Nangia-Makker P, Tait L, Balan V, Hogan V, Pienta KJ, Raz A. Regulation of prostate cancer progression by galectin-3. Am J Pathol 2009; 174:1515 - 23; http://dx.doi.org/10.2353/ajpath.2009.080816; PMID: 19286570
- Aravena C, Beltrán AR, Cornejo M, Torres V, Díaz ES, Guzmán-Gutiérrez E, Pardo F, Leiva A, Sobrevia L, Ramírez MA. Potential role of sodium-proton exchangers in the low concentration arsenic trioxide-increased intracellular pH and cell proliferation. PLoS One 2012; 7:e51451; http://dx.doi.org/10.1371/journal.pone.0051451; PMID: 23236503
- Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci U S A 1996; 93:6737 - 42; http://dx.doi.org/10.1073/pnas.93.13.6737; PMID: 8692888
- Akahani S, Nangia-Makker P, Inohara H, Kim HR, Raz A. Galectin-3: a novel antiapoptotic molecule with a functional BH1 (NWGR) domain of Bcl-2 family. Cancer Res 1997; 57:5272 - 6; PMID: 9393748
- Hanada M, Aimé-Sempé C, Sato T, Reed JC. Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J Biol Chem 1995; 270:11962 - 9; PMID: 7744846
- Takenaka Y, Fukumori T, Yoshii T, Oka N, Inohara H, Kim HR, Bresalier RS, Raz A. Nuclear export of phosphorylated galectin-3 regulates its antiapoptotic activity in response to chemotherapeutic drugs. Mol Cell Biol 2004; 24:4395 - 406; http://dx.doi.org/10.1128/MCB.24.10.4395-4406.2004; PMID: 15121858
- Wang Y, Nangia-Makker P, Balan V, Hogan V, Raz A. Calpain activation through galectin-3 inhibition sensitizes prostate cancer cells to cisplatin treatment. Cell Death Dis 2010; 1:e101; http://dx.doi.org/10.1038/cddis.2010.79; PMID: 21368866