Abstract
The present studies focused on defining the mechanisms by which anoikis-resistant (AR) mammary carcinoma cells can be reverted to a therapy-sensitive phenotype. AR mammary carcinoma cells had reduced expression of the toxic BH3 domain proteins BAX, BAK, NOXA, and PUMA. In AR cells expression of the protective BCL-2 family proteins BCL-XL and MCL-1 was increased. AR cells were resistant to cell killing by multiple anti-tumor cell therapies, including ERBB1/2 inhibitor + MCL-1 inhibitor treatment, and had a reduced autophagic flux response to these therapies, despite similarly exhibiting increased levels of LC3II processing. Knockdown of MCL-1 and BCL-XL caused necro-apoptosis in AR cells to a greater extent than in parental cells. Pre-treatment of anoikis-resistant cells with histone deacetylase inhibitors (HDACIs) for 24 h increased the levels of toxic BH3 domain proteins, reduced MCL-1 levels, and restored/re-sensitized the cell death response of AR tumor cells to multiple toxic therapies. In vivo, pre-treatment of AR breast tumors in the brain with valproate restored the chemo-sensitivity of the tumors and prolonged animal survival. These data argue that one mechanism to enhance the anti-tumor effect of chemotherapy could be HDACI pre-treatment.
Introduction
Dissociation of cells from an extracellular matrix leads to the rapid induction of cell death processes, most frequently termed anoikis (Greek for homelessness).Citation1 Formation of ducts during development of the mammary gland involves the induction of anoikis in the lumen and anoikis resistance in these luminal cells is believed to be a part of the biology of early stage breast cancers.Citation2-Citation5 Cancer cells, by their nature, are relatively speaking more able to suppress the induction of anoikis which permits them to remain viable under anchorage independent conditions.Citation6 And, anoikis resistance in vitro is known to correlate with in vivo metastatic potential.Citation7
Many studies of anoikis resistance have focused on protein and lipid kinases modulating the activity of apoptotic pathways. In particular the activity of growth factor receptors (e.g., ERBB1/2), non-receptor tyrosine kinases (e.g., SRC), and signal transduction pathways (e.g., AKT and ERK1/2) have been linked to anoikis resistance.Citation8-Citation10 Inhibitors of each of these kinases have been shown, in part, to revert anoikis resistance in a variety of tumor cell types. Downstream of these pathways resistance has been linked to altered expression of the toxic BH3 domain protein BIM and the regulation of mitochondrial function. Nevertheless, new approaches to revert anoikis resistance than can actually be translated to the clinic are still needed.
HDAC inhibitors (HDACIs) are a structurally diverse class of agents, e.g., vorinostat (SAHA; Zolinza) and sodium valproate, (Depakote). These agents block histone de-acetylation and neutralization of positively charged lysine residues on histone tails, thereby modifying chromatin structure/condensation and transcription.Citation11-Citation13 However, the mode of HDACI action is in fact multi-factorial with an additional ~20 targets, including disruption of co-repressor complexes, induction of oxidative injury, upregulation of death receptor and ligand expression, generation of lipid second messengers, interference with chaperone protein function, modulation of NFκB activity, and the induction of DNA damage.Citation14 As we have shown previously, induction of DNA damage and the generation of ceramide and ROS production is a common molecular mechanism involved in HDACs-induced anti-tumor activity.Citation15,Citation16 HDACIs have been shown to have selective toxicity in tumor cells compared with non-transformed cells which may be due to altered gene expression and/or the generation of ROS and the threshold at which ROS causes cell death in non-transformed and transformed cells.
In our several prior studies combining the ERBB1/2 inhibitor lapatinib and the MCL-1 inhibitor obatoclax we have demonstrated that: lapatinib and obatoclax interact to kill through a toxic form of autophagy dependent on the toxic BH3 domain proteins NOXA and BAK; that based on the cell system lapatinib and obatoclax necro-apoptotic/autophagic killing occurs through inhibition of ERBB1/2/3/4 signaling in a cell type dependent manner and with parallel inhibition of both BCL-XL and MCL-1; and that killing requires both ROS generation and endoplasmic reticulum stress signaling.Citation17-Citation19
The present studies were initially designed to develop multiple anoikis-resistant breast and glioma stem cells, and examine anoikis resistance mechanisms toward lapatinib + obatoclax treatment in these cells. We show that anoikis-resistant breast and brain cancer cells have reduced expression of multiple toxic BH3 domain proteins, including BAK and NOXA. BIM did not appear to be a key player in survival regulation. Re-expression of these proteins restored the sensitivity of tumor cells to cancer therapies, including lapatinib + obatoclax treatment; and to treatment of cells with lapatinib + CDK9 inhibitor that also reduces MCL-1 expression. Treatment of anoikis-resistant tumor cells with HDAC inhibitors increased expression of multiple toxic BH3 domain proteins and restored the sensitivity of tumor cells to cancer therapies in vitro. In vivo, to our surprise, AR cells were more sensitive to therapy than in vitro, suggesting that as a tumor in vivo some reverting from the AR phenotype occurs.
Results
Prior studies from this laboratory have demonstrated that lapatinib + obatoclax treatment kills breast and brain cancer cells through a NOXA- and BAK-dependent form of autophagy with ROS generation also playing a key role in the killing.Citation17-Citation19 The lapatinib + obatoclax treatment form of killing at the cellular level was necro-apoptotic as judged using TUNEL, DAPI, Geimsa, and trypan blue staining (i.e., death was best measured using trypan blue inclusion as a definitive measure of death).Citation17-Citation19
It has been recently claimed that many manuscripts in the cancer therapeutics field that contain data with drugs/kinase inhibitors which cannot be reproduced at low clinically relevant drug concentrations.Citation20 Initial viability studies in the present manuscript built upon our prior work showing lapatinib and obatoclax interacted to kill breast and brain cancer cells in vitro within a short time frame (~12 h) using ~1 μM lapatinib.Citation17-Citation19 In human plasma the Cmax of lapatinib is ~10 μM but for tumors localized in the brain, due to the blood brain barrier, the Cmax for the drug is only ~1 μM. Obatoclax has a Cmax of ~2 μM and clearly crosses the blood brain barrier to a much greater extent based on one side effect which is transient euphoria due to interaction with neuronal opiod receptors.
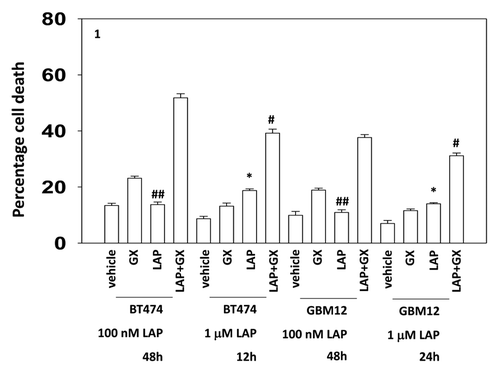
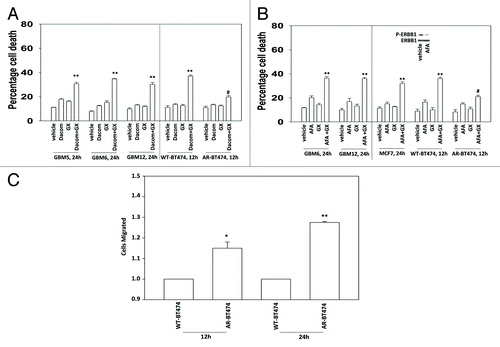
Thus we compared the interaction between a relatively low dose of lapatinib and that of a 10-fold greater dose, together with the time required to cause similar levels of killing. Low dose lapatinib interacted with obatoclax in ERBB2+ BT474 breast and ERBB1+ GBM12 brain cancer cells to cause similar levels of killing when compared with higher dose lapatinib at the 12 h and 48 h time points (for BT474) and at the 24 h and 48 h time points (for GBM12), respectively (). Of note was that low dose lapatinib as a single agent was not particularly toxic in either of these ERBB2/ERBB1 addicted tumor cell types whereas a 10-fold higher dose of the drug caused a significant measurable level of single agent killing.
