
Abstract
Clear cell RCC is the most common, and more likely to metastasize, of the three main histological types of RCC. Pathologic stage is the most important prognostic indicator and nuclear grade can predict outcome within stages of localized RCC. Epithelial tumors are thought to accumulate a series of genetic and epigenetic changes as they progress through well-defined clinical and histopathological changes. MicroRNAs (miRNAs) are involved in the regulation of mRNA expression from many human genes and miRNA expression is dysregulated in cancer. To better understand the contribution of dysregulated miRNA expression to the progression and biology of ccRCC, we examined the differences in expression levels of 723 human miRNAs through a series of analyses by stage, grade, and disease progression status in a large series of 94 ccRCC. We found a consistent signature that included significant upregulation of miR-21-5p, 142-3p, let-7g-5p, let-7i-5p and 424-5p, as well as downregulation of miR-204-5p, to be associated with ccRCC of high stage, or high grade, or progression. Discrete signatures associated with each of stage, grade, or progression were also identified. The let-7 family was significantly downregulated in ccRCC compared with normal renal parenchyma. Expression of the 6 most significantly differentially expressed miRNAs between ccRCC was verified by stem-loop qRT-PCR. Pathways predicted as targets of the most significantly dysregulated miRNAs included signaling, epithelial cancers, metabolism, and epithelial to mesenchymal transition. Our studies help to further elucidate the biology underlying the progression of ccRCC and identify miRNAs for potential translational application.
Introduction
In the United States, approximately 58 000 new cases of, and 13 000 deaths from, RCC are estimated in 2013.Citation1 Among the subtypes of RCC, clear cell carcinoma (ccRCC) is the most abundant (∼75%) and is over-represented (∼90%) in series of metastatic RCC.Citation2 The vast majority of localized (stage I or II), and a proportion of locally advanced (stage III), ccRCC are curable by surgical resection whereas for metastatic ccRCC (stage IV) available treatment options are of limited effectiveness. Immunotherapy has proven to be effective in a very small subset of patients and tumors invariably develop resistance to the now standard anti-VEGF and anti-mTOR therapeutic agents.Citation3
Prognostic assessment is important for risk stratification to inform management of localized or locally advanced RCC after surgical resection. Pathologic stage, based on the size and extent of invasion by the tumor, is the most accurate indicator of prognosis. For localized RCC, high grade tumors are considered at greater risk of progression. Molecular progression models suggest that most solid neoplasms accumulate a series of genetic and epigenetic alterations as they progress through well-defined clinical and histopathological changes.Citation4,Citation5 The precise molecular events that underlie tumor progression from a lower to higher pathologic stage or grade and from local to metastatic disease are, for the most part, unclear. Tumor alterations reported to be correlated with stage, grade and/or prognosis in RCC include overexpression of CA-IX and survivin, deletion of chromosome 9p and 14q, gene mRNA expression levels,Citation6,Citation7 and point mutation of BAP1.Citation8 A better understanding of the biology that underlies the progression of ccRCC could lead to more accurate prognosis and also to identification of new therapeutic targets to alter the natural history of metastatic disease.
MicroRNAs (miRNAs) are a class of small (∼22 nt) non-coding RNAs that regulate post-transcriptional gene expression through the epigenetic mechanism of RNA interference. miRNAs base pair 2–8 nucleotides of their sequence to the 3′-UTR of complementary mRNA transcripts and facilitate degradation or inhibit translation of multiple target mRNAs. miRNAs are thought to be involved in the regulation of mRNA from most human genesCitation9 and are implicated in most biological processes in normal cells. Dysregulation of expression of miRNAs can occur in tumor cells and affect differentiation, proliferation, and apoptosis.Citation10 miRNAs overexpressed in cancer cells compared with normal cells have been termed oncogenic and miRNAs that show reduced expression in tumor cells as tumor suppressors. miRNAs demonstrated to have a functional role in cancer have been referred to as oncomirs.Citation11 Global miRNA expression studies have identified miRNAs consistently dysregulated across many types of cancer, and other dysregulated miRNAs more specifically associated with a particular cell type or organ site of cancer, as of potential clinical relevance.Citation10
In this report, we have performed a global miRNA expression microarray study on a large series of ccRCC grouped by stage, grade, or disease progression in order to identify miRNAs putatively involved in the biology of RCC progression. Our study implicates a number of miRNAs, in particular miR-21-5p, 142-3p, let-7g-5p, let-7i-5p, 424-5p, and 204-5p, in the progression of ccRCC, as these miRNAs are significantly dysregulated irrespective of whether stage alone, grade alone, stage and grade together, or progression are used as criteria to group ccRCC for analysis. Pathway analysis suggests that the significantly dysregulated miRNAs target pathways in signaling, cancer, metabolism, and epithelial to mesenchymal transition (EMT) in clear cell renal carcinoma cells. The miRNA signatures may have utility for prediction of outcome and in the identification of therapeutic targets for ccRCC.
