
Abstract
Acute lymphoblastic leukemia (ALL) is a malignant disorder of lymphoid progenitor cells that are committed to the B- or the T-cell lineage. The pathogenesis of ALL is heterogeneous and may be at least in part caused by genetic alterations. Although the modern sequencing technologies make it possible to rapidly discover novel genetic and epigenetic alterations and molecular targets for therapeutic intervention for ALL, conventional chemotherapy is still the most important therapeutic approach. Relapses and high morbidity and mortality remain major challenges particularly in adult patients with ALL. Therefore, development of novel chemotherapeutic agents remains in demand for ALL patients. In the course of seeking novel agents against ALL, we screened a library of small molecules and identified that PQJS380, a S-(E)-4-([7S,10S]-4-ethyl-7-isopropyl-2,5,8,12-tetraoxo-9-oxa-3,6,13,18-tetraaza-bicycle[13,2,1] octadec-1-en-10-yl)but-3-enyl octanethioate, showed potent anti-leukemia activity. PQJS380 inhibited the proliferation with IC50 values of 14.25 nM and 5 nM in REH and NALM-6 cells, respectively. PQJS380 had 10-fold higher molar potency than the front-line ALL drugs Ara-C and VP-16. The median IC50 value for leukemia blast cells from 17 patients with ALL was 52 nM. PQJS380 induced G1-phase arrest in REH cells, and S-phase in NALM-6 cells, respectively. Treatment of PQJS380 led to apoptosis in ALL cell lines (REH and NALM-6) and primary ALL cells. Our data supported that PQJS380 may be a promising lead compound for ALL treatment even though the precise targets remain to be elucidated.
Introduction
Acute lymphoblastic leukemia (ALL) is a malignant disorder of lymphoid progenitor cells which are committed to the B- or the T-cell lineage. For ALL patients, the normal hematopoiesis is suppressed because of an accumulation of primitive or immature lymphocytes in hematopoietic tissues (bone marrow, spleen, and lymph node).Citation1 ALL occurs in both children and adults. ALL is the most common cancer diagnosed in children and represents ∼25% of cancer diagnoses among children younger than 15 y.Citation2 The pathogenesis of ALL is heterogeneous and may be associated with genetic alterations,Citation1,Citation2 gene fusions,Citation3-Citation5 gene translocations,Citation6-Citation10 deletions, and cooperative mutations. Discovery of genetic changes may facilitate targeted therapy. For instance, Bcr-Abl is observed in ∼30% adult ALL patients, which produced effective treatment with tyrosine kinase inhibitors such as imatinib, ponatinib, and bosutinib.Citation5 ETV6-RUNX1 is detectable in ∼25% of childhood B-ALL individuals,Citation3 while overexpression of c-MYC is associated with poor prognosis in French–American–British (FAB)-L3-type ALL. Mutation in Notch1 has been recently identified in ∼50% T-ALL patients.Citation4 All of this progress offers enormous promise for targeted therapy against ALL individuals.
Although the modern sequencing technologies make it possible to better and rapidly discover novel genetic and epigenetic alterations and attractive targets for therapeutic intervention for ALL, conventional chemotherapeutic agents are still one of the most important therapeutic approaches. Actually, chemotherapy leads to ∼80% and ∼40% of 5-y survival rate for children and adults ALL patients, respectively.Citation11-Citation14 Some long-term survivors have healthy and productive lives.Citation9
Relapses and high morbidity and mortality remain major challenges particularly in adult patients with ALL. Therefore, development of novel chemotherapeutic agents remains in demand for ALL patients.
In the course of seeking novel agents against ALL, we screened a small-scale library of compounds built in our laboratory, and identified that PQJS380, a small molecule compound, showed a potent anti-leukemia activity. Results showed that PQJS380 inhibited the proliferation, and induced apoptosis in ALL cell lines (REH and NALM-6) and primary cells from specimens of patients with ALL. Our data suggested that PQJS380 may be a promising lead compound for ALL treatment.
Results
PQJS380 inhibited growth of acute lymphoblastic leukemia cells
We first examined the effects of PQJS380 on growth of acute lymphoblastic leukemia cells. Cell viability was measured by MTS assay in REH and NALM-6 cells treated with increasing concentrations of PQJS380. PQJS380 inhibited the growth of both ALL lines of cells in a concentration-dependent manner (). Concentrations of PQJS380 resulting in 50% inhibition of REH and NALM-6 cells growth (IC50) were 14.25 nM and 5 nM, respectively. Comparison of PQJS380 with the front-line ALL drugs, cytosine arabinose (Ara-C) and etoposide (VP-16) revealed that PQJS380 had 10-fold activity more effective than Ara-C and VP-16 ().
Figure 1. PQJS380 inhibited growth of acute lymphoblastic leukemia cells. (A) PQJS380 inhibited cell viability (percent relative to control) in acute lymphoblastic leukemia (ALL) cell lines REH and NALM-6. The ALL cells were exposed to escalating concentrations of PQJS380 and conventional chemotherapeutic agents (Ara-C and VP-16) for 72 h; the cell viability was detected by MTS assay. (B) PQJS380 decreased clonogenicity of ALL cells. The REH and NALM-6 cells were exposed to different concentrations of PQJS380, Ara-C, and VP-16 for 48 h, and harvested, washed, plated in soft agar, and continued to grow in absence of these compounds.
