
Abstract
Imatinib, an ABL tyrosine kinase inhibitor (TKI), has shown clinical efficacy against chronic myeloid leukemia (CML). However, a substantial number of patients develop resistance to imatinib treatment due to the emergence of clones carrying mutations in the protein BCR-ABL. The phosphoinositide 3 kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway regulates various processes, including cell proliferation, cell survival, and antiapoptosis activity. In this study, we investigated the efficacy of NVP-BEZ235, a dual PI3K and mTOR inhibitor, using BCR-ABL-positive cell lines. Treatment with NVP-BEZ235 for 48 h inhibited cell growth and induced apoptosis. The phosphorylation of the AKT kinase, eukaryotic initiation factor 4-binding protein 1 (4E-BP1), and p70 S6 kinase were decreased after NVP-BEZ235 treatment. The combination of NVP-BEZ235 with a BCR-ABL kinase inhibitor, imatinib, or nilotinib, induced a more pronounced colony growth inhibition, whereas the combination of NVP-BEZ235 and nilotinib was more effective in inducing apoptosis and reducing the phosphorylation of AKT, 4E-BP1, and S6 kinase. NVP-BEZ235 in combination with nilotinib also inhibited tumor growth in a xenograft model and inhibited the growth of primary T315I mutant cells and ponatinib-resistant cells. Taken together, these results suggest that administration of the dual PI3K and mTOR inhibitor NVP-BEZ235 may be an effective strategy against BCR-ABL mutant cells and may enhance the cytotoxic effects of nilotinib in ABL TKI-resistant BCR-ABL mutant cells.
Introduction
Chronic myeloid leukemia (CML) is a myeloproliferative disorder of hematopoietic stem cells, characterized by the presence of the Philadelphia chromosome (Ph).Citation1,Citation2 BCR-ABL kinase is largely responsible for the pathogenesis of CML. BCR-ABL activates the phosphatidylinositol-3 kinase (PI3K)/AKT, Ras, and Janus kinase (JAK)/signal transducers and activator of transcription (STAT) downstream signaling pathways; therefore, these molecules may have leukemogenic activity.Citation3-Citation5 Although imatinib is used for the treatment of CML and Ph-positive acute lymphoblastic leukemia (ALL),Citation6,Citation7 some patients develop secondary resistance to imatinib therapy. Several imatinib resistance mechanisms have been reported in vitro, which involve reactivation of the BCR-ABL signaling pathway. These include BCR-ABL gene amplification, BCR-ABL point mutation, and overexpression of LYN, a SRC-family kinase protein.Citation8,Citation9 Second-line BCR-ABL kinase inhibitors such as nilotinib and dasatinib are clinically available. The majority of imatinib-resistant or imatinib-intolerant patients responded to treatment with these second-generation BCR-ABL kinase inhibitors.Citation10 However, therapy with second-generation BCR-ABL kinase inhibitors or transplantation is less successful in patients with advanced or blast-phase CML than in those with chronic phase.Citation11 Furthermore, if quiescent leukemia stem cells are not eradicated, current BCR-ABL tyrosine kinase inhibitor will not cure the disease.Citation12 Alternative treatment modalities, such as BCR-ABL targeting tyrosine kinase inhibitors (TKIs) combined with tolerated agents that target pathways downstream of BCR-ABL could prevent the emergence of resistant clones.
PI3K is involved in the phosphorylation of membrane inositol lipids. AKT, also referred as to as protein kinase B, is a 57-kDa serine/threonine kinase that is the cellular homolog of the v-AKT oncogene, and it has 3 isoforms that share a high degree of sequence homology. All the 3 members (AKT1, AKT2, and AKT3) belong to the AGC family of protein kinases.Citation13 AKT1 and AKT2 are ubiquitously expressed and AKT3 is found in nervous tissue.Citation14 The PI3K/AKT signaling pathways play an important role in normal cellular processes such as proliferation, survival, and differentiation.Citation15 Mammalian target of rapamycin (mTOR) is a 289-kDa serine/threonine protein kinase belonging to the PI3K-related kinase family. It is established that the upregulation and activation of PI3K/AKT/mTOR signaling is important for conferring a growth advantage to leukemia cells, including CML cells. Moreover, activation of the PI3K/AKT/mTOR pathway negatively influences the tumor response to conventional antileukemic treatments.Citation16
In this study, we investigated the effects of NVP-BEZ235 on Ph-positive leukemia cells. NVP-BEZ235 is a dual inhibitor of PI3K and mTOR.Citation17 We clarified the mechanisms of action of NVP-BEZ235 in BCR-ABL–expressing cells and determined its efficacy alone and in combination with ABL kinase inhibitors such as nilotinib or imatinib. We found that the co-treatment with NVP-BEZ235 and BCR-ABL kinase inhibitors is effective against imatinib-resistant BCR-ABL-positive leukemia cells.