Results
Performance of assay
We analyzed the expression of 723 miRNAs in 95 primary clear cell RCC (ccRCC) and 5 normal renal parenchyma (NRP) specimens by the Agilent v2 human microRNA microarray. We examined the quality of microarray raw data for the 95 ccRCC and 5 NRP samples by performing a pairwise Pearson correlation analysis among all 100 samples. One ccRCC specimen was an obvious outlier in technical quality (Fig. S1) and was removed from further study. There was very little variation seen between the 5 NRP at the bottom right of the heatmap (Fig. S1).
miRNA expression profiles can distinguish early and advanced ccRCC
To more clearly differentiate miRNA expression signatures for biological progression, we first compared typical “early” ccRCC with typical “advanced” ccRCC. We selected 29 tumors of low grade and low stage (grade 1 or 2 and stage 1 or 2) that had no evidence of progression for a median 64.19 mo (mean 67.46, range 25 to 123) from the date of surgery. We also selected 29 tumors of high grade and high stage (grade 3 or 4 and stage 3 or 4) () with a median follow-up of 14.49 mo (mean 26.77, range 0.3 to 95.24 mo) from the date of surgery. During follow-up, 28 of 29 HGHS ccRCC patients died. Two HGHS patients had no evidence of disease after surgery including the one patient that is alive and another patient that died of causes other than ccRCC. The HGHS group included 21 stage IV at diagnosis and some patients who received sunitinib. Thirty-six of the total 94 ccRCC in our study were excluded from this analysis: 27 because they were of low stage but high grade or vice versa, 5 with previous history of RCC at the time of diagnosis, and 4 of low grade and stage that progressed.
Table 1. Clinicopathological data for the 94 ccRCC
To identify differentially expressed miRNAs between the LGLS and HGHS groups we performed statistical analysis by Limma. The full miRNA expression data for this, and the following, comparisons can be found in the Supplemental Materials. shows the miRNAs differentially expressed between the LGLS and HGHS ccRCC with a FDR less than 0.2. We noted that for several miRNAs more than one (of generally two) probes for each individual miRNA on the array was within the FDR cut-off for significant differential expression. There were 9 upregulated probes from 7 miRNAs (miR-21-5p, 142-3p, 193b-3p, let-7i-5p, let-7g-5p, 223-3p, and 15b-5p) and 10 downregulated probes from 8 miRNAs (miR-204-5p, 145-5p, 30a-3p, 502-3p, 99b-5p, 320a, 30c-2-3p, 151a-3p). Unsupervised two-dimensional hierarchical clustering using a data matrix of the 19 probes from 15 miRNAs with significant differential expression showed a separation into two main groups of ccRCC (). A majority of LGLS ccRCC occupied the cluster on the left side (24 LGLS of 31 total samples), while the cluster on the right contained mainly HGHS ccRCC (22 HGHS of 27 total). The difference between the proportions of LGLS and HGHS in the two highest-level clusters was significant (the Fisher exact test P < 0.0001, two-sided).