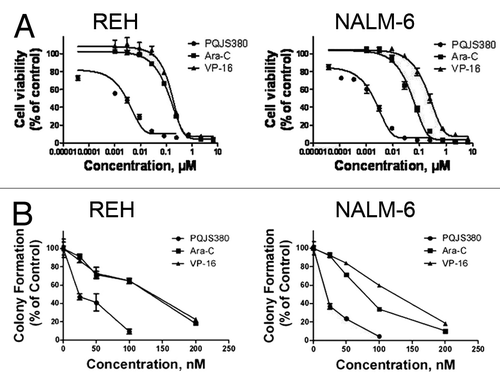
To evaluate the effect of PQJS380 on anchorage-independent proliferation of ALL cells, soft agar clonogenic assay was performed. The results showed that PQJS380 effectively inhibited colony formation of REH and NALM-6 cells with IC50 values of 36.4 nM and 23.5 nM, respectively (). Moreover, the data showed that PQJS380 displayed a higher potency in inhibiting clonogenicity in REH and NALM-6 cells than Ara-C and VP-16 ().
PQJS380 induced cell cycle arrest in acute lymphoblastic leukemia cells
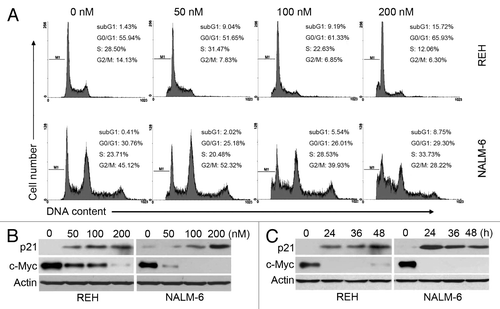
We next explored the effects of PQJS380 on cell cycle progression in ALL cells; cell-cycle distribution results analyzed by FACS indicated that PQJS380 induced G1-phase arrest in REH cells, and S-phase arrest in NALM-6 cells (). Furthermore, we detected cell cycle related proteins by western blotting. The data showed that PQJS380 decreased the levels of c-MYC (a transcriptional repressor of p21) and increased the levels of p21 (a CDK inhibitor) ().
Figure 2. PQJS380 induced cell cycle arrest in acute lymphoblastic leukemia cells. (A) Cell cycle distribution was analyzed after exposing to PQJS380 for 48 h by flow cytometer in REH and NALM-6 cells. (B and C) Effects of PQJS380 on p21 and c-Myc in REH and NALM-6 cells. Cells treated with increasing concentrations of PQJS380 for 48 h (B) or 200 nM PQJS380 for indicated durations (C), the protein levels of p21 and c-Myc were detected by western blotting.
PQJS380 induced cell apoptosis in acute lymphoblastic leukemia cells
To determine the killing effects of PQJS380 on ALL cells, trypan blue exclusion assay was conducted. A total of 2 × 105/mL REH and NALM-6 cells were seeded and treated with PQJS380 at concentrations of 0, 25, 50, 100, and 200 nM, respectively. Cell numbers were assessed by cell counting with a hemocytometer after trypan blue staining after treatment of PQJS380 for 24, 30, 36, and 48 h, respectively. The results revealed that PQJS380 obviously induced cell death of REH and NAML-6 cells in a concentration-dependent and time-dependent manner ().
Figure 3. PQJS380 induces cell death in acute lymphoblastic leukemia cells. (A and B) REH (A) and NALM-6 (B) cells were treated with increasing concentrations of PQJS380 for 24, 30, 36, and 48 h, and the ratio of dead cell was determined by trypan blue exclusion assay using a hemocytometer. (C and D) The ALL cells were treated with increasing concentrations of PQJS380 for 48 h (C) or 200 nM PQJS380 for various times (D), and the apoptotic cells were analyzed by flow cytometer after double staining with Annexin V/ PI. *P < 0.05, **P < 0.01, ***P < 0.0001, one-way ANOVA, post hoc comparisons. Columns, mean; error bars, SE.
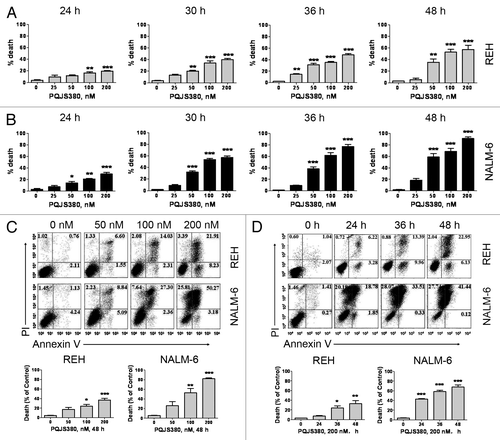
To assess the ability of PQJS380 to induce apoptosis in ALL cells, we performed FACS analysis after Annexin V/PI double staining. The data demonstrated that remarkable apoptosis was observed in both REH and NALM-6 cells in dose-dependent and time-dependent manner after PQJS380 treatment ().