Results
Effect of NVP-BEZ235 and imatinib or nilotinib on Ba/F3 BCR-ABL random mutagenesis cells
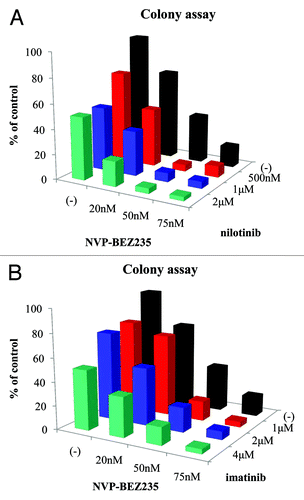
BCR-ABL point mutations are major mechanism of imatinib resistance.Citation8,Citation9 In many leukemia cases, patient samples contain multiple genetically distinct leukemia-initiating cell subclones and reconstitution with the predominant diagnosis clone is associated with more aggressive growth properties.Citation18 In particular, compound mutations are common in patients with sequencing evidence for double BCR-ABL1 mutations and frequently reflect a highly complex clonal network in Ph-positive leukemia patients.Citation19 Therefore, we used the in vitro screen of randomly mutagenized BCR-ABL Ba/F3 cells that confer resistance to ABL tyrosine kinase inhibitor in this study. We evaluated the various NVP-BEZ235 combinations employing random mutagenesis of Ba/F3 BCR-ABL cells and analyzed the antiproliferative activity by long-term culture colony assay. After exposure to indicated concentrations of NVP-BEZ235, the percentage of colony formation was lower than that in the control (). The inhibition rate calculated by colony assay was 62% with 50 nM NVP-BEZ235 and 84% with 75 nM. In this study, we examined the efficacy of the combination treatment of nilotinib or imatinib with NVP-BEZ235. We found that the proliferation of Ba/F3 BCR-ABL random mutagenesis cells was partially reduced by treatment with imatinib or nilotinib alone compared with the control. We found that the percentage of colony formation was significantly inhibited by the co-treatment with NVP-BEZ235 and nilotinib or imatinib. We also found that colony growth was completely inhibited by treatment with 50 nM NVP-BEZ235 and 500 nM nilotinib. Statistical analysis was also performed. Two-way ANOVA analysis revealed these drugs combined effects were additive.
Figure 1. Effects of NVP-BEZ235 and imatinib or nilotinib on Ba/F3 BCR-ABL random mutagenesis cells. Ba/F3 BCR-ABL random mutagenesis cells were treated with the indicated concentration of NVP-BEZ235 and/or nilotinib (A) or imatinib (B) for 14 d. The colony-formation assay was performed as described in Materials and Methods. The data are representative of 3 independent sets of experiments. Statistical analysis was also performed by using two-way ANOVA. Two-way ANOVA analysis revealed combined effects of two drugs were additive.
NVP-BEZ235 inhibits growth and induces apoptosis in BCR-ABL-positive cells
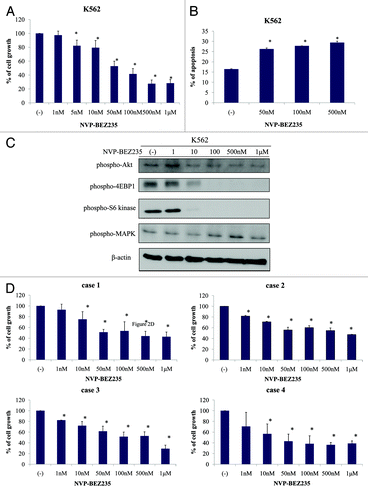
We next evaluated the effect of NVP-BEZ235 on BCR-ABL-positive cell line K562 using the cell proliferation assay. K562 cells were incubated for 48 h with increasing concentrations of NVP-BEZ235. As shown in , the cell growth of K562 was inhibited in a dose-dependent manner. Effective inhibitory concentrations for this cell line were found in the nanomolar range. We next examined whether NVP-BEZ235 induced apoptosis in K562 cells. As indicated in , the percentage of Annexin V-positive cells increased after 48 h NVP-BEZ235 treatment in a dose-dependent manner. We also evaluated the intracellular signaling after NVP-BEZ235 treatment. 4E-BP1 and S6 kinases are the downstream targets of PI3K/AKT and mTOR pathways, and these are correlated with protein transcription.Citation20 In immunoblot analysis, we found that the phosphorylation level of AKT, 4E-BP1, and S6 kinase decreased after 24 h NVP-BEZ235 treatment in a dose-dependent manner. However, the phosphorylation of MAPK was not found to be reduced (). We next examined the effect on Ph-positive primary cells. We found that the cellular growth of Ph-positive primary cells was inhibited by NVP-BEZ235 in a dose dependent manner ().