Table 2. miRNAs with significant differential expression across analyses of stage, grade, and disease progression
Figure 1. miRNA expression can distinguish early and advanced ccRCC. (A) Unsupervised two-dimensional hierarchical clustering of 58 ccRCC samples using a data matrix of the 19 probes with an FDR < 0.2 and P < 0.05 and >1.5-fold change of the level of expression between 29 LGLS and 29 HGHS ccRCC. Tumor samples are displayed on the horizontal axis and individual miRNAs on the vertical axis. Tumor samples are designated L for LGLS and H for HGHS. The dendrogram on the horizontal axis indicates the two highest level clusters of tumors. (B) Real-time RT-PCR was performed on all the 58 ccRCC samples run on the array to amplify the six most differentially expressed miRNAs indicated by microarray analysis. Relative quantification was achieved by plotting 2(−∆CT) values of all the early and advanced groups of samples.
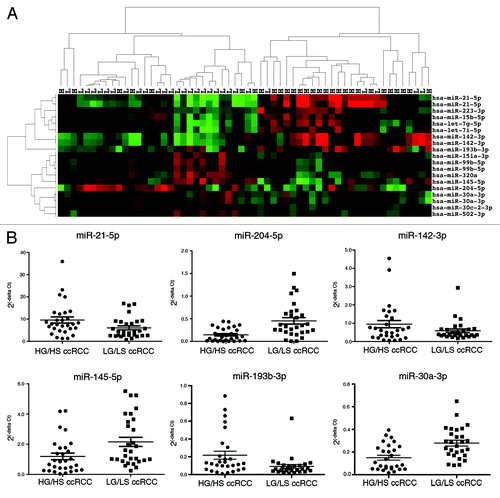
To verify the microarray expression data, we performed stem-loop reverse transcriptionCitation12 followed by real-time qRT-PCR for the three most upregulated (miR-21-5p, miR-142-3p, and miR-193b-3p) and the three most downregulated (miR-204-5p, miR-145-5p, and miR-30a-3p) miRNAs in HGHS ccRCC identified from the statistical analysis. Real-time qRT-PCR results from the 58 ccRCC () showed the same directional trend in miRNA expression and were similar to the microarray data.
miRNA expression separates a majority of high stage from low stage ccRCC
We next examined ccRCC by stage irrespective of nuclear grade but after exclusion of 13 low stage (I–II) ccRCC patients whom later progressed to metastatic (stage IV) disease. We considered the 13 tumors to be biologically more advanced than indicated by the clinical staging of the disease at the time of diagnosis and surgical resection and, therefore, to be potential confounders. We thus had a total of 81 samples for this curated analysis including the 58 samples used for the previous analysis. Among the 81 samples, 46 samples were of low stage (I–II) and 35 samples were of high stage (III–IV). Limma analysis found 11 probes from 8 miRNAs to be significantly upregulated (miR-21-5p, 142-3p, 424-5p, 223-3p, 148a-3p, let-7g-5p, let-7i-5p, and 31-5p) and 2 miRNAs significantly downregulated (miR-204-5p and 99a-5p) in high stage (HS) ccRCC. The most upregulated miRNAs, miR-21-5p and miR-142-3p, and the most downregulated miRNA, miR-204-5p, in HS ccRCC () were the same as in the prior LGLS vs. HGHS ccRCC analysis. Three miRNAs were significantly upregulated (miR-424-5p, 148a-3p, and miR-31-5p) and a single probe for miR-99a-5p significantly downregulated in HS tumors but not in HGHS tumors from the prior analysis. Two-dimensional unsupervised hierarchical clustering with a data matrix of the 13 probes showed most of the LS samples (36 LS of 45 total samples) in the cluster at the right side of the heatmap, and many of the HS samples (26 HS of 36 total samples) in the cluster on the left side of the heatmap (). The difference in proportions was significant (the Fisher exact test P < 0.0001, two-sided).
Figure 2. miRNA expression separates a majority of high stage from low stage ccRCC. Unsupervised two-dimensional hierarchical clustering with a data matrix of 13 probes in 81 annotated ccRCC (46 LS and 35 HS).