Figure 1. Low and high dose lapatinib increase obatoclax toxicity. BT474 and GBM12 cells in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM or 100 nM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated as indicated 12, 24, and 48 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in WT cells ##P > 0.05 compared with vehicle treated cells; *P < 0.05 greater than vehicle control.
For breast cancer cells to safely migrate through the vasculature from the thorax to the brain requires that they become anoikis-resistant. Hence we next determined changes in protein expression in mammary carcinoma cells rigorously selected to grow as unattached anoikis-resistant (AR) cell cultures (see Methods section for details). AR-BT474, AR-MCF7, and AR-SKBR3 cells all had reduced expression of the toxic BH3 domain proteins BAX, BAK, NOXA, and PUMA, and the tumor suppressor PTEN, and elevated expression of MCL-1, BCL-XL, and Beclin1 (). Similar data were obtained using multiple isolates of AR cells (see Methods, data not shown). AR cells also had increased levels of P-AKT, P-ERK, P-p70 S6K, and reduced expression of PTEN. Total levels of ERBB2 were modestly enhanced in AR-BT474 cells whereas those of ERBB1 were highly elevated, and cell surface levels of ERBB1 were increased in AR cells ().
Figure 2A–D. Development of anoikis resistance in breast cancer cells. (A) BT474 cells (wild type; anoikis-resistant) were plated and 24 h later cells were isolated for immunoblotting against the indicated proteins and phospho-proteins (n = 3). (B) MCF7 cells (wild type; anoikis-resistant) were plated and 24 h later cells were isolated for immunoblotting against the indicated proteins and phospho-proteins (n = 3). (C) SKBR3 cells (wild type; anoikis-resistant) were plated and 24 h later cells were isolated for immunoblotting against the indicated proteins and phospho-proteins (n = 3). (D) BT474 cells (wild type; anoikis-resistant) were assessed for the expression of ERBB1 and ERBB2, and the impact of lapatinib (1 μM) on the phosphorylation of these proteins (n = 3); BT474 (wild type; anoikis-resistant) cells were fixed but not permeabilized and the levels of cell surface ERBB1 and ERBB2 determined.
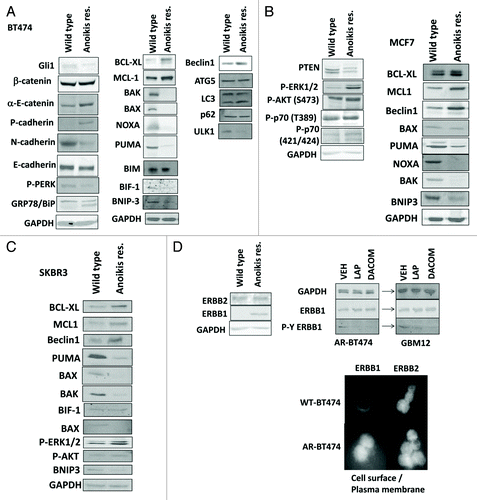
Despite higher ERBB1 levels, AR mammary tumor cells were generally more resistant to being killed by lapatinib + obatoclax treatment than matched wild type parental breast cancer cells (). This finding is not surprising per se as prior studies have also delineated that lapatinib + obatoclax treatment activates BAX and BAK, and increases NOXA and PUMA levels in wild-type parental cells that are causal in killing; proteins whose expression is largely lost in AR cells.Citation17-Citation19 Similar drug combination data with lapatinib + obatoclax treatment were obtained in colony formation assays (). Comparable lapatinib + obatoclax treatment killing data was also obtained in primary human GBM cells weakly adapted to substratum attachment free growth (; see data in for fully adapted anoikis-resistant and stem cell-derived GBM cells). Parallel toxicity drug combination data to that using the competitive ERBB1/ERBB2 inhibitor lapatinib was also obtained using the relatively novel ERBB1/ERBB2/ERBB4 suicide inhibitors dacomitinib and afatinib (). AR mammary tumor cells were more invasive than their parental wild-type counterparts and grew 57 ± 5% faster than parental wild type cells ().
Figure 2E–I.(E) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in WT cells. (F) MCF7 cells (WT, wild type; AR, anoikis-resistant,) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated 24 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in WT cells. (G) SKBR3 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in WT cells. (H) BT474 cells (WT, wild type; AR, anoikis-resistant) in sextuplicate were plated as single cells in soft agar, thus growing in 3 dimensions. Twelve hours after plating cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. After 12 h the media was changed and replaced with drug free media. Colonies were permitted to form for the following 10 d, before fixing, staining and counting (± SEM, n = 3) #P < 0.05 less than corresponding value in vehicle-treated cells; ##P < 0.05 less than value in lapatinib treated cells. (I) GBM6 and GBM12 cells (WT, wild type; AR, partially anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated 24 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in WT cells.
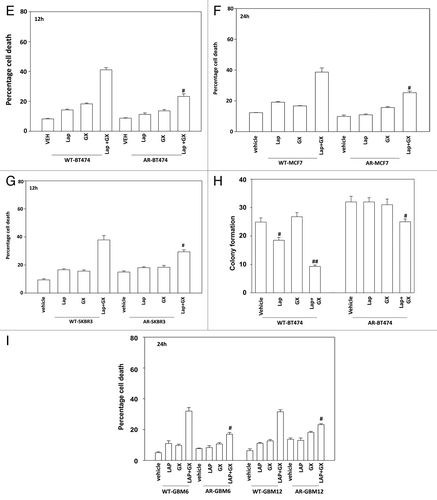
Figure 5. HDAC inhibitors restore expression of toxic BH3 domain proteins in anoikis-resistant breast cancer cells. (A) BT474 (WT, wild type; AR, anoikis-resistant) cells were treated for 24 h with vehicle or with either sodium valproate (750 μM) or vorinostat (750 nM). Cells were isolated and immunoblotting performed to determine expression of PUMA, BAX, BAX, and NOXA (n = 3). (B) BT474 and SKBR3 (WT, wild type; AR, anoikis-resistant) cells were treated for 36 h with vehicle or with either sodium valproate (750 μM) or vorinostat (750 nM). Cells were isolated and viability determined by trypan blue (± SEM, n = 3). (C) BT474 (WT, wild type; AR, anoikis-resistant; LAP-R, lapatinib-resistant) cells were treated for 24 h with vehicle or with either sodium valproate (750 μM) or vorinostat (750 nM). Cells were washed and the media was replaced with drug free media. Cells were then treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in vehicle-treated cells. (D) GBM6 and GBM12 cells (WT, wild type; STEM, stem cells) cells were treated for 24 h with vehicle or with sodium valproate (750 μM). Cells were washed and the media was replaced with drug-free media. Cells were then treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 24 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in vehicle-treated cells. (E) BT474 (WT, wild type; AR, anoikis-resistant) cells were transfected with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down BAK, BAK, NOXA, and PUMA, as indicated. cells were treated for 24 h with vehicle or with sodium valproate (750 μM). Cells were washed and the media was replaced with drug free media. Twenty-four hours after transfection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in valproate treated siSCR cells.