PQJS380 induced apoptosis through both intrinsic and extrinsic pathways in acute lymphoblastic leukemia cells
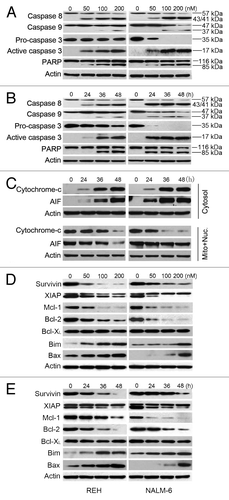
We next analyzed apoptosis related proteins. Results demonstrated that PQJS380 induced a concentration-dependent and time-dependent specific cleavage of PARP and caspase-8, -9, and -3 in both REH and NALM-6 cells (), which suggests that PQJS380 induced apoptosis of ALL cells probably through both intrinsic and extrinsic pathways. To further elucidate the mechanism of intrinsic apoptosis pathway, we tested the release of cytochrome c and AIF from mitochondria into the cytosol. REH and NALM-6 cells were exposed with 200 nM PQJS380 for various durations, and western blotting analysis showed that PQJS380 induced a substantial time-dependent release of cytochrome c and AIF into cytosol from mitochondria (), which implies that PQJS380 triggered the intrinsic (mitochondrial) pathway of apoptosis. Moreover, we detected the effects of PQJS380 on members of Bcl-2 family. Western blotting analysis illustrated that PQJS380 decreased anti-apoptotic protein levels (survivin, XIAP, Mcl-1, and Bcl-2), and increased pro-apoptosis protein levels such as Bim and Bax ().
Figure 4. PQJS380 triggered intrinsic and extrinsic apoptosis in acute lymphoblastic leukemia cells. (A and B) Dynamic analysis of western blotting of PARP and caspase-3, -8, and -9 in REH and NALM-6 cells treated with increasing concentrations of PQJS380 for 48 h (A), or with 200 nM PQJS380 for indicated time points (B). (C) REH and NALM-6 cells were treated with 200 nM PQJS380 for 0, 24, 36, and 48 h, respectively, the levels of cytochrome c and AIF in the cytosolic and mitochondrial fractions were measured by western blotting analysis, respectively. (D and E) Western blotting analysis of apoptotic proteins in ALL cells treated with increasing concentrations of PQJS380 for 48 h (D), or with 200 nM PQJS380 for indicated time points (E).
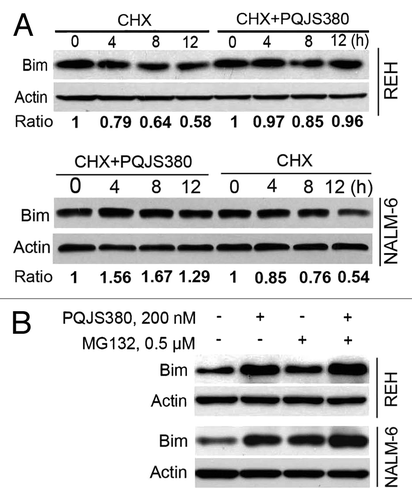
PQJS380 increased Bim protein through inhibiting proteasome degradation system
Because Bim was found to be an important mediator of apoptosis in ALL cells, we therefore focused on Bim. To address the reason of the increased expression of Bim by PQJS380, we performed a chase experiment with CHX, a ubiquitous protein synthesis inhibitor. We found that Bim was downregulated in a time-dependent manner (0, 4, 8, and 12 h) in REH and NAML-6 cells treated alone with CHX (100 μg/mL). However, pretreatment with PQJS380 (30 nM) can apparently reverse this tendency (), suggesting the turnover rate of Bim was decreased by PQJS380 treatment. Further, we measured the stability of Bim with MG132, a proteasome degradation system inhibitor. The results showed that MG132 inhibited Bim degradation, while the combinational treatment of PQJS380 and MG132 didn’t induce an additive/synergistic increase of Bim than PQJS380 or MG-132 singly alone. The results implied that PQJS380 might increase the stability of Bim protein through blocking the proteasome degradation pathway.
Figure 5. PQJS380 increased stability of Bim protein. (A) PQJS380 delayed the turnover rate of Bim protein. REH and NALM-6 cells were pretreated with or without 30 nM PQJS380 for 2 h, then exposed to 100 μg/mL cycloheximide (CHX) for various durations, and then underwent western blotting analysis. The immunoblots were quantified by densitometry. The scale of blots were normalized to that of relevant actin, and then normalized to control. (B) PQJS380 increased the protein levels of Bim through ubiquitin-proteasome pathway. REH and NALM-6 cells were treated with 200 nM PQJS380 for 24 h in presence or absence of pretreatment with 0.5 μM MG132 for 2 h, and then underwent western blotting analysis for Bim protein levels.