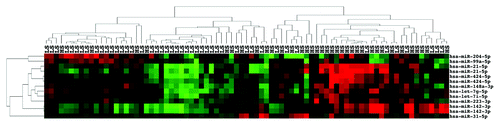
We then examined all 94 ccRCC divided into 59 low stage or 35 high stage () irrespective of nuclear grade or disease progression status. There were noticeably fewer significantly dysregulated miRNAs: two probes for miR-21-5p and a probe for miR-223-3p were significantly upregulated and none downregulated in HS ccRCC (). This finding strengthened our idea that the clinical stage at the time of surgical resection underestimated the metastatic potential of the subset of low stage ccRCC that subsequently progressed. We include this analysis simply for comparison to the curated stage analysis above. Clustering showed 41 LS of 51 total samples in the left cluster and 25 HS of 43 samples in the right cluster (the Fisher exact test P = 0.0002, two-sided) (Fig. S2).
miRNA expression can also differentiate high grade from low grade ccRCC
In a similar manner, we examined miRNA expression by tumor grade. For this analysis, we grouped all 44 low grade (I–II) ccRCC samples () together in one group and all 50 high grade (III–IV) samples in the other group, irrespective of clinical stage, for a total of 94 ccRCC. Significant differentially expressed miRNAs between LG and HG ccRCC are listed in . Ten probes from 7 miRNAs were significantly upregulated (miR-21-5p, 142-3p, 193-3p, 15b-5p, let-7i-5p, 146b-5p, and let-7g-5p) in the HG ccRCC. Only miR-146b-5p was significantly upregulated in HG but not in HGHS or HS ccRCC. Two miRNAs, 15b-5p and 193b-3p, significantly upregulated in HG and HGHS were not significantly upregulated in HS ccRCC. Seven probes from 5 miRNAs were significantly downregulated (miR-145-5p, 204–5p, 30a-5p, 99b-5p, and 181a-5p) in HG ccRCC. Two of the 5 miRNAs (miR-30a-5p and 181a-5p) were not significantly downregulated in the previous analyses. Only miR-204-5p was significantly downregulated in HGHS and HS. Unsupervised two-dimensional hierarchical clustering of a data matrix of the 17 probes grouped a majority of HG samples (35 HG of 48 total) in the right side cluster while most of the LG samples (31 LG of 46 total) were in the cluster on the left side (). The proportion of LG to HG in the two clusters was significant (the Fisher exact test P = 0.0002, two-sided).
Figure 3. miRNA expression can differentiate high grade from low grade ccRCC. (A) Unsupervised two-dimensional hierarchical clustering with a data matrix of 17 probes in 94 ccRCC (44 LG and 50 HG). (B) Unsupervised two-dimensional hierarchical clustering with a data matrix of 37 probes in 23 ccRCC (7 grade I and 16 grade IV). (C) Unsupervised two-dimensional hierarchical clustering with a data matrix of 7 probes in 59 ccRCC (38 LGLS and 21 HGLS). LL, low stage, low grade; LH, low stage, high grade.
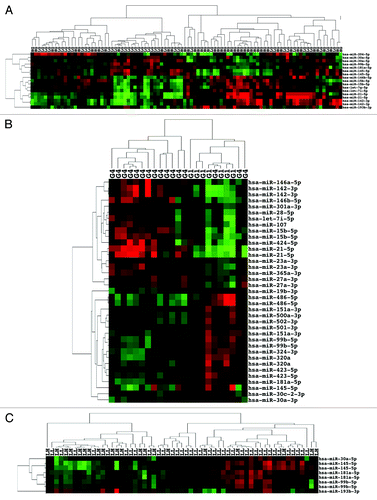
We next examined differentially expressed miRNAs in the 7 grade I vs. 16 grade IV tumors among the 94 ccRCC as we reasoned that a comparison of the lowest and highest categories of grade would show the clearest differences in expression. Eighteen probes from 15 miRNAs were significantly upregulated and 19 probes from 14 miRNAs were significantly downregulated in grade IV ccRCC (). Cluster analysis of the 37 probes () showed a left side cluster of 13 grade IV ccRCC and a right side cluster of mainly grade I tumors (7 grade I and 3 grade IV). The difference in proportions was significant (the Fisher exact test P = 0.0005, two-sided).