Figure 2. NVP-BEZ235 inhibits cell growth and induces apoptosis in Ph-positive cells. (A) Cells were cultured at a concentration of 8 × 104/mL in the presence or absence of NVP-BEZ235 for 48 h. Viable cells were evaluated as described in Materials and Methods. (B) K562 cells were treated with NVP-BEZ235 at the indicated concentration for 48 h. The percentage of apoptotic cells was examined as described in Materials and Methods. (C) K562 cells were treated with NVP-BEZ235 for 24 h; total extracts were analyzed by immunoblot analysis with anti-phospho Akt, 4E-BP1, S6 kinase, MAPK, and anti-actin antibodies. The data presented here are representative of 3 separate experiments. (D) Ph-positive primary cells were cultured at a concentration of 2 × 105/mL in the presence or absence of NVP-BEZ235 for 48 h. Viable cells were evaluated as indicated in Materials and Methods.
Co-treatment with NVP-BEZ235 and nilotinib inhibits growth and induces apoptosis of K562 and primary cells
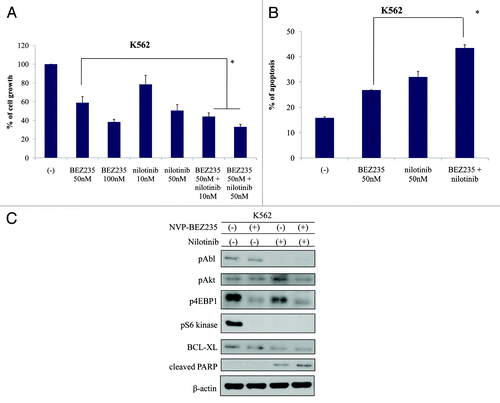
To examine the inhibitory effect of NVP-BEZ235 and nilotinib on the proliferation of CML cells, we evaluated the growth of K562 cells before and after NVP-BEZ235 and/or nilotinib treatment. The cell growth of K562 was inhibited by NVP-BEZ235 and nilotinib treatment (). As indicated in , the percentage of Annexin V-positive cells increased after NVP-BEZ235 and nilotinib treatment. We next examined the intracellular signaling before and after NVP-BEZ235 and/or nilotinib treatment in K562 cells. The results indicated that the phosphorylation of BCR-ABL was reduced after 24 h treatment with nilotinib. The phosphorylation of AKT, 4E-BP1, and S6 kinase reduced after co-treatment with NVP-BEZ235 and nilotinib. Moreover, BCL-XL was downregulated and cleaved PARP was upregulated after treatment with NVP-BEZ235 and nilotinib ().
Figure 3. Effect of NVP-BEZ235 and nilotinib treatment on K562 cells. (A and B) Cells were cultured in the presence or absence of NVP-BEZ235 or nilotinib for 48 h. Viable cells and apoptotic cells were evaluated as described in Materials and Methods. *P < 0.05 for nilotinib and NVP-BEZ235 treatment vs. treatment with 50 nM nilotinib alone in the same cell line. (C) K562 cells were treated with NVP-BEZ235 and/or nilotinib for 24 h; total cellular lysates were immunoblotted with anti-phospho Abl, Akt, 4E-BP1, S6 kinase, BCL-XL, PARP, and actin Abs.