PQJS380 induced growth inhibition and apoptosis in primary ALL cells
We next assessed the efficacy of PQJS380 on the primary blast leukemia cells from 17 patients with ALL and 9 patients with AML patients. PQJS380 inhibited the cell viability of primary ALL and AML cells in a concentration-dependent manner (). The median IC50 value for all 17 ALL patients was 52 nM, which was more effective than front-line ALL drugs, Ara-C and VP-16 (). Of 17 ALL cases tested, 13 ALL primary cases were shown sensitive to PQJS380 with median IC50 value of 27 nM (ranging from 1 to 70 nM), while the blast cells from the other 4 cases of ALL patients appeared resistant to PQJS380 with median IC50 value of 10 350 nM (ranging from 7000 to 26 500 nM) (). The median IC50 value for all 9 AML patients tested was 557 nM. There were 5 cases of AML that were sensitive to PQJS380 with IC50 values ranging from 70 to 557 nM, and there were 4 cases of AML that were resistant to PQJS380 with IC50 values 2000∼18 500 nM respectively ().
Figure 6. PQJS380 induced apoptosis in acute lymphoblastic leukemia primary cells. (A) PQJS380 inhibited growth (percent relative to control) in primary ALL cells. Primary cells were exposed to escalating concentrations of PQJS380 for 72 h, and cell viability was assayed by MTS. (B) Primary ALL cells were exposed to the indicated concentrations of PQJS380 for 48 h, and then underwent Annexin V/ PI double staining by flow cytometry. The representative data from the acute lymphoblastic leukemia patients were shown.
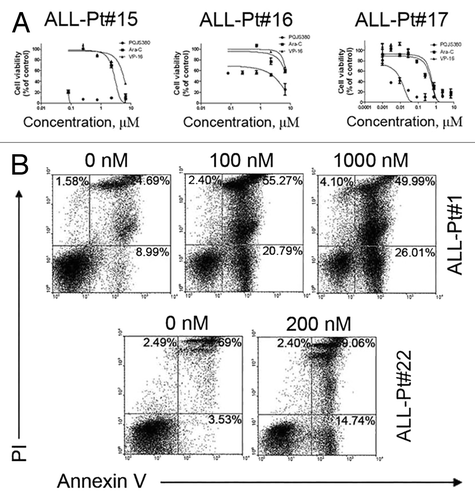
Table 1. IC50 values of primary leukemia cells from patients with leukemia to PQJS380
PQJS380 significantly induced apoptotic cell death in primary ALL cells (). The primary cells from two representative ALL patients were stained with Annexin V/PI and analyzed by flow cytometry after PQJS380 treatment for 48 h. The results were consistent with the in vitro inhibitory effect of PQJS380 in REH and NALM-6 cells.
In order to examine the specificity of PQJS380 for leukemia cells, mononuclear blood cells from 3 health volunteers were treated with PQJS380, Ara-C, and VP-16, respectively; the cell viability was assayed. The results revealed that PQJS380 lowered the cell viability in normal cells with IC50 values of 73.67 nM, which was more toxic than that Ara-C (IC50 = 1776 nM) and VP-16 (IC50 = 1563 nM) (data not shown).
Discussion
In present manuscript, we investigated the anti-leukemia activities of PQJS380 in acute lymphoblastic leukemia cells. We discovered that PQJS380 inhibited cell viability and colony formation capacity in REH and NALM-6 cells and primary cells from ALL patients, suggesting its potent anti-tumor effects.
Our results showed that PQJS380 displayed a potent activity to inhibit the proliferation of ALL cells. PQJS380 induced G1-phase arrest in REH cells and S-phase arrest in NAML-6 cells. Previous reports showed c-MYC regulated cell cycle via its inhibitory effects on p21Citation15-Citation18 which is known as an inhibitor of cyclin-dependent kinase (CKI). Consistent with these studies, our data revealed PQJS380 blocked cell cycle progression in G1 phase and S phase through upregulation of p21 and downregulation of c-MYC, and thus decreased the transition rate of cells out of G1 into S phase in REH cells and out of S into G2/M phase in NAML-6 cells. Because L3 ALL is associated with a variety of translocations that involve translocation of the c-MYC proto-oncogene to the immunoglobulin gene locus t(2;8), t(8;12), and t(8;22), the inhibitory effect of PQSJ380 on c-MYC might potentially be of importance for FAB L3 ALL treatment.Citation8 Unfortunately, our patients did not include L3-type ALL patient. Future work may test.
Induction of apoptosis is an important mechanism for chemotherapeutic agents to eliminate tumor cells.Citation19-Citation21 Our data showed that PQJS380 induced substantial apoptosis as determined by Annexin V/PI double staining in REH and NALM-6 cells and primary ALL cells. Moreover, specific cleavage in PARP which deemed as a hallmark of apoptosis further validated the apoptosis by PQJS380 in REH and NALM-6 cells. Caspases, as the major components of apoptosis, played a critical role in this procedure. We demonstrated that PQJS380 induced activation of caspase-8, -9, and -3 and release of cytochrome c and AIF, suggesting PQJS380-induced apoptosis was through both extrinsic and intrinsic pathways in REH and NALM-6 cells.