Lastly, since in the setting of a clinical trial high grade, low stage ccRCC are considered at greater risk of progression than low grade, low stage ccRCC, we also examined 38 LGLS vs. 21 HGLS ccRCC. miR-193b-3p was significantly upregulated and miR-145, 181a-5p, 99b-5p, and 30a significantly downregulated in HGLS ccRCC (). Cluster analysis of the 8 probes () shows the left cluster with a small majority of HGLS tumors (15 LH of 24 total) and the right cluster containing mostly LGLS tumors (29 LL of 35 total). The difference in proportions was significant (the Fisher exact test P = 0.0007, two-sided).
miRNA expression signatures of biological aggressiveness and progression to metastatic disease
In our set of 94 ccRCC, there were 70 stage I–III ccRCC with a median follow up of 57.39 mo (mean 54.09, range 1.5–123.1 mo). During this period, 19 patients progressed to metastatic disease at a median of 12.02 mo (mean 18.56, range 1.51–56.47 mo). The remaining 51 patients had no evidence of disease at a median follow-up of 72.17 mo (mean 67.33, range 11.4–123.13 mo). We examined miRNA expression in the group of 51 stage I–III with no progression (NP) vs. the 19 stage I–III that progressed (P). Twenty-five probes from 17 miRNAs were significantly upregulated in the P group. The most upregulated miRNAs were miR-21-5p and miR-142-3p. Significantly upregulated miRNAs not identified in previous analyses included miR-451a, 155-5p, let-7f-5p, 96-5p, 16-5p, 142-5p, 221-3p, and 146b-5p (). Of the 21 probes from 16 miRNAs significantly downregulated in the P group: miR-204-5p, 320a, 145-5p, 99b-5p, and 151a-3p were the most downregulated. miRNAs not identified as significantly downregulated in previous analyses were miR-638, 210, 139-5p, 361-5p, 193a-5p, 885-3p, let-7b-5p, and 181c-3p (). Two-dimensional hierarchical unsupervised clustering of a data matrix of the 46 probes () grouped 45 total tumors in the left cluster, 6 of which were P tumors and 25 total in the cluster on the right, 13 of which were in the P group. This difference in proportions was significant (the Fisher exact test P = 0.0008, two-sided).
Figure 4. miRNA expression can distinguish de novo metastatic ccRCC as well as de novo non-metastatic ccRCC that subsequently progressed. (A) Unsupervised two-dimensional hierarchical clustering with a data matrix of 46 probes in 70 ccRCC (51 NP and 19 P). (B) Unsupervised two-dimensional hierarchical clustering with a data matrix of 24 probes in 94 ccRCC (51 NM and 43 M).
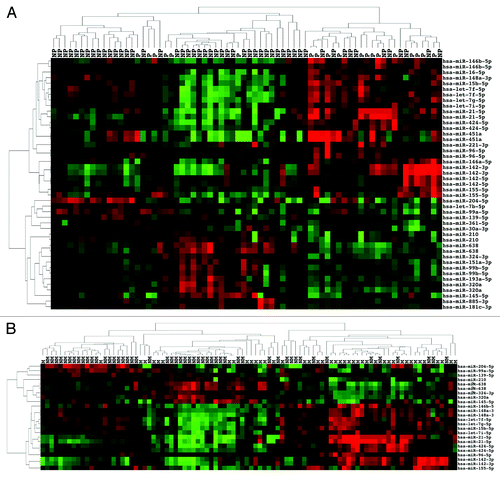
To further identify miRNAs associated with biological aggressiveness, we divided the 19 cases of progression into 10 cases of progression within 12 mo or 9 cases of progression after 12 mo from the date of surgical resection. We excluded the latter group from analysis. We therefore examined 51 stage I–III ccRCC with no evidence of progression (less aggressive, LA) vs. 34 more aggressive (MA) ccRCC comprised of the 24 stage IV at diagnosis tumors and also the 10 stage I–III at diagnosis tumors that progressed within 1 y as these tumors were likely understaged. All of the 16 significantly upregulated and 8 downregulated miRNAs identified by this analysis had appeared in prior analyses (). Clustering of the 35 probes (Fig. S3) grouped 23 MA of 53 total in the left cluster and 11 MA of 32 total in the right cluster but the differences in proportions was not significant (the Fisher exact test P = 0.4956, two-sided).