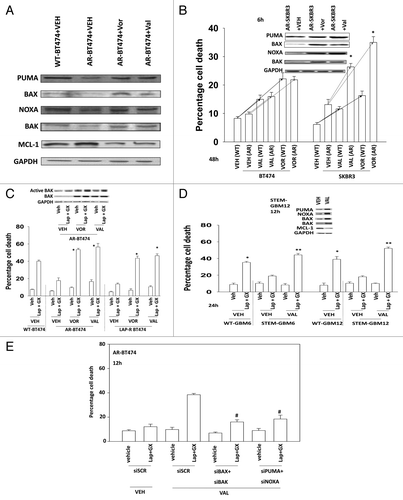
Figure 3. Lapatinib-resistant BT474 cells overexpress MCL-1 and BCL-XL and have reduced expression of toxic BH3 domain proteins. (A) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), dacomitinib (dacom., 100 nM), obatoclax (GX, 50 nM), or the drug combination. Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than others values in (dacom. + GX) cells; **P < 0.05 greater than VEH cells. (B) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), afatinib (AFA, 100 nM), obatoclax (GX, 50 nM) or the drug combination. Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than others values in (AFA. + GX) cells; **P < 0.05 greater than VEH cells. (C) BT474 cells (WT, wild type; AR, anoikis-resistant) were plated in a Millipore Millicell. The migration of cells was determined after 12 h (± SEM, n = 3) *P < 0.05 greater than corresponding value in WT cells.
Based on our data in AR cells, we re-examined the roles of BH3 domain proteins in wild type parental cells. As previously noted knock down of BAX, BAK, NOXA, and PUMA suppressed lapatinib + obatoclax lethality in wild-type BT474 cells ().Citation17-Citation19 Re-expression of toxic BH3 domain proteins BAX, BAK, NOXA, and PUMA restored lapatinib + obatoclax the responsiveness of AR cells; cells that have low expression of such proteins (). Knockdown of MCL-1, BCL-XL, or c-FLIP-s also partially restored lapatinib + obatoclax responsiveness of AR cells (). Expression of dominant negative MEK1 or of dominant negative AKT also partially restored lapatinib + obatoclax responsiveness of AR cells (, cf. data in ).
Figure 4. Regulation of lapatinib + obatoclax lethality by toxic BH3 domain proteins. (A) BT474 (wild type) cells were transfected with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down BAK, BAK, NOXA, and PUMA, as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in siSCR cells; ##P < 0.05 less than corresponding single knock down cells. (B) BT474 (WT, wild type; AR, anoikis-resistant) cells were transfected with empty vector plasmid (CMV) or plasmids to express BAX, BAK, NOXA, and PUMA as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in CMV cells; **P < 0.05 greater than corresponding single expression cells. (C) BT474 (AR, anoikis-resistant) cells were transfected with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down MCL-1, c-FLIP-s, and BCL-XL, as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in siSCR cells; %P < 0.05 less than corresponding value in siMCL-1 cells. (D) SKBR3 (AR, anoikis-resistant) cells were transfected with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down MCL-1 and BCL-XL, as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in siSCR cells; %P < 0.05 less than corresponding value in siMCL-1 cells. (E) BT474 (AR, anoikis-resistant) cells were infected with empty vector adenovirus (CMV) or with adenoviruses to express dominant negative MEK1 or dominant negative AKT, as indicated. Thirty-six hours after infection cells were treated with vehicle (VEH, DMSO) or with lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in CMV cells.
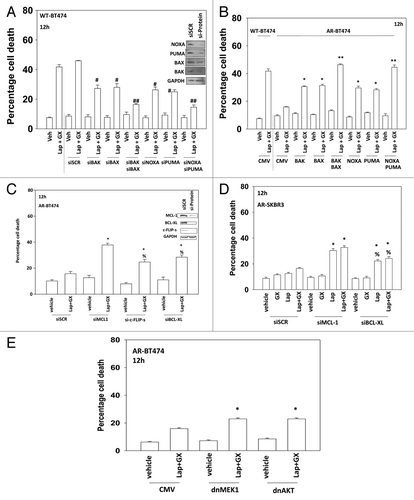
Epigenetic mechanisms are known to reduce the expression of toxic BH3 domain proteins, including BAX, BAK, NOXA, and PUMA and in a variety of tumor cell systems histone deacetylase inhibitors have been shown to individually increase expression of these proteins.Citation21-Citation26 Coordinate expression of multiple BH3 domain proteins in the same system, and focused on a translational outcome has not been previously examined. HDAC inhibitor treatment for 24 h increased BAX, BAK, NOXA, and PUMA expression in AR-BT474 mammary carcinoma cells to a level comparable to that expressed in wild type parental cells (). The HDAC inhibitor treatment reduced MCL-1 levels. To our surprise in AR-SKBR3 cells, treatment with an HDAC inhibitor alone caused significant amounts of tumor cell killing making similar studies to those in near impossible (). In agreement with these findings, pre-treatment of AR-BT474 cells or lapatinib resistant BT474 cells (LAP-R) with HDAC inhibitors promoted lapatinib + obatoclax lethality (). (NB: lapatinib-resistant BT474 cells expressed higher levels of MCL-1 and BCL-XL and lower levels of BAX and BAK [data not shown; near identical to that in seen in LAP-R HCT116 cells]).Citation27,Citation28 In GBM wild-type and neurosphere-derived stem cells (WT-GBM6/STEM-GBM6; WT-GBM12/STEM-GBM12) pre-treatment with valproate also promoted lapatinib + obatoclax lethality (). Valproate treatment also increased the expression of BAX, BAK, NOXA, and PUMA and reduced MCL-1 levels in the STEM-GBM cells (, upper inset). Knockdown of BAX + BAK or of PUMA + NOXA in AR-BT474 cells suppressed the stimulatory effect of HDAC inhibitor pre-treatment on lapatinib + obatoclax toxicity in AR cells ().
Figure 6. CDK inhibitors reduce expression of protective BCL-XL and MCL-1 proteins in anoikis-resistant breast cancer cells. (A) MCF7 and BT474 (WT, wild type; AR, anoikis-resistant) cells were treated with vehicle or with dinaciclib (10 nM), afatinib (100 nM), lapatinib (1 μM), or the drugs in combination as indicated. Cells were isolated after 24 h and viability determined by trypan blue (± SEM, n = 3). *P < 0.05 greater than corresponding value in vehicle-treated cells; #P < 0.05 less than corresponding value in wild-type treated cells. Upper inset panel: the expression of MCL-1 12 h after exposure to the indicated drugs. (B) BT474 (AR, anoikis-resistant) cells were treated for 36 h with vehicle or with either sodium valproate (750 μM) or vorinostat (750 nM). Cells were then treated with vehicle or with dinaciclib (10 nM), afatinib (100 nM), lapatinib (1 μM), or the drugs in combination as indicated. Cells were isolated after 24 h and viability determined by trypan blue (± SEM, n = 3). *P < 0.05 greater than corresponding value in vehicle-treated cells within own group; **P < 0.05 greater than corresponding value in vehicle-treated cells within whole study. (C) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. Thirty minutes after drug treatment cells were mock exposed or irradiated (4 Gy). Cells were isolated 6 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in lapatinib alone cells; **P < 0.05 greater than corresponding unirradiated cells; %P < 0.05 less than corresponding value in WT cells. (D) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. As indicated cells were treated with vehicle control or with doxorubicin (0.75 μM). Cells were isolated 6 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in lapatinib alone cells; **P < 0.05 greater than corresponding non-DOX treated cells; %P < 0.05 less than corresponding value in WT cells. (E) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated for 24 h with vehicle or with sodium valproate (750 μM). Cells were washed and the media was replaced with drug free media. Cells were then treated with vehicle (VEH, DMSO) or with [lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM)] and with as indicated doxorubicin (0.75 μM) or irradiated (n.b. 2 Gy). Cells were isolated 24 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 difference between value and LAP+GX greater than corresponding value in vehicle only-treated AR-BT474 cells.