Our results revealed that PQJS380 led to disturbance in the balance of Bcl-2 family, e.g., downregulation of Bcl-2 and Mcl-1, and upregulation of Bax and Bim. Bim is a pro-apoptotic protein and its aberrant expression was widely reported in hematopoietic cells.Citation22-Citation24 Blockage of proteasome degradation pathway might be involved in upregulation of Bim by PQJS380. Although Bim was explored in the present study, we do not exclude possible roles of Bcl-2, survivin, Mcl-1, Bax, etc. Bcl-2 is an anti-apoptotic Bcl-2 family protein and exerts its anti-apoptotic effect by binding to Bax and Bak, preventing their formation of pores in the mitochondrial membrane.Citation25,Citation26 Bcl-2 was showed overexpressed in various hematopoietic malignancies and conferred resistance to chemotherapy.Citation27,Citation28 Therefore, targeting Bcl-2 may be a promising strategy for hematopoietic malignancies.Citation29 Downregulation of Bcl-2 by PQJS380 would prompt impair of mitochondrial and facilitate the cell death. Mcl-1 is another anti-apoptotic Bcl-2 protein, and often overexpressed in leukemia cells as well. Silencing Mcl-1 by siRNA or antisense oligonucleotides sensitized leukemia cells to chemotherapy.Citation30 Bax is a major member of the Bcl-2 family in regulating cell survival and apoptosis. Low expression of Bax correlates with poor response to therapy and shorter overall survival in patients with solid tumors.Citation31,Citation32 Small interfering RNA (siRNA) targeting of Bax clearly attenuated mitochondrial membrane potential loss and cell death in human cervical cancer cells.Citation33 Moreover, Bax expression levels and the Bax/Bcl-2 ratio are significantly lower in ALL samples at relapse as compared with samples at initial diagnosis.Citation34 In our study, increased Bax and BH3-only protein Bim induced by PQJS380 might facilitate induction of apoptosis in REH and NALM-6 cells.
XIAP and survivin were also downregulated by PQJS380. XIAP is a most potent anti-apoptotic IAP (inhibitor of apoptosis proteins) protein and it inhibits apoptosis by preventing activation of caspases such as caspase-3, -7, and -9.Citation6 Survivin is another IAP protein, and its high level expression was thought to correlate with poor prognosis in childhood precursor B-cell ALL.Citation35 High levels of survivin inhibited the activation of caspase-3 by interacting with anti-apoptotic Smac protein.Citation36 Activation of caspase-3 and -9 were presumably facilitated by loss of inhibitory effects of XIAP and survivin on caspase-3 and -9 after treatment with PQJS380 in REH and NALM-6 cells. As a whole, these apoptosis-related proteins discussed above would function synergistically to induce apoptosis, and the changes of multiple apoptotic proteins suggested that PQJS380 might be a multi-target compound.
In summary, we first reported anti-leukemia activities of PQJS380 in REH and NALM-6 cells. Our results demonstrated PQJS380 inhibited cell growth and induced apoptosis in REH, NALM-6 and primary ALL cells. All these data indicated that PQJS380 may be a promising lead compound against ALL cells. The safety and the precise molecule target(s) of this compound remain to be investigated.
Materials and Methods
Chemicals and antibodies
Compound PQJS380 (purity >98.5%), a S-(E)-4-([7S,10S]-4-ethyl-7-isopropyl-2,5,8,12-tetraoxo-9-oxa-3,6,13,18-tetraaza-bicycle[13,2,1] octadec-1-en-10-yl)but-3-enyl octanethioate, was synthesized in our laboratory, and dissolved in DMSO (Sigma-Aldrich) at a stock concentration of 20 mM and stored at –20 °C. Cytosine arabinose (Ara-C) and etoposide (VP-16) were purchased from Sigma-Aldrich. RPMI 1640 medium and fetal bovine serum (FBS) were purchased from Invitrogen Corporation. Annexin V-FITC, propidium iodide (PI), and cycloheximide (CHX) were purchased from Sigma-Aldrich (Sigma). MG132 was from Calbiochem. Antibodies against apoptosis-inducing factor (AIF, H300), Bax, Bcl-XL, and Mcl-1 (S-19) were from Santa Cruz Biotechnology. Mouse antibodies against poly(adenosine diphosphate [ADP]-ribose) polymerase (PARP, clone 4C10-5), caspase-3, X-linked inhibitor of apoptosis protein (XIAP), and cytochrome c (clone 6H2.B4) were from BD Biosciences. Antibodies against Bcl-2 were from Upstate Technology. Mouse monoclonal antibody against β-actin was from Sigma-Aldrich. Antibodies against caspase-8 and caspase-9 were from Cell Signaling Technology. Anti-Bim was from Upstate Technology. Anti-survivin was from Novus Biologicals.
Cell culture
Pre-B acute lymphoblastic leukemia cells REH and NALM-6 were grown in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), as described previously.Citation37
Cell viability assay
Suspended in 100 μL RPMI 1640 medium, 2 × 104 cells were seeded in each well of 96-well (CellTiter 96 Aqueous One Solution Cell Proliferation assay; Promega)Citation38,Citation39 were exposed to different concentrations of PQJS380, Ara-C, and VP-16 for 72 h. Control cells were added equivalent RPMI 1640 medium containing DMSO (<0.1%). Three hours prior to culture termination, 20 μL of MTS/PMS (20:1) mixed solution was added to each well of culture. Absorbance was read on a 96-well plate reader at 490 nm wavelength. Fifty percent inhibition of PQJS380 to cell growth (IC50) was determined by GraphPad Prism 5.0 software.