Effects of NVP-BEZ235 and nilotinib on Ba/F3 BCR-ABL random mutagenesis cells in a xenograft model
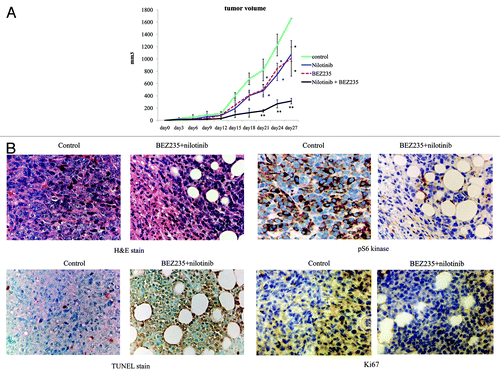
To assess the activity of NVP-BEZ235, we tested CML tumor formation in mice. Therefore, we injected nude mice subcutaneously with 1 × 107 Ba/F3 BCR-ABL random mutagenesis cells. The next day, mice were separated into 4 groups (control, nilotinib, NVP-BEZ235, and nilotinib + NVP-BEZ235). Control mice were treated with 0.9% NaCl daily. Tumor size was evaluated every 3 d. An orally administered dose of 30 mg/kg/day of nilotinib or NVP-BEZ235 inhibited tumor growth and reduced tumor volume compared with control mice. Moreover, it was observed that the tumor volume in the nilotinib + NVP-BEZ235 group decreased significantly (P < 0.001) (). The tumor from mice treated with nilotinib and NVP-BEZ235 displayed higher necrosis levels compared with that from vehicle-treated mice. We also performed immunohistochemical analysis. TdT-mediated dUTP nick-end labeling (TUNEL) assay showed that the number of apoptotic cells was higher and the expression level of the proliferation maker Ki-67 was lower in the nilotinib and NVP-BEZ235 treatment group than in the other groups (). Furthermore, we found that the phosphorylation of S6 kinase was significantly lower in the nilotinib and NVP-BEZ235 combination treatment group compared with that in the control mice. These results suggest that nilotinib and NVP-BEZ235 treatment effectively suppress tumor growth in vivo and that the tumor inhibition achieved by the combinatorial treatment was superior to that achieved by nilotinib or NVP-BEZ235 alone.
Figure 4. Effect of NVP-BEZ235 and nilotinib on Ba/F3 BCR-ABL random mutagenesis cells in xenograft model. (A) In vivo studies were performed as described in Materials and Methods. (B) Tumor cells treated with or without NVP-BEZ235 and nilotinib for 24 d were examined by immunohistochemical analysis as described in Materials and Methods. Original magnification: 400×. H&E, hematoxylin and eosin; TUNEL, TdT-mediated dUTP nick-end labeling. *P < 0.01, **P < 0.001 compared with control.
Co-treatment with NVP-BEZ235 and nilotinib inhibits the growth of wild type and mutant BCR-ABL-positive cells
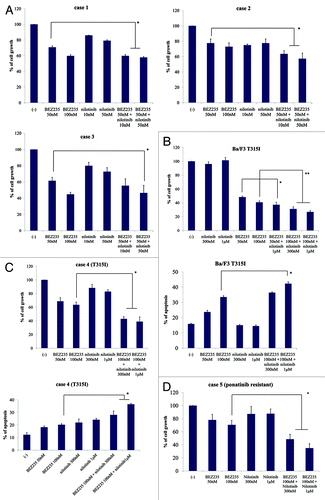
Because co-treatment with NVP-BEZ235 and nilotinib inhibited colony formation, we investigated whether NVP-BEZ235 and nilotinib treatment could inhibit Ph-positive primary cells as well. The results showed that 48 h NVP-BEZ235 and nilotinib co-treatment suppressed the growth of Ph-positive primary cells (). We next investigated the effect of the treatment on T315I point mutant primary cells. We found that NVP-BEZ235 and nilotinib inhibited cell growth and induced apoptosis of T315I-positive cells (). In addition, we found that NVP-BEZ235 and nilotinib combination treatment inhibited the growth of ponatinib (AP24534)-resistant primary cells (). These results indicated that the combination of NVP-BEZ235 and nilotinib treatment is effective against Ph-positive primary cells, including ABL TKI-resistant cells.
Figure 5. Co-treatment with NVP-BEZ235 and nilotinib inhibits cell growth and induces apoptosis of wild-type and T315I mutant BCR-ABL-positive cells. (A) Wild-type primary cells were cultured at a concentration of 2 × 105/mL in the presence or absence of NVP-BEZ235 or nilotinib for 48 h. Viable cells were evaluated. (B) Ba/F3 T315I cells were cultured in the presence or absence of NVP-BEZ235 or nilotinib for 48 h. Viable cells and percentage of apoptotic cells was examined. (C) T315I mutant primary cells were cultured in the presence or absence of NVP-BEZ235 or nilotinib for 48 h. Viable cells and percentage of apoptotic cells was examined. (D) Ponatinib-resistant primary cells were treated with NVP-BEZ235 and/or nilotinib at the indicated concentration for 48 h. Viable cells were evaluated as described in Materials and Methods. *P < 0.05, nilotinib and NVP-BEZ235 treatment vs. treatment with 1 μM nilotinib alone in the same cell line.