![Figure 6. CDK inhibitors reduce expression of protective BCL-XL and MCL-1 proteins in anoikis-resistant breast cancer cells. (A) MCF7 and BT474 (WT, wild type; AR, anoikis-resistant) cells were treated with vehicle or with dinaciclib (10 nM), afatinib (100 nM), lapatinib (1 μM), or the drugs in combination as indicated. Cells were isolated after 24 h and viability determined by trypan blue (± SEM, n = 3). *P < 0.05 greater than corresponding value in vehicle-treated cells; #P < 0.05 less than corresponding value in wild-type treated cells. Upper inset panel: the expression of MCL-1 12 h after exposure to the indicated drugs. (B) BT474 (AR, anoikis-resistant) cells were treated for 36 h with vehicle or with either sodium valproate (750 μM) or vorinostat (750 nM). Cells were then treated with vehicle or with dinaciclib (10 nM), afatinib (100 nM), lapatinib (1 μM), or the drugs in combination as indicated. Cells were isolated after 24 h and viability determined by trypan blue (± SEM, n = 3). *P < 0.05 greater than corresponding value in vehicle-treated cells within own group; **P < 0.05 greater than corresponding value in vehicle-treated cells within whole study. (C) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. Thirty minutes after drug treatment cells were mock exposed or irradiated (4 Gy). Cells were isolated 6 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in lapatinib alone cells; **P < 0.05 greater than corresponding unirradiated cells; %P < 0.05 less than corresponding value in WT cells. (D) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. As indicated cells were treated with vehicle control or with doxorubicin (0.75 μM). Cells were isolated 6 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 greater than corresponding value in lapatinib alone cells; **P < 0.05 greater than corresponding non-DOX treated cells; %P < 0.05 less than corresponding value in WT cells. (E) BT474 cells (WT, wild type; AR, anoikis-resistant) in triplicate were treated for 24 h with vehicle or with sodium valproate (750 μM). Cells were washed and the media was replaced with drug free media. Cells were then treated with vehicle (VEH, DMSO) or with [lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM)] and with as indicated doxorubicin (0.75 μM) or irradiated (n.b. 2 Gy). Cells were isolated 24 h later and viability determined by trypan blue (± SEM, n = 3) *P < 0.05 difference between value and LAP+GX greater than corresponding value in vehicle only-treated AR-BT474 cells.](/cms/asset/3da1f9c2-184a-46e4-8b49-2211f5bd31ab/kcbt_a_10926234_f0007.gif)
There are multiple therapeutic approaches beyond the use of obatoclax that can be rationally applied to suppress the expression/function of MCL-1 and BCL-XL in tumor cells. In prior studies, when combined with lapatinib, we have shown that cyclin-dependent kinase (CDK) inhibitors (flavopiridol, roscovitine) can substitute for obatoclax to kill breast cancer cells; CDK inhibitors, through inhibition of CDK9, reduce the protein levels of MCL-1 rather than as obatoclax which directly inhibits the function of the protein.Citation29 Hence, we determined whether a novel clinically relevant CDK9 inhibitor, dinaciclib, could enhance the lethality of the lapatinib as well as the novel ERBB1/ERBB2/ERBB4 suicide inhibitor afatinib in wild-type and AR BT474 cells. The combination of dinaciclib with either ERBB1/2 inhibitor (lapatinib or afatinib) resulted in a greater than additive induction of mammary tumor cell killing within 12 h, including in AR cells (). Very similar data were obtained using the chemically distinct CDK9 inhibitor flavopiridol (data not shown). This correlated with reduced expression of MCL-1 (, upper inset panel). Pre-treatment of AR BT474 cells with valproate resulted in a greater lethal effect of a subsequent afatinib + dinaciclib/lapatinib + dinaciclib treatment (). Very similar data to that in BT474 cells was obtained in MCF7 cells (data not shown). Treatment of breast cancer cells with lapatinib + obatoclax enhanced their sensitivity to ionizing radiation and to doxorubicin, an effect that was reduced in anoikis-resistant cells (). Pre-treatment of AR cells with valproate restored to a significant extent the chemo- and radio-sensitization effect observed in wild-type cells ().
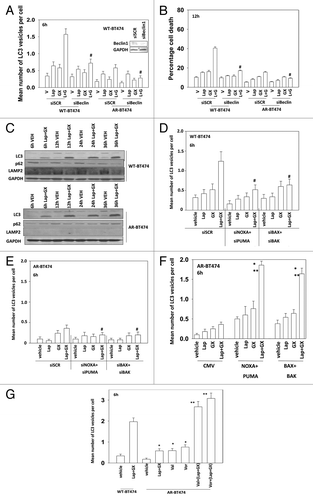
The induction of autophagy and the stalling of autophagy followed by release and fluxing plays a major role in mediating the toxicity of lapatinib + obatoclax treatment.Citation17-Citation19 As expression of Beclin1 was elevated in BT474-AR cells we next examined the induction of autophagy by lapatinib + obatoclax treatment. Treatment of wild-type parental BT474 cells expressing LC3-GFP with lapatinib + obatoclax increased the percentage of intense GFP-positive staining cells (). In BT474-AR cells, despite expressing more Beclin1, much less induction of autophagy induction was observed by this method (cf. ). Knockdown of Beclin1 expression suppressed autophagy and lapatinib + obatoclax-induced cell killing (). In wild-type parental BT474 cells increased intense GFP staining correlated with increased LC3II levels and decreased p62 and LAMP2 levels (). In BT474-AR cells although LC3II levels were increased no decrease in p62 levels was observed and LAMP2 expression was undetectable, suggestive of a stalled autophagy process and reduced acidic endosome levels in AR cells. Knockdown of BAX+BAK or of NOXA+PUMA suppressed the induction of autophagy in wild-type parental cells but had little effect in AR cells (). Expression of BAX+BAK or NOXA+PUMA restored the autophagic response of AR cells (). Pre-treatment of AR cells with HDAC inhibitors facilitated the induction of autophagy by a subsequent lapatinib + obatoclax treatment ().