Western blotting analysis
For detecting cellular protein, total cell lysates were prepared. Control or PQJS380-treated cells were pelleted by centrifugation, and were lysed in a suitable volume of RIPA buffer (PBS with 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate supplemented with 1 mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail,Citation40 one tablet per 50 mL) on ice for 30 min. After 6 to 8 times of ultrasonication, the cell lysates were centrifuged at 14 000 rpm for 10 min at 4 °C. The supernatants were quantitated with the Bio-Rad protein assay dye reagent (Bio-Rad), then subjected to SDS-PAGE.
To detect the cytochrome c and apoptosis-inducing factor (AIF) in cytosol, the cytosolic fractions were extracted with digitonin buffer (0.015% [w/v] digitonin, 300 mM sucrose, 10 mM PIPES [pH 6.8], 100 mM NaCl, 3 mM MgCl2, 5 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride) supplemented with freshly added phosphatase inhibitors and protease inhibitors, as described previously.Citation7 After incubation on ice for 10 min, the lysates were centrifuged at 14 000 rpm for 10 min. Supernatants containing cytosolic protein were transferred to a new 1.5 mL EP tube. The remained pallets were further extracted for mitochondrial fractions. Protein concentration was determined as mentioned above. Protein samples were subjected to SDS-PAGE or stored in aliquots at –80 °C.
Apoptosis assay
Apoptosis was determined by fluorescence activated cell sorter (FACS) using annexin V/PI double staining. ALL cells REH and NALM-6 were cultured in the presence of different concentrations of PQJS380 for indicated time points. The cell pellets were collected, washed in 1× PBS, then washed in 0.5 mL binding buffer (10 mM 4-[2-hydroxyethyl]-1-piperazineethane-sulfonic acid, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2). Next, cells were resuspended in 100 μL binding buffer containing 0.3% (v/v) annexin V–FITC, and incubated at room temperature for 15 min in the dark. After incubation, cells were washed in 0.5 mL binding buffer twice, and resuspended in 0.5 mL binding buffer. PI was added just before FACS analysis. The resultant data were analyzed with WinMDI 2.9 software.
Cell cycle analysis
REH and NALM-6 cells were cultured with or without PQJS380 for 48 h. Cells were pelleted, washed in 1× PBS, and fixed with 70% cold ethanol overnight at 4 °C. The cells were then washed twice in cold PBS buffer. Before FACS analysis, the cells were labeled with PI buffer (1× PBS, 50 μg/mL PI, 2.5 μg/mL RNase) at room temperature for 1 h in the dark. Cell cycle distribution was determined by FACS using WinMDI 2.9 software.
Soft agar clonogenic assay
Soft agar clonogenic assay was used to evaluate anchorage-independent growth of ALL cells. After treatment with indicated concentrations of PQJS380 for 48 h, 2 × 105/mL REH and NALM-6 cells were then washed with PBS and seeded in drug-free RPMI 1640 medium containing 0.3% agar and 20% FBS in 24-well plates. The plates were cultured at 37 °C, 5% CO2 incubator for 10–14 d, colonies containing ≥50 cells were counted using an inverted phase-contrast microscope.
Primary cells from leukemia patients
Bone marrow or peripheral blood samples were obtained from 26 patients with 17 ALL and 9 acute myelogenous leukemia (AML) from the Second Affiliated Hospital of Sun Yat-sen University after informed consent according to the institutional guidelines and Declaration of Helsinki principles. Mononuclear cells were separated using Histopaque gradient centrifugation (density 1.077 g/mL; Sigma-Aldrich). Contaminating red cells were lysed in 0.8% ammonium chloride solution for 10 min. After a wash with PBS, cells were suspended in RPMI 1640 supplemented with 10% FBS.
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software) was used to conduct the statistical analysis. Comparison among multiple groups employed one-way ANOVA with post hoc intergroup comparisons. P < 0.05 was considered statistical significance.
| Abbreviations: | ||
| ALL | = | acute lymphoblastic leukemia |
| AML | = | acute myeloid leukemia |
| SDS | = | sodium dodecyl sulfate |
| PI | = | propidium iodide |
| PARP | = | poly ADP-ribose polymerase |
| XIAP | = | X-linked inhibitor of apoptosis proteins |
| FBS | = | fetal bovine serum |
| DMSO | = | dimethyl sulfoxide |
| CHX | = | cycloheximide |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants the National Basic Research Program of China (973 2009CB825506 to J Pan, 2009CB940900 to J Sheng), from National Natural Science Funds of China for Distinguished Young Scholars (81025021) to J Pan, the Research Foundation of Education Bureau of Guangdong Province, China (cxzd1103 to J Pan), the Research Foundation of Guangzhou Bureau of Science and Technology, China (to J Pan), National Natural Science Fund of China (90713036 to J Pan, 21172220 S Jiang), the National High Technology Research and Development Program of China (863 2008AA02Z420 to J Pan), and the Fundamental Research Funds for the Central Universities to J Pan.