Discussion
Imatinib resistance caused by ABL kinase domain mutation activates the downstream targets of BCR-ABL, such as the PI3K/Akt/mTOR pathways, which have recently been implicated in the survival and expansion of leukemic cells. Therefore, the targeting of the PI3K/Akt/mTOR signaling pathway by specific kinase inhibitors have been the focus of extensive treatment for Ph-positive leukemia cells including T315I mutation.
A number of PI3K/Akt/mTOR signaling inhibitors are under investigation. Among these, NVP-BEZ235 has shown antitumor activity against various tumor types,Citation21-Citation23 and it is now being evaluated in clinical trials. NVP-BEZ235 has shown in vivo anticancer efficacy in models of lung cancer, breast cancer, glioma, and myeloma.Citation23-Citation26 Another dual PI3K/mTOR inhibitor, PI-103 and Mdm2 inhibitor, Nutlin-3 enhances the antileukemic activity-mediated p53 pathway in acute myeloid leukemia (AML) cells.Citation27 The simultaneous inhibition of PI3K/Akt/mTOR and MAPK signaling is effective against epidermal growth factor receptor (EGFR) mutant lung cancer.Citation28 Although it has been reported that the co-treatment of the PI3K inhibitors LY294002 or wortmannin with imatinib enhances the antileukemic activity in CML,Citation29 these compounds cannot be used in clinical setting due to their severe toxicity. Rapamycin has been shown to be effective in vitro against imatinib-resistant CML cellsCitation30 and a phase I–II study of everolimus (RAD001), a derivative of rapamycin, in combination with imatinib in imatinib-resistant CML patients.
In the present study, we evaluated the potential antileukemic effect and the action mechanism of NVP-BEZ235 against BCR-ABL-positive cells. NVP-BEZ235 displayed a statistically significant growth inhibition of BCR-ABL mutant cell () and the combined use of NVP-BEZ235 and nilotinib induced apoptosis in BCR-ABL-positive cells, Ph-positive primary cells including T315I mutation, and ponatinib-resistant cells (). In this study, we could not demonstrate the synergistic effect when the two agents were combined in colony assay. Our screen links mutations in these residues to drug resistance. Because the number of clones isolated from this type of screen, many of the novel ABL TKI-resistant variants recovered. These TKI-resistant variants suggest these might be weak cooperating mutations whose modest effects might not be resolved in short-term cell growth assays. We also demonstrated that co-treatment with nilotinib and NVP-BEZ235 enhanced antitumor activity in vivo and was less toxic in a xenograft model. The challenge for the development of an effective Ph-positive leukemia therapy is to develop an alternative treatment strategy that does not rely solely on kinase domain inhibition. In this study, we showed that the targeting of PI3K/Akt/mTOR signaling network with pharmacological inhibitors in combination with ABL TKIs is an efficacious treatment of Ph-positive leukemia.
In summary, the experimental data presented in this report suggests that NVP-BEZ235, a PI3K/AKT-mTOR inhibitor, potentiates nilotinib-induced apoptosis of BCR-ABL-positive leukemia cells through S6 kinase inhibition. Combination regimens involving novel and conventional anti-BCR-ABL agents may be a key strategy to improve clinical benefits and reduce the development of imatinib-resistant clones. This molecular model will provide useful information for development of therapeutic strategies against Ph-positive cells.
Materials and Methods
Cell, reagents, and animals
A BCR-ABL-positive cell line K562 was obtained from the American Type Culture Collection. Ba/F3 BCR-ABL, BCR-ABL point mutant cells, and BCR-ABL random mutagenesis cells were described previously.Citation31,Citation32 A full-length BCR-ABL cDNA was cloned into pMSCV-IRES-GFP retroviral vector, generating MSCV-IRES-GFP-B/A. XL-1 Red competent Escherichia coli cells (Stratagene) deficient in DNA repair mechanisms were used for transformation. The plasmid libraries were transfected onto HEK 293T cells and the viral supernatant was collected. Murine Ba/F3 cells were then transfected with the mutated vector. Ba/F3 cells expressing the different BCR-ABL constructs became interleukin-3 (IL-3)-independent and were grown in Roswell Park Memorial Institute (RPMI) without IL-3. These cells were cultured in the RPMI1640 medium containing 10% fetal bovine serum (FBS). Nude mice were obtained from Clea Japan and maintained under standard conditions according to the institutional guidelines. NVP-BEZ235, nilotinib and imatinib were obtained from Novartis Pharma AG. Stock solutions of NVP-BEZ235 and nilotinib were prepared in dimethyl sulfoxide (DMSO), and imatinib was dissolved in distilled water prior to dilution to the desired concentration in the growth medium. Anti-phospho Abl, anti-phospho CRK-L, S6 kinase, eukaryotic initiation factor 4-binding protein 1 (4E-BP1), mitogen-activated Protein kinase (MAPK), anti-cleaved caspase 3, and poly (ADP-ribose) polymerase (PARP) antibodies (Abs) were purchased from Cell Signaling. Other reagents were obtained from Sigma.