Figure 7. HDAC inhibitor treatment restores the autophagy response of AR-BT474 cells. (A) BT474 (WT, wild type,; AR, anoikis-resistant) cells were transfected with a plasmid to express LC3-GFP and with scrambled siRNA (siSCR, 20 nM) or an siRNA molecule to knock down Beclin1. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. Six hours after drug exposure cells were examined under a fluorescent microscope and the number of intense GFP punctae per cell determined in at least 50 cells (± SEM, n = 3) #P < 0.05 less than corresponding value in siSCR cells. (B) BT474 (WT, wild type; AR, anoikis-resistant) cells were transfected with scrambled siRNA (siSCR, 20 nM) or an siRNA molecule to knock down Beclin1. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM), or the drug combination. Cells were isolated 12 h later and viability determined by trypan blue (± SEM, n = 3) #P < 0.05 less than corresponding value in siSCR cells. (C) BT474 (WT, wild type; AR, anoikis-resistant) cells were treated with vehicle (VEH, DMSO) or lapatinib (lap, 1 μM) and obatoclax (GX, 50 nM). Cells were isolated 6, 12, 24, and 36 h after drug exposure. Immunoblotting was performed to determine the expression of the indicated proteins (n = 3). (D) BT474 (WT, wild type) cells were transfected with a plasmid to express LC3-GFP and with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down BAK, BAX, NOXA, and PUMA, as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Six hours after drug exposure cells were examined under a fluorescent microscope and the number of intense GFP punctae per cell determined in at least 50 cells (± SEM, n = 3) #P < 0.05 less than corresponding value in siSCR cells. (E) BT474 (AR, anoikis-resistant) cells were transfected with a plasmid to express LC3-GFP and with scrambled siRNA (siSCR, 20 nM) or siRNA molecules to knock down BAK, BAX, NOXA, and PUMA, as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Six hours after drug exposure cells were examined under a fluorescent microscope and the number of intense GFP punctae per cell determined in at least 50–100 cells (± SEM, n = 3) #P < 0.05 less than the corresponding value in siSCR cells. (F) BT474 (AR, anoikis-resistant) cells were transfected with a plasmid to express LC3-GFP and with empty vector plasmid (CMV) or plasmids to express BAX, BAK, NOXA, and PUMA as indicated. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Six hours after drug exposure cells were examined under a fluorescent microscope and the number of intense GFP punctae per cell determined in at least 50 cells (± SEM, n = 3) *P < 0.05 greater than corresponding value in CMV cells. (G) BT474 (WT, wild type; AR, anoikis-resistant) cells were transfected with a plasmid to express LC3-GFP. Thirty-six hours after transfection cells were treated with vehicle (VEH, DMSO), lapatinib (lap, 1 μM), obatoclax (GX, 50 nM) or the drug combination. Six hours after drug exposure cells were examined under a fluorescent microscope and the number of intense GFP punctae per cell determined in at least 50 cells (± SEM, n = 3) *P < 0.05 greater than value in vehicle cells; **P < 0.05 greater than corresponding value in Lap+GX cells.
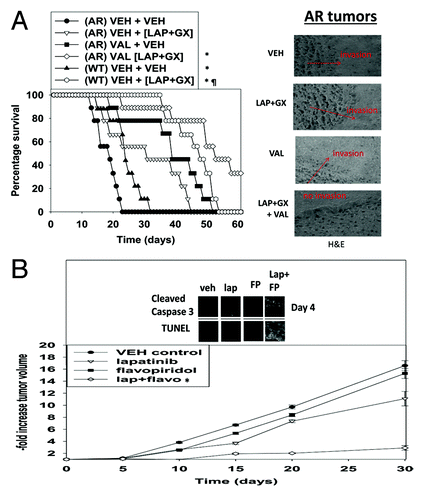
We next examined the effect of a 24 h valproate pre-treatment of established AR-BT474 intra-cranial tumors on their response to a subsequent lapatinib + obatoclax treatment. AR-BT474 tumors grew more rapidly than WT-BT474 tumors, as judged by a more rapid animal nadir, in general agreement with our in vitro data examining these cells (). Treatment of WT-BT474 tumors with lapatinib + obatoclax significantly prolonged animal survival compared with vehicle control and, to our surprise, treatment of AR-BT474 tumors with the drug combination also significantly promoted animal survival. Valproate treatment of AR-BT474 tumors caused a profound prolongation of survival, and treatment of AR-BT474 tumors with valproate followed by lapatinib + obatoclax also resulted in a significant enhancement in survival. In agreement with our in vitro data using CDK inhibitors wherein CDK9 inhibitors enhanced lapatinib toxicity; the CDK inhibitor flavopiridol also enhanced the anti-tumor effect of lapatinib in vivo ().
Figure 8. Valproate pre-treatment of AR-BT474 cells growing in mouse brains enhances animal survival. (A) Intracerebral injection of WT-BT474 and AR-BT474 cells was performed over 10 min (1 × 106 cells, each). Seven days after tumor cell implantation, animals were segregated into treatment groups. For animal administration sodium valproate was dissolved in saline. Animals were treated with saline vehicle or sodium valproate to a final concentration of 100 mg/kg QD for 2 d. For animal administration, lapatinib and obatoclax were first dissolved in DMSO, and an equal volume of 50:50 Cremophor EL/ethanol (Sigma-Aldrich) was added. After mixing, a 1:10 dilution was made with sterile PBS. Animals were treated with vehicle (PBS/Cremophor EL/ethanol/DMSO), lapatinib, obatoclax, or a combination of lapatinib and obatoclax using oral gavage to a final concentration of 5 mg/kg QD body mass for obatoclax and 100 mg/kg BID for lapatinib for 3 d. *P < 0.05 greater survival than in AR-BT474 cells with vehicle; ¶P < 0.05 greater survival than vehicle treated WT-BT474 cells. Inset: H&E staining of tumors at the time of animal death. (B) WT-BT474 cells (1 × 106 cells, each animal) were injected into the fourth mammary fat pad. Fourteen days after tumor cell implantation tumors of ~100 mm3 had formed, and animals were segregated into treatment groups. For animal administration, lapatinib and flavopiridol were first dissolved in DMSO, and an equal volume of 50:50 Cremophor EL/ethanol (Sigma-Aldrich) was added. After mixing, a 1:10 dilution was made with sterile PBS. Animals were treated with vehicle (PBS/Cremophor EL/ethanol/DMSO), lapatinib, flavopiridol, or a combination of the drugs using oral gavage to a final concentration of 25 mg/kg QD body mass for flavopiridol and 50 mg/kg BID for lapatinib for 3 d. *P < 0.05 greater survival than in vehicle-treated cells.
Discussion
The present studies were designed to define the mechanisms of anoikis resistance (AR) in breast cancer cells and how these resistance mechanisms could potentially be reversed to achieve a better therapeutic outcome, in particular in their response to the drug combination lapatinib + obatoclax.Citation17-Citation19
AR breast cancer cells had multiple defects in their ability to undergo cell death processes. AR cells lacked expression of multiple toxic BH3 domain proteins and had increased expression of c-FLIP-s, resulting in an inherent reduction in the ability of either the intrinsic or extrinsic apoptosis pathways from being engaged. In addition, AR cells had reduced expression of PTEN and increased activity within the PI3K/AKT and ERK1/2 pathways, pathways known to protect tumor cells from noxious stresses. Genetic restoration of PTEN expression or inhibition of the PI3K/AKT and ERK1/2 pathways partially restored the sensitivity of AR cells to lapatinib + obatoclax treatment.
Knockdown of c-FLIP-s in AR cells weakly enhanced the toxic response to lapatinib + obatoclax treatment. In contrast, exogenous re-expression of toxic BH3 domain proteins largely restored the toxic response to lapatinib + obatoclax treatment. Individually BAX, BAK, NOXA, and PUMA restoration all resulted in a significant restoration of lapatinib + obatoclax toxicity, with the combination producing a greater death response to drug treatment. We did not find that BIM expression was noticeably altered in our system and that BIM knock down as a single protein did not alter the therapeutic response to lapatinib + obatoclax treatment (unpublished observations).
HDAC inhibitors cause altered gene expression as well as oxidative damage to cells, and both processes can contribute to their lethality. Prior studies in a variety of different cell types have shown that HDAC inhibitors can increase expression of toxic BH3 domain proteins and reduce those of protective MCL-1.Citation21-Citation26 In AR breast cells that had reduced expression of several toxic BH3 domain proteins, a 24 h pre-treatment with clinically relevant concentrations of HDAC inhibitors resulted in elevated (re-)expression of BH3 domain proteins. Furthermore, increased expression of these BH3 domain proteins restored the lethality of a subsequent lapatinib + obatoclax treatment or reverted in part the resistance of AR doxorubicin-resistant BT474 cells.