References
- Pui C-H, Evans WE. Acute lymphoblastic leukemia. N Engl J Med 1998; 339:605 - 15; http://dx.doi.org/10.1056/NEJM199808273390907; PMID: 9718381
- Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell 2002; 1:417 - 20; http://dx.doi.org/10.1016/S1535-6108(02)00081-8; PMID: 12124171
- Loh ML, Rubnitz JE. TEL/AML1-positive pediatric leukemia: prognostic significance and therapeutic approaches. Curr Opin Hematol 2002; 9:345 - 52; http://dx.doi.org/10.1097/00062752-200207000-00013; PMID: 12042710
- Speck NA, Gilliland DG. Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2:502 - 13; http://dx.doi.org/10.1038/nrc840; PMID: 12094236
- Ferrando AA, Look AT. Gene expression profiling in T-cell acute lymphoblastic leukemia. Semin Hematol 2003; 40:274 - 80; http://dx.doi.org/10.1016/S0037-1963(03)00195-1; PMID: 14582078
- Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene 2001; 20:5695 - 707; http://dx.doi.org/10.1038/sj.onc.1204639; PMID: 11607819
- Ernst P, Wang J, Korsmeyer SJ. The role of MLL in hematopoiesis and leukemia. Curr Opin Hematol 2002; 9:282 - 7; http://dx.doi.org/10.1097/00062752-200207000-00004; PMID: 12042701
- Ayton PM, Cleary ML. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev 2003; 17:2298 - 307; http://dx.doi.org/10.1101/gad.1111603; PMID: 12952893
- Pui C-H, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet 2008; 371:1030 - 43; http://dx.doi.org/10.1016/S0140-6736(08)60457-2; PMID: 18358930
- Harrison CJ. Cytogenetics of paediatric and adolescent acute lymphoblastic leukaemia. Br J Haematol 2009; 144:147 - 56; http://dx.doi.org/10.1111/j.1365-2141.2008.07417.x; PMID: 19006567
- Schrappe M, Reiter A, Ludwig W-D, Harbott J, Zimmermann M, Hiddemann W, Niemeyer C, Henze G, Feldges A, Zintl F, et al, German-Austrian-Swiss ALL-BFM Study Group. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: results of trial ALL-BFM 90. Blood 2000; 95:3310 - 22; PMID: 10828010
- Silverman LB, Gelber RD, Dalton VK, Asselin BL, Barr RD, Clavell LA, Hurwitz CA, Moghrabi A, Samson Y, Schorin MA, et al. Improved outcome for children with acute lymphoblastic leukemia: results of Dana-Farber Consortium Protocol 91-01. Blood 2001; 97:1211 - 8; http://dx.doi.org/10.1182/blood.V97.5.1211; PMID: 11222362
- Linker C, Damon L, Ries C, Navarro W. Intensified and shortened cyclical chemotherapy for adult acute lymphoblastic leukemia. J Clin Oncol 2002; 20:2464 - 71; http://dx.doi.org/10.1200/JCO.2002.07.116; PMID: 12011123
- Pui C-H, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med 2006; 354:166 - 78; http://dx.doi.org/10.1056/NEJMra052603; PMID: 16407512
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002; 111:241 - 50; http://dx.doi.org/10.1016/S0092-8674(02)01014-0; PMID: 12408868
- Gartel AL, Shchors K. Mechanisms of c-myc-mediated transcriptional repression of growth arrest genes. Exp Cell Res 2003; 283:17 - 21; http://dx.doi.org/10.1016/S0014-4827(02)00020-4; PMID: 12565816
- Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene 2003; 22:351 - 60; http://dx.doi.org/10.1038/sj.onc.1206145; PMID: 12545156
- Si J, Yu X, Zhang Y, DeWille JW. Myc interacts with Max and Miz1 to repress C/EBPdelta promoter activity and gene expression. Mol Cancer 2010; 9:92; http://dx.doi.org/10.1186/1476-4598-9-92; PMID: 20426839
- Yan W, Samson M, Jégou B, Toppari J. Bcl-w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl-w and Bak/Bcl-w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol Endocrinol 2000; 14:682 - 99; http://dx.doi.org/10.1210/me.14.5.682; PMID: 10809232
- Weber A, Paschen SA, Heger K, Wilfling F, Frankenberg T, Bauerschmitt H, Seiffert BM, Kirschnek S, Wagner H, Häcker G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J Cell Biol 2007; 177:625 - 36; http://dx.doi.org/10.1083/jcb.200610148; PMID: 17517961
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47 - 59; http://dx.doi.org/10.1038/nrm2308; PMID: 18097445
- Garbe C, Leiter U. Melanoma epidemiology and trends. Clin Dermatol 2009; 27:3 - 9; http://dx.doi.org/10.1016/j.clindermatol.2008.09.001; PMID: 19095149
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 74; http://dx.doi.org/10.1016/j.cell.2011.02.013; PMID: 21376230