Cell proliferation assay
Cell proliferation analysis was performed as previously described.Citation33,Citation34 Briefly, the cells were seeded in 24- or 96-well plates at a density of 1 × 105 or 1 × 104 cells/well. The cells were treated with the inhibitors at the following concentrations: NVP-BEZ235, 1–100 nM; nilotinib, from 10 nM to 1 μM. Viable cells were counted by trypan blue exclusion or fluorescence-activated cell sorting (FACS).
FACS analysis
The cells were treated with NVP-BEZ235 and/or nilotinib at the concentrations described above for 48 h. Annexin V/propidium iodide apoptosis assay was performed according to the manufacturer’s protocol (Becton, Dickinson and Company). The cells were mixed gently and were analyzed immediately by flow cytometry.
Western blotting
The cells were treated with NVP-BEZ235 at indicated times and concentrations, and washed once with ice-cold phosphate-buffered saline (PBS). The cells were then lysed in lysis buffer for 20 min on ice. The lysates were centrifuged at 12 000 rpm for 20 min at 4 °C. The protein content of the lysates was determined using a protein assay kit (Bio-Rad Laboratories). Equivalent amounts of proteins of the cell lysates were boiled with 2× SDS sample buffer for 5 min. The proteins were loaded onto polyacrylamide gels and then transferred to polyvinylidine difluoride (PVDF) membranes (Millipore). The membranes were blocked by 1% bovine serum albumin (BSA) in PBS-Tween 20 (PBST) and probed with the indicated primary antibodies at appropriated dilution for 2 h at room temperature (RT). Blots were probed with secondary antibodies conjugated with horseradish peroxidase and developed using the enhanced chemiluminescence (ECL, Amersham Phamacia Biotech) system with ECL-film according to the manufacture’s specifications.
In vivo studies
The mice were injected subcutaneously with 1 × 107 Ba/F3 BCR-ABL mutagenesis cells. The mice were treated orally with 30 mg/kg NVP-BEZ235 or 30 mg/kg nilotinib, or a combination of the 2 agents daily. Control mice were treated with 0.9% NaCl of the vehicle. Tumor sizes were observed at various time points. The average tumor weight per mouse (n = 5) was calculated and was used to analyze the group mean tumor weight ± standard error (s.e.).
Immunohistochemical analysis
Tumor cells were collected at indicated time points and fixed in paraformaldehyde. Paraffin-embedded tissues were stained with hematoxylin and eosin (H&E) and TdT-mediated dUTP nick-end labeling (TUNEL), and treated with phospho S6 kinase and Ki-67 antibodies.
Primary BCR-ABL-positive cells
This study was performed according to approved laboratory protocols and in accordance with the Declaration of Helsinki. Peripheral blood was collected in heparinized tubes and mononuclear cells were separated using LymphoSepare (Immuno-Biological Laboratories). These cells were cultured in RPMI1640 medium containing 10% FBS.
Colony-formation assay
Ba/F3 BCR-ABL random mutagenesis cells were seeded in 6-well culture plates in MethoCult GFH4434 medium (Stem Cell Technology) containing the indicated concentration of nilotinib and/or NVP-BEZ235. Cells were incubated for more than 14 d in a humidified atmosphere with 5% CO2 at 37 °C, and the colonies were counted with a reversed microscope. Cell aggregates containing 40 or more cells were considered colonies. Each experiment was performed in triplicate.