Prior studies from this laboratory have shown that lapatinib + obatoclax toxicity was associated with a toxic form of autophagy that required expression of NOXA and BAK.Citation17-Citation19 In response to lapatinib + obatoclax treatment AR cells exhibited similar levels of LC3II production as judged by western blotting of the faster migrating form of LC3 but neither exhibited strong staining for increased levels of LC3-GFP vesicles nor did they show decreased p62 or LAMP2 expression after drug treatment. AR cells had reduced expression of LAMP2 that correlated with reduced autophagic flux. Treatment of AR cells with an HDAC inhibitor facilitated lapatinib + obatoclax-induced LC3-GFP vesicularization in a BH3 domain protein-dependent manner.
A priori it would be predicted that for tumor cells to migrate from the mammary environment through the vascular system to the brain requires some degree of anoikis resistance development. Previously we demonstrated that the survival of mice carrying wild type parental BT474 tumors in their brains was increased following lapatinib + obatoclax treatment.Citation18 Based on our in vitro data it would be predicted that anoikis-resistant tumors would be less sensitive to lapatinib + obatoclax treatment, as we in fact observed to be the case based on animal survival. Pre-treatment of established anoikis-resistant BT474 tumors with sodium valproate also rapidly increased the expression of multiple toxic BH3 domain proteins in vivo (unpublished data). Collectively our in vitro and in vivo data argue that pre-treatment of mammary tumor cells with HDAC inhibitors increases the levels of multiple pro-apoptotic proteins which predisposes the cells to being killed by lapatinib + obatoclax treatment.
In addition to obatoclax we have also determined whether other BCL-2/BCL-XL/MCL-1 inhibitors enhance lapatinib toxicity. AT-101, in a manner similar to obatoclax, inhibits the protective functions of BCL-2/BCL-XL/MCL-1. AT-101 enhanced lapatinib toxicity to a similar extent as obatoclax in mammary and brain cancer cells (Unpublished observations). However, we have previously shown that inhibitors of BCL-2/BCL-XL but not of MCL-1 (i.e., ABT-263, low Sabutoclax concentrations) do not enhance lapatinib toxicity, arguing that inhibition of MCL-1 is essential to promote killing by this RTK inhibitor. In a similar fashion, CDK9 inhibitors that inhibit MCL-1 and BCL-XL expression also interacted with lapatinib/afatinib to kill mammary carcinoma cells, and combined with valproate to kill AR cells. At present there is no information as to whether AT-101, dinaciclin, or afatinib can cross the blood–brain barrier and alter the biology of CNS localized tumors.
Over the past several years obatoclax has completed multiple Phase I and Phase II evaluations.Citation30-Citation35 Obatoclax has been administered intravenously either over 3 h (30 mg or 20 mg/m2 dose) for days 1–3 of a 2 week cycle or over 24 h (60 mg dose) once every 2 weeks. Toxicities that were considered to be related to obatoclax infusion have been somnolence, euphoric mood, insomnia, and ataxia, which were mostly grade 1 and transient resolving within 1 h post-infusion with no long-term post-infusion sequelae. It has been argued that in addition to BCL-2 proteins obatoclax also crosses the blood–brain barrier and binds to opiod receptors. For the 3 h infusion peak plasma concentrations were approximately 2 μM that rapidly declined by 24 h to less than 100 nM. Thus our use of obatoclax in the 50–100 nM range over a 24 h incubation/time course in this and our other manuscripts has clinical relevance. Because of CNS toxicity the development of obatoclax as a therapeutic has stopped; based on our encouraging data in CNS localized tumors, we would argue that this agent may still have a utility as a cancer therapeutic drug.
In conclusion, anoikis-resistant mammary carcinoma cells have a reduced ability to undergo cell death processes that correlates with increased expression of protective BCL-2 proteins and decreased expression of multiple toxic BH3 domain proteins. Expression of toxic BH3 domain proteins can be at least partially restored by treatment of cells with HDAC inhibitors. Pre-treatment of anoikis-resistant breast cancer cells with an HDAC inhibitor enhances lapatinib + obatoclax lethality in vitro and in vivo. Based on prior data and the present studies, it appears logical to combine lapatinib with an inhibitor of BCL-2/BCL-XL/MCL-1 or with a CDK9 inhibitor in a phase I trial of recurrent cancer patients.
Materials and Methods
Materials
Vorinostat (SAHA) and sodium valproate were supplied by Calbiochem as a powder, dissolved in sterile DMSO, and stored frozen under light-protected conditions at –80 °C. Other kinase inhibitors and obatoclax were from Selleckchem. Trypsin-EDTA, DMEM, and RPMI medium, and penicillin–streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies). Cytoselect cell viability and cytotoxicity assay kit was from Cell Biolabs. The Sceptor cell viability system was from Millipore. Other reagents were of the highest quality commercially available. See refs. Citation15, Citation17–Citation19, and Citation27–Citation29.
Methods
Cell culture and in vitro exposure of cells to drugs
All breast cancer lines were cultured at 37 °C (5% (v/v CO2) in vitro using RPMI supplemented with 10% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. Glioma cells were sub-cultured in 5% (v/v) fetal calf serum. For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 36 h after plating were treated with various drugs, as indicated. In vitro small molecule inhibitor treatments were from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). For adenoviral infection, cells were infected 12 h after plating and the expression of the recombinant viral transgene allowed to occur for at least 12 h prior to any additional experimental procedure. Cells were not cultured in reduced serum media during any study.
Generation of anoikis-resistant breast and brain cancer stem cells
Mammary carcinoma cells were plated on poly hydroxyl ethyl-methacrylate (poly-HEMA) coated 10 cm tissue culture plates (2 × 106 cell/dish). Cells were cultured for 1 week, and viable cells isolated and re-plated on a fresh poly-HEMA coated plate. Examination of free floating anoikis-resistant mammary carcinoma cells took place at least 4 weeks after the initial plating. Multiple isolations of BT474 anoikis-resistant cells were made, with all exhibiting a very similar growth and invasion biology and protein expression pattern (data not shown). GBM cells were re-suspended in RPMI media containing 20 ng/ml of EGF; 20 ng/ml bFGF. Cells were grown on poly-HEMA coated plates as described above to facilitate anchorage free tumor cell growth. CD133+ glioma cells were isolated by fluorescence-activated cell sorting analysis and continued in stem cell growth media until stable isolates of neurospheres were isolated. Neurospheres of GBM6- and GBM12-derived stem cells readily proliferated and displayed a non-adherent growth pattern.
Recombinant adenoviral vectors: Infection in vitro
We generated and purchased previously noted recombinant adenoviruses to express dominant negative caspase 9, dnAKT, dnMEK1, c-FLIP-s, and BCL-XL (Vector Biolabs). Cells were infected with these adenoviruses at an approximate m.o.i. of 50. Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.Citation17-Citation19
Detection of cell death by Trypan Blue assays
Cells were harvested by trypsinization with Trypsin/EDTA for ~10 min at 37 °C. As some apoptotic cells detached from the culture substratum into the medium, these cells were also collected by centrifugation of the medium at 1500 rpm for 5 min. The pooled cell pellets were resuspended and mixed with trypan blue dye. Trypan blue stain, in which blue dye incorporating cells were scored as being dead was performed by counting of cells using a light microscope and a hemacytometer. Five hundred cells from randomly chosen fields were counted and the number of dead cells was counted and expressed as a percentage of the total number of cells counted. Cell killing was confirmed using the Sceptor instrument which measured tumor cell size/sub G1 DNA as an indication of tumor cell viability.
Plasmid transfection
Plasmid DNA (0.5 μg/total plasmid transfected) was diluted into 50 μl of RPMI growth media that lacked supplementation with FBS or with penicillin-streptomycin. Lipofectamine 2000 reagent (1 μl) (Invitrogen) was diluted into 50 μl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The two solutions were then mixed together and incubated at room temperature for 30 min. The total mix was added to each well (4-well glass slide or 12-well plate) containing 200 μl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The cells were incubated for 4 h at 37 °C, after which time the media was replaced with RPMI growth media containing 5% (v/v) FBS and 1× pen-strep.