- Zhang LN, Li JY, Xu W. A review of the role of Puma, Noxa and Bim in the tumorigenesis, therapy and drug resistance of chronic lymphocytic leukemia. Cancer Gene Ther 2013; 20:1 - 7; http://dx.doi.org/10.1038/cgt.2012.84; PMID: 23175245
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 2001; 8:705 - 11; http://dx.doi.org/10.1016/S1097-2765(01)00320-3; PMID: 11583631
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292:727 - 30; http://dx.doi.org/10.1126/science.1059108; PMID: 11326099
- Kitada S, Pedersen IM, Schimmer AD, Reed JC. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene 2002; 21:3459 - 74; http://dx.doi.org/10.1038/sj.onc.1205327; PMID: 12032782
- Mounier N, Briere J, Gisselbrecht C, Emile JF, Lederlin P, Sebban C, Berger F, Bosly A, Morel P, Tilly H, et al. Rituximab plus CHOP (R-CHOP) overcomes bcl-2--associated resistance to chemotherapy in elderly patients with diffuse large B-cell lymphoma (DLBCL). Blood 2003; 101:4279 - 84; http://dx.doi.org/10.1182/blood-2002-11-3442; PMID: 12576316
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677 - 81; http://dx.doi.org/10.1038/nature03579; PMID: 15902208
- Aichberger KJ, Mayerhofer M, Gleixner KV, Krauth MT, Gruze A, Pickl WF, Wacheck V, Selzer E, Müllauer L, Agis H, et al. Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood 2007; 109:3031 - 41; PMID: 17110460
- Kang SY, Han JH, Lee KJ, Choi JH, Park JI, Kim HI, Lee HW, Jang JH, Park JS, Kim HC, et al. Low expression of Bax predicts poor prognosis in patients with locally advanced esophageal cancer treated with definitive chemoradiotherapy. Clin Cancer Res 2007; 13:4146 - 53; http://dx.doi.org/10.1158/1078-0432.CCR-06-3063; PMID: 17634542
- Jeong SH, Lee HW, Han JH, Kang SY, Choi JH, Jung YM, Choi H, Oh YT, Park KJ, Hwang SC, et al. Low expression of Bax predicts poor prognosis in resected non-small cell lung cancer patients with non-squamous histology. Jpn J Clin Oncol 2008; 38:661 - 9; http://dx.doi.org/10.1093/jjco/hyn089; PMID: 18772168
- Byun JY, Kim MJ, Eum DY, Yoon CH, Seo WD, Park KH, Hyun JW, Lee YS, Lee JS, Yoon MY, et al. Reactive oxygen species-dependent activation of Bax and poly(ADP-ribose) polymerase-1 is required for mitochondrial cell death induced by triterpenoid pristimerin in human cervical cancer cells. Mol Pharmacol 2009; 76:734 - 44; http://dx.doi.org/10.1124/mol.109.056259; PMID: 19574249
- Prokop A, Wieder T, Sturm I, Essmann F, Seeger K, Wuchter C, Ludwig WD, Henze G, Dörken B, Daniel PT. Relapse in childhood acute lymphoblastic leukemia is associated with a decrease of the Bax/Bcl-2 ratio and loss of spontaneous caspase-3 processing in vivo. Leukemia 2000; 14:1606 - 13; http://dx.doi.org/10.1038/sj.leu.2401866; PMID: 10995007
- Troeger A, Siepermann M, Escherich G, Meisel R, Willers R, Gudowius S, Moritz T, Laws HJ, Hanenberg H, Goebel U, et al. Survivin and its prognostic significance in pediatric acute B-cell precursor lymphoblastic leukemia. Haematologica 2007; 92:1043 - 50; http://dx.doi.org/10.3324/haematol.10675; PMID: 17640858
- Vogler M, Giagkousiklidis S, Genze F, Gschwend JE, Debatin KM, Fulda S. Inhibition of clonogenic tumor growth: a novel function of Smac contributing to its antitumor activity. Oncogene 2005; 24:7190 - 202; http://dx.doi.org/10.1038/sj.onc.1208876; PMID: 16091752
- Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, Lu Z, Chen Q, Lai Y, Pan J. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 2009; 15:1686 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-08-2141; PMID: 19240172
- Pan J, Quintás-Cardama A, Kantarjian HM, Akin C, Manshouri T, Lamb P, Cortes JE, Tefferi A, Giles FJ, Verstovsek S. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 2007; 109:315 - 22; http://dx.doi.org/10.1182/blood-2006-04-013805; PMID: 16912224
- Jin Y, Chen Q, Shi X, Lu Z, Cheng C, Lai Y, Zheng Q, Pan J. Activity of triptolide against human mast cells harboring the kinase domain mutant KIT. Cancer Sci 2009; 100:1335 - 43; http://dx.doi.org/10.1111/j.1349-7006.2009.01159.x; PMID: 19383029
- Gotlib J, Berubé C, Growney JD, Chen CC, George TI, Williams C, Kajiguchi T, Ruan J, Lilleberg SL, Durocher JA, et al. Activity of the tyrosine kinase inhibitor PKC412 in a patient with mast cell leukemia with the D816V KIT mutation. Blood 2005; 106:2865 - 70; http://dx.doi.org/10.1182/blood-2005-04-1568; PMID: 15972446