Statistical analysis
Differences between treatments in terms of responses were determined using the Student t test. P < 0.05 was considered statistically significant. Data for comparison of multiple groups are presented as mean ± SD and were analyzed by ANOVA.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Novartis for providing the drugs (NVP-BEZ235, nilotinib, and imatinib). This work was supported by a High-Tech Research Center Project for private universities—a matching fund subsidy from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)—and by the University-Industry Joint Research Project for private universities—a matching fund subsidy from the MEXT. This work was also supported by Grants-in-Aid for Scientific Research from the MEXT.
References
- Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science 1960; 142:1497
- Sawyers CL. Chronic myeloid leukemia. N Engl J Med 1999; 340:1330 - 40; http://dx.doi.org/10.1056/NEJM199904293401706; PMID: 10219069
- Tauchi T, Okabe S, Miyazawa K, Ohyashiki K. The tetramerization domain-independent Ras activation by BCR-ABL oncoprotein in hematopoietic cells. Int J Oncol 1998; 12:1269 - 76; PMID: 9592185
- Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 1995; 86:726 - 36; PMID: 7606002
- Chai SK, Nichols GL, Rothman P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J Immunol 1997; 159:4720 - 8; PMID: 9366395
- Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med 2001; 344:1038 - 42; http://dx.doi.org/10.1056/NEJM200104053441402; PMID: 11287973
- Druker BJ, Guilhot F, O’Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM, et al, IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355:2408 - 17; http://dx.doi.org/10.1056/NEJMoa062867; PMID: 17151364
- Kantarjian HM, Talpaz M, Giles F, O’Brien S, Cortes J. New insights into the pathophysiology of chronic myeloid leukemia and imatinib resistance. Ann Intern Med 2006; 145:913 - 23; http://dx.doi.org/10.7326/0003-4819-145-12-200612190-00008; PMID: 17179059
- Hochhaus A, Kreil S, Corbin AS, La Rosée P, Müller MC, Lahaye T, Hanfstein B, Schoch C, Cross NC, Berger U, et al. Molecular and chromosomal mechanisms of resistance to imatinib (STI571) therapy. Leukemia 2002; 16:2190 - 6; http://dx.doi.org/10.1038/sj.leu.2402741; PMID: 12399961
- Baccarani M, Cortes J, Pane F, Niederwieser D, Saglio G, Apperley J, Cervantes F, Deininger M, Gratwohl A, Guilhot F, et al, European LeukemiaNet. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol 2009; 27:6041 - 51; http://dx.doi.org/10.1200/JCO.2009.25.0779; PMID: 19884523
- Garg RJ, Kantarjian H, O’Brien S, Quintás-Cardama A, Faderl S, Estrov Z, Cortes J. The use of nilotinib or dasatinib after failure to 2 prior tyrosine kinase inhibitors: long-term follow-up. Blood 2009; 114:4361 - 8; http://dx.doi.org/10.1182/blood-2009-05-221531; PMID: 19729517
- Pavlovsky C, Kantarjian H, Cortes JE. First-line therapy for chronic myeloid leukemia: Past, present, and future. Am J Hematol 2009; 84:287 - 93; http://dx.doi.org/10.1002/ajh.21380; PMID: 19306355
- Vasudevan KM, Garraway LA. AKT signaling in physiology and disease. Curr Top Microbiol Immunol 2010; 347:105 - 33; http://dx.doi.org/10.1007/82_2010_66; PMID: 20549472
- Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans 2007; 35:231 - 5; http://dx.doi.org/10.1042/BST0350231; PMID: 17371246
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29:233 - 42; http://dx.doi.org/10.1016/j.tibs.2004.03.006; PMID: 15130559
- LoPiccolo J, Blumenthal GM, Bernstein WB, Dennis PA. Targeting the PI3K/Akt/mTOR pathway: effective combinations and clinical considerations. Drug Resist Updat 2008; 11:32 - 50; http://dx.doi.org/10.1016/j.drup.2007.11.003; PMID: 18166498
- Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chène P, De Pover A, Schoemaker K, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther 2008; 7:1851 - 63; http://dx.doi.org/10.1158/1535-7163.MCT-08-0017; PMID: 18606717
- Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature 2011; 469:362 - 7; http://dx.doi.org/10.1038/nature09733; PMID: 21248843