Microscopy for LC3-GFP expression
Where indicated, LC3-GFP transfected cells were 12 h after transfection infected with drugs then cultured for 24 h. LC3-GFP transfected cells were visualized at the indicated time points on the Zeiss Axiovert 200 microscope using the FITC filter.
Cell migration assay
Place chambers in a 24-well plate. Add 100 μl of serum-free media to chambers. Place 200 μl of cells (2.5 × 105/ml in serum-free media) into each chamber. On a separate row in the plate, place 750 μl culture media (with serum): this is referred to as the lower chamber. Place chambers into wells containing serum media (lower chamber). Incubate at 37 °C for between 6–24 h (experiment dependent). Remove media from chamber and discard. Wash 2× with PBS. Place chamber into clean well. Fix with paraformaldehyde. Leave for 2 min at room temperature. Remove and wash 2× with PBS. Place chamber into clean well and add 100% methanol. Leave for 20 min. Remove and wash 2× with PBS and place in another clean well. Add Geimsa stain, cover and leave at room temperature for 15 min. Remove stain and wash 2× with PBS. Wipe off non-migrated cells with cotton swab (inside of chamber) and count cells under microscope.
Intracerebral and flank inoculations of BT474 cells
Athymic female NCr-nu/nu mice (National Cancer Institute) weighing 20 g were used for this study. Mice were maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, the US Department of Health and Human Services, and the National Institutes of Health. Mice were anesthetized via intraperitoneal administration of ketamine (40 mg/kg) and xylazine (3 mg/kg) and immobilized in a stereotactic frame (David Kopf Instruments). A 24-gauge needle attached to a Hamilton syringe was inserted into the right basal ganglia to a depth of 3.5 mm and then withdrawn 0.5 mm to make space for tumor cell accumulation. The entry point at the skull was 2 mm lateral and 1 mm dorsal to the bregma. Intracerebral injection of BT474 cells in 2 μl of PBS was performed over 10 min (1 × 106 cells). The skull opening was enclosed with sterile bone wax, and the skin incision was closed using sterile surgical staples. Seven days after tumor cell implantation, animals were segregated into treatment groups. For animal administration sodium valproate was dissolved in saline. Animals were treated with saline vehicle or sodium valproate to a final concentration of 100 mg/kg QD for one day. For animal administration, lapatinib and obatoclax were first dissolved in DMSO, and an equal volume of 50:50 Cremophor EL/ethanol (Sigma-Aldrich) was added. After mixing, a 1:10 dilution was made with sterile PBS. Animals were treated with vehicle (PBS/Cremophor EL/ethanol/DMSO), lapatinib, obatoclax, or a combination of lapatinib and obatoclax using oral gavage to a final concentration of 5 mg/kg QD body mass for obatoclax and 100 mg/kg BID for lapatinib for 3 d.
WT-BT474 cells (1 × 106 cells, each animal) were injected into the fourth mammary fat pad. Fourteen days after tumor cell implantation tumors of ~100 mm3 had formed, and animals were segregated into treatment groups. For animal administration, lapatinib and flavopiridol were first dissolved in DMSO, and an equal volume of 50:50 Cremophor EL/ethanol (Sigma-Aldrich) was added. After mixing, a 1:10 dilution was made with sterile PBS. Animals were treated with vehicle (PBS/Cremophor EL/ethanol/DMSO), lapatinib, flavopiridol, or a combination of the drugs using oral gavage to a final concentration of 25 mg/kg QD body mass for flavopiridol and 50 mg/kg BID for lapatinib for 3 d.
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a two tailed Student t test. Differences with a P value of < 0.05 were considered statistically significant. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
| Abbreviations: | ||
| Δψ | = | mitochondrial membrane potential |
| AVOs | = | acidic vesicular organelles |
| LC3 | = | microtubule-associated protein light chain 3 |
| ROS | = | reactive oxygen species |
| NAC | = | N-acetylcysteine |
| DAPI | = | 40-6-diamidino-2-phenylindole |
| LAMP-2 | = | lysosomal associated membrane protein 2 |
| BCL-2 | = | B cell CLL/lymphoma-2 |
| BCL-xL | = | BCL-2-related gene long isoform |
| MCL-1 | = | myeloid cell leukemia-1 |
| BAD | = | BCL-2 antagonist of cell death |
| BID | = | BCL-2-interacting domain death agonist |
| BIM | = | BCL-2-interacting mediator of cell death |
| BAK | = | BCL-2 antagonist killer |
| BAX | = | BCL-2-associated x protein |
| MOMP | = | mitochondrial outer membrane permeabilization |
| ER | = | estrogen receptor |
Acknowledgments
Studies in this manuscript were funded by Department of Defense Breast Cancer Idea award W81XWH-10-1-0009, National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases (DK52825), and National Cancer Institute (CA141703 and CA150218).
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
References
- Nagaprashantha L, Vartak N, Awasthi S, Awasthi S, Singhal SS. Novel anti-cancer compounds for developing combinatorial therapies to target anoikis-resistant tumors. Pharm Res 2012; 29:621 - 36; http://dx.doi.org/10.1007/s11095-011-0645-9; PMID: 22203324
- Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS. BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell 2007; 12:221 - 34; http://dx.doi.org/10.1016/j.devcel.2006.12.003; PMID: 17276340
- Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS. Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci U S A 2007; 104:3787 - 92; http://dx.doi.org/10.1073/pnas.0700115104; PMID: 17360431
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19:797 - 806; http://dx.doi.org/10.1091/mbc.E07-10-1092; PMID: 18094039
- Whelan KA, Caldwell SA, Shahriari KS, Jackson SR, Franchetti LD, Johannes GJ, Reginato MJ. Hypoxia suppression of Bim and Bmf blocks anoikis and luminal clearing during mammary morphogenesis. Mol Biol Cell 2010; 21:3829 - 37; http://dx.doi.org/10.1091/mbc.E10-04-0353; PMID: 20861305
- Zhong X, Rescorla FJ. Cell surface adhesion molecules and adhesion-initiated signaling: understanding of anoikis resistance mechanisms and therapeutic opportunities. Cell Signal 2012; 24:393 - 401; http://dx.doi.org/10.1016/j.cellsig.2011.10.005; PMID: 22024283
- Sodek KL, Murphy KJ, Brown TJ, Ringuette MJ. Cell-cell and cell-matrix dynamics in intraperitoneal cancer metastasis. Cancer Metastasis Rev 2012; 31:397 - 414; http://dx.doi.org/10.1007/s10555-012-9351-2; PMID: 22527451
- Beauséjour M, Noël D, Thibodeau S, Bouchard V, Harnois C, Beaulieu JF, Demers MJ, Vachon PH. Integrin/Fak/Src-mediated regulation of cell survival and anoikis in human intestinal epithelial crypt cells: selective engagement and roles of PI3-K isoform complexes. Apoptosis 2012; 17:566 - 78; http://dx.doi.org/10.1007/s10495-012-0713-6; PMID: 22402981