Lastly, we compared non-metastatic (NM) disease (51 stage I–III without progression) vs. 43 ccRCC with either de novo metastasis (24 patients) or metastasis during follow-up (19 patients) of a median 12.02 mo (mean 18.56, range 1.51–56.47 mo). Eleven miRNAs were significantly upregulated and 8 significantly downregulated in the metastatic (M) group (). All the significantly dysregulated miRNAs had appeared in previous analyses. Clustering of 24 probes () grouped most NM ccRCC in the left cluster (39 NM of 54 total) and most M ccRCC in the right cluster (28 M of 40 total). The difference in proportions was significant (the Fisher exact test P < 0.0001, two-sided).
Differences in miRNA expression between ccRCC and normal renal parenchyma
Finally, we examined miRNA expression in the 94 ccRCC compared with the 5 NRP. Seventy probes from 44 miRNAs were significantly upregulated in the ccRCC. The most upregulated miRNAs included miR-210, 21-3p, 494, 21-5p, 1225-5p, 194-5p, and 142-3p. One-hundred-and-six probes from 64 miRNAs were significantly downregulated in ccRCC. The most downregulated were miR-141-3p, 200c-3p, 10a-5p, 199a-3p, 10b-5p, and 200b-3p. All members of the let-7 family were significantly downregulated in ccRCC except for let-7b that was downregulated at a level that only approached statistical significance. Unsupervised clustering of the 99 renal specimens (Fig. S4) indicated two highest level groups each with several subgroups. The 5 NRP formed a tight cluster at the side of one of the highest level groups.
Discussion
Pathogenesis of ccRCC
Among the main histological subtypes of RCC, clear cell is the most common and the most likely to metastasize.Citation2 Pathologic stage based on the size of the tumor and the extent of invasion is the most important prognostic indicator, followed by grade. However, after surgical removal of the primary RCC, a subset of 10–28% of individuals with organ-confined (pT1 and T2) RCC progress usually within 3–5 y.Citation13 Similarly, patients with more advanced disease (pT3) have disparate characteristics such as invasion of the perirenal or sinus fat or vascular invasion that affect therapeutic outcomes. Additionally, there is a growing movement to manage renal masses more conservatively, including non-operative management. In the population under active surveillance for a small renal mass at present there are no recognized non-pathologic markers that might predict aggressive or indolent tumor biology. Better predictors of tumor behavior are therefore needed to more appropriately guide management of the individual patient. Furthermore, the advent of newer targeted therapies has not benefited all patients with metastatic ccRCC and those tumors that do respond invariably develop resistance. Hence, a better understanding of the biological pathways disrupted in ccRCC and of the genes that regulate these pathways is necessary to develop therapeutics targeted to the disease. Through the epigenetic regulation of gene mRNA expression microRNAs have an important role in the control of pathways in many crucial cellular processes. We therefore studied the expression of miRNAs to gain insight into the biology and, in particular, progression of ccRCC.