- Khorashad JS, Kelley TW, Szankasi P, Mason CC, Soverini S, Adrian LT, Eide CA, Zabriskie MS, Lange T, Estrada JC, et al. BCR-ABL1 compound mutations in tyrosine kinase inhibitor-resistant CML: frequency and clonal relationships. Blood 2013; 121:489 - 98; http://dx.doi.org/10.1182/blood-2012-05-431379; PMID: 23223358
- Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene 2000; 19:6680 - 6; http://dx.doi.org/10.1038/sj.onc.1204091; PMID: 11426655
- Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, García-Echeverría C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 2008; 68:6598 - 607; http://dx.doi.org/10.1158/0008-5472.CAN-08-1044; PMID: 18701483
- Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, et al. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res 2008; 68:8022 - 30; http://dx.doi.org/10.1158/0008-5472.CAN-08-1385; PMID: 18829560
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, Maira M, McNamara K, Perera SA, Song Y, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008; 14:1351 - 6; http://dx.doi.org/10.1038/nm.1890; PMID: 19029981
- Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, Garcia-Echevrria C, Yung WK. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther 2009; 8:2204 - 10; http://dx.doi.org/10.1158/1535-7163.MCT-09-0160; PMID: 19671762
- Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG, Lin L, et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res 2009; 69:3955 - 62; http://dx.doi.org/10.1158/0008-5472.CAN-08-4450; PMID: 19366795
- McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, Jakubikova J, Maira SM, Garcia-Echeverria C, Schlossman R, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer Res 2009; 69:5835 - 42; http://dx.doi.org/10.1158/0008-5472.CAN-08-4285; PMID: 19584292
- Kojima K, Shimanuki M, Shikami M, Samudio IJ, Ruvolo V, Corn P, Hanaoka N, Konopleva M, Andreeff M, Nakakuma H. The dual PI3 kinase/mTOR inhibitor PI-103 prevents p53 induction by Mdm2 inhibition but enhances p53-mediated mitochondrial apoptosis in p53 wild-type AML. Leukemia 2008; 22:1728 - 36; http://dx.doi.org/10.1038/leu.2008.158; PMID: 18548093
- Faber AC, Li D, Song Y, Liang MC, Yeap BY, Bronson RT, Lifshits E, Chen Z, Maira SM, García-Echeverría C, et al. Differential induction of apoptosis in HER2 and EGFR addicted cancers following PI3K inhibition. Proc Natl Acad Sci U S A 2009; 106:19503 - 8; http://dx.doi.org/10.1073/pnas.0905056106; PMID: 19850869
- Klejman A, Rushen L, Morrione A, Slupianek A, Skorski T. Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene 2002; 21:5868 - 76; http://dx.doi.org/10.1038/sj.onc.1205724; PMID: 12185586
- Sillaber C, Mayerhofer M, Böhm A, Vales A, Gruze A, Aichberger KJ, Esterbauer H, Pfeilstöcker M, Sperr WR, Pickl WF, et al. Evaluation of antileukaemic effects of rapamycin in patients with imatinib-resistant chronic myeloid leukaemia. Eur J Clin Invest 2008; 38:43 - 52; http://dx.doi.org/10.1111/j.1365-2362.2007.01892.x; PMID: 18173550
- Kimura S, Naito H, Segawa H, Kuroda J, Yuasa T, Sato K, Yokota A, Kamitsuji Y, Kawata E, Ashihara E, et al. NS-187, a potent and selective dual Bcr-Abl/Lyn tyrosine kinase inhibitor, is a novel agent for imatinib-resistant leukemia. Blood 2005; 106:3948 - 54; http://dx.doi.org/10.1182/blood-2005-06-2209; PMID: 16105974
- Ray A, Cowan-Jacob SW, Manley PW, Mestan J, Griffin JD. Identification of BCR-ABL point mutations conferring resistance to the Abl kinase inhibitor AMN107 (nilotinib) by a random mutagenesis study. Blood 2007; 109:5011 - 5; http://dx.doi.org/10.1182/blood-2006-01-015347; PMID: 17303698
- Okabe S, Tauchi T, Nakajima A, Sashida G, Gotoh A, Broxmeyer HE, Ohyashiki JH, Ohyashiki K. Depsipeptide (FK228) preferentially induces apoptosis in BCR/ABL-expressing cell lines and cells from patients with chronic myelogenous leukemia in blast crisis. Stem Cells Dev 2007; 16:503 - 14; http://dx.doi.org/10.1089/scd.2007.9994; PMID: 17610380
- Okabe S, Tauchi T, Tanaka Y, Katagiri S, Ohyashiki K. Effects of the hedgehog inhibitor GDC-0449, alone or in combination with dasatinib, on BCR-ABL-positive leukemia cells. Stem Cells Dev 2012; 21:2939 - 48; http://dx.doi.org/10.1089/scd.2012.0016; PMID: 22642671