- Haenssen KK, Caldwell SA, Shahriari KS, Jackson SR, Whelan KA, Klein-Szanto AJ, Reginato MJ. ErbB2 requires integrin alpha5 for anoikis resistance via Src regulation of receptor activity in human mammary epithelial cells. J Cell Sci 2010; 123:1373 - 82; http://dx.doi.org/10.1242/jcs.050906; PMID: 20332114
- Zheng Y, Gierut J, Wang Z, Miao J, Asara JM, Tyner AL. Protein tyrosine kinase 6 protects cells from anoikis by directly phosphorylating focal adhesion kinase and activating AKT. Oncogene 2012; PMID: 23027128
- Atadja PW. HDAC inhibitors and cancer therapy. Prog Drug Res 2011; 67:175 - 95; PMID: 21141730
- Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem 2012; 4:1439 - 60; http://dx.doi.org/10.4155/fmc.12.80; PMID: 22857533
- Lee JH, Choy ML, Marks PA. Mechanisms of resistance to histone deacetylase inhibitors. Adv Cancer Res 2012; 116:39 - 86; PMID: 23088868
- Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2012; 31:537 - 51; PMID: 21725353
- Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Häussinger D, Reinehr R, Larner A, Spiegel S, Fisher PB, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res 2010; 70:6313 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-10-0999; PMID: 20631069
- Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, Dent P, Grant S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol 2006; 69:216 - 25; PMID: 16189296
- Cruickshanks N, Hamed HA, Bareford MD, Poklepovic A, Fisher PB, Grant S, Dent P. Lapatinib and obatoclax kill tumor cells through blockade of ERBB1/3/4 and through inhibition of BCL-XL and MCL-1. Mol Pharmacol 2012; 81:748 - 58; http://dx.doi.org/10.1124/mol.112.077586; PMID: 22357666
- Cruickshanks N, Tang Y, Booth L, Hamed H, Grant S, Dent P. Lapatinib and obatoclax kill breast cancer cells through reactive oxygen species-dependent endoplasmic reticulum stress. Mol Pharmacol 2012; 82:1217 - 29; http://dx.doi.org/10.1124/mol.112.081539; PMID: 22989520
- Tang Y, Hamed HA, Cruickshanks N, Fisher PB, Grant S, Dent P. Obatoclax and lapatinib interact to induce toxic autophagy through NOXA. Mol Pharmacol 2012; 81:527 - 40; http://dx.doi.org/10.1124/mol.111.076851; PMID: 22219388
- Smith MA, Houghton P. A proposal regarding reporting of in vitro testing results. Clin Cancer Res 2013; 19:2828 - 33; http://dx.doi.org/10.1158/1078-0432.CCR-13-0043; PMID: 23580781
- Bouzar AB, Boxus M, Defoiche J, Berchem G, Macallan D, Pettengell R, Willis F, Burny A, Lagneaux L, Bron D, et al. Valproate synergizes with purine nucleoside analogues to induce apoptosis of B-chronic lymphocytic leukaemia cells. Br J Haematol 2009; 144:41 - 52; http://dx.doi.org/10.1111/j.1365-2141.2008.07426.x; PMID: 19006566
- Pompeia C, Hodge DR, Plass C, Wu YZ, Marquez VE, Kelley JA, Farrar WL. Microarray analysis of epigenetic silencing of gene expression in the KAS-6/1 multiple myeloma cell line. Cancer Res 2004; 64:3465 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-03-3970; PMID: 15150099
- Premkumar DR, Jane EP, Agostino NR, DiDomenico JD, Pollack IF. Bortezomib-induced sensitization of malignant human glioma cells to vorinostat-induced apoptosis depends on reactive oxygen species production, mitochondrial dysfunction, Noxa upregulation, Mcl-1 cleavage, and DNA damage. Mol Carcinog 2013; 52:118 - 33; http://dx.doi.org/10.1002/mc.21835; PMID: 22086447
- Barbone D, Cheung P, Battula S, Busacca S, Gray SG, Longley DB, Bueno R, Sugarbaker DJ, Fennell DA, Broaddus VC. Vorinostat eliminates multicellular resistance of mesothelioma 3D spheroids via restoration of Noxa expression. PLoS One 2012; 7:e52753; http://dx.doi.org/10.1371/journal.pone.0052753; PMID: 23300762
- Feng L, Pan M, Sun J, Lu H, Shen Q, Zhang S, Jiang T, Liu L, Jin W, Chen Y, et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J Mol Med (Berl) 2013; 91:49 - 58; http://dx.doi.org/10.1007/s00109-012-0932-x; PMID: 22763818
- Fritsche P, Seidler B, Schüler S, Schnieke A, Göttlicher M, Schmid RM, Saur D, Schneider G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009; 58:1399 - 409; http://dx.doi.org/10.1136/gut.2009.180711; PMID: 19528037
- Martin AP, Miller A, Emad L, Rahmani M, Walker T, Mitchell C, Hagan MP, Park MA, Yacoub A, Fisher PB, et al. Lapatinib resistance in HCT116 cells is mediated by elevated MCL-1 expression and decreased BAK activation and not by ERBB receptor kinase mutation. Mol Pharmacol 2008; 74:807 - 22; http://dx.doi.org/10.1124/mol.108.047365; PMID: 18544666
- Martin AP, Mitchell C, Rahmani M, Nephew KP, Grant S, Dent P. Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol Ther 2009; 8:2084 - 96; http://dx.doi.org/10.4161/cbt.8.21.9895; PMID: 19823038
- Mitchell C, Yacoub A, Hossein H, Martin AP, Bareford MD, Eulitt P, Yang C, Nephew KP, Dent P. Inhibition of MCL-1 in breast cancer cells promotes cell death in vitro and in vivo. Cancer Biol Ther 2010; 10:903 - 17; http://dx.doi.org/10.4161/cbt.10.9.13273; PMID: 20855960
- Hwang JJ, Kuruvilla J, Mendelson D, Pishvaian MJ, Deeken JF, Siu LL, Berger MS, Viallet J, Marshall JL. Phase I dose finding studies of obatoclax (GX15-070), a small molecule pan-BCL-2 family antagonist, in patients with advanced solid tumors or lymphoma. Clin Cancer Res 2010; 16:4038 - 45; http://dx.doi.org/10.1158/1078-0432.CCR-10-0822; PMID: 20538761
- Chiappori AA, Schreeder MT, Moezi MM, Stephenson JJ, Blakely J, Salgia R, Chu QS, Ross HJ, Subramaniam DS, Schnyder J, et al. A phase I trial of pan-Bcl-2 antagonist obatoclax administered as a 3-h or a 24-h infusion in combination with carboplatin and etoposide in patients with extensive-stage small cell lung cancer. Br J Cancer 2012; 106:839 - 45; http://dx.doi.org/10.1038/bjc.2012.21; PMID: 22333598
- O’Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, Viallet J, Cheson BD. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood 2009; 113:299 - 305; http://dx.doi.org/10.1182/blood-2008-02-137943; PMID: 18931344
- Paik PK, Rudin CM, Brown A, Rizvi NA, Takebe N, Travis W, James L, Ginsberg MS, Juergens R, Markus S, et al. A phase I study of obatoclax mesylate, a Bcl-2 antagonist, plus topotecan in solid tumor malignancies. Cancer Chemother Pharmacol 2010; 66:1079 - 85; http://dx.doi.org/10.1007/s00280-010-1265-5; PMID: 20165849
- Parikh SA, Kantarjian H, Schimmer A, Walsh W, Asatiani E, El-Shami K, Winton E, Verstovsek S. Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin Lymphoma Myeloma Leuk 2010; 10:285 - 9; http://dx.doi.org/10.3816/CLML.2010.n.059; PMID: 20709666
- Schimmer AD, O’Brien S, Kantarjian H, Brandwein J, Cheson BD, Minden MD, Yee K, Ravandi F, Giles F, Schuh A, et al. A phase I study of the pan bcl-2 family inhibitor obatoclax mesylate in patients with advanced hematologic malignancies. Clin Cancer Res 2008; 14:8295 - 301; http://dx.doi.org/10.1158/1078-0432.CCR-08-0999; PMID: 19088047