miRNA expression by stage, grade, and progression in ccRCC
We performed a series of analyses of miRNA expression by stage, grade, and/or disease progression on 94 representative ccRCC. A series of analyses could be helpful because each parameter of prognosis, i.e., stage, grade, or disease progression, is imperfect. For example, stage can be confounded by sub-clinical metastasis, nuclear grading is susceptible to measurable inter-observer variability, and propensity to progression might be masked by time of surgery. Overall, 39 probes from 25 miRNAs were significantly upregulated and 37 probes from 26 miRNAs downregulated in one or more of the nine patient groups of less favorable prognosis or outcome among the 94 ccRCC (). The direction of the miRNA expression change was consistent across all nine analyses in our study. As might be expected, there was considerable overlap between the miRNAs identified in different analyses since higher stage and higher grade are associated with each other and with progression to metastatic disease. Several particular microRNAs (miR-21-5p, 142-3p, let-7g-5p, let-7i-5p, and 424-5p) were consistently significantly upregulated, while miR-204-5p was consistently significantly downregulated, in ccRCC of high stage, or high grade, or in non-metastatic ccRCC at diagnosis that progressed as well as other of the analyses. Another set of microRNAs (miR-223-3p, 148a-3p, and 31-5p) were upregulated and miR-99a-5p downregulated only in analyses where stage or progression was a parameter but no association with grade was apparent. Conversely, dysregulation of a different group of microRNAs was associated with high grade and sometimes progression but not in analysis by stage alone. This group included upregulation of miR-15b-5p, 193b-3p, 146-5p, 23a-3p, and 27a-3p as well as downregulation of miR-30a-3p, 145-5p, 151a-3p, 99b-5p, and 320a among others. A further set of microRNAs was significantly upregulated (miR-155-5p, 451a, let-7f-5p, 96-5p, 16-5p, 142-5p, and 221-3p) or downregulated (miR-638, 210, and others) in stage I–III ccRCC with subsequent metastasis and/or de novo metastatic ccRCC but not by analyses where the parameter of interest was high stage or high grade.
As can be seen from the chromosomal location, given in , of the most upregulated and downregulated miRNAS, there does not appear to be a strong association with the chromosomal regions 5q, 7q, 12p, 20q, 8q, and 3q that commonly show gain or 3p, 14q, 8p, 9p 6q, and 10q that are often lost in ccRCC.Citation14,Citation15
Biological roles of miRNAs with dysregulated expression in ccRCC
miR-21 is reported to be upregulated in RCC compared with normal renal cells and an association between higher expression with higher stage and grade of RCC has been described.Citation16 miR-21 is well studied as upregulated, thought to function as an oncomir in proliferation, invasion, and apoptosis, and is often associated with advanced disease in many other cancer types including lung, breast, and prostate.Citation17 miR-21 has been shown to target tumor suppressor genes including the PTEN pathway, the RAS pathway through PDCD4,Citation18 and the p53 network.Citation19
miR-142-3p is not as well studied; however, using a mouse model of cancer, Olson et al. found miR-142-3p to be specifically upregulated during angiogenic islets formation, an intermediate stage in cancer metastasis. This suggests a specific role for miR-142-3p in angiogenesis.Citation20 miR-142-3p is reported upregulated in RCC compared with normal renal cellsCitation21,Citation22 but has not been previously implicated in the progression of ccRCC. miR-142-3p is also upregulated in T-cell acute lymphoblastic leukemia.Citation23 TargetScanHuman release 6.2 (http://www.targetscan.org/) predicts APC as a conserved target of miR-142-3p and the Wnt signaling pathway is known to be a major target of aberrant hypermethylation in ccRCC and is likely involved in the pathogenesis of ccRCC.Citation3 Another upregulated miRNA in our study, miR 424-5p, has also been reported to promote angiogenesis.Citation24
An interesting finding in our study was upregulation of let-7g-5p and let-7-i-5p in ccRCC of high grade or stage, or stage I-III ccRCC that progressed. The let-7 miRNA family is generally considered to be downregulated, and by implication to have a tumor suppressor role, in cancer cells compared with normal cells.Citation17,Citation25,Citation26 However there are several reports of upregulation in certain cancer typesCitation25 and let-7f is known to promote angiogenesisCitation27 which provides a basis for upregulation in tumor progression. We also found downregulation of all the let-7 family miRNAs in ccRCC compared with NRP but consistent upregulation of let-7g-5p and let-7-i-5p in ccRCC of high stage, or of high grade, or with progression, compared with less advanced ccRCC. The let-7 family of miRNAs are very similar in sequence but both the Agilent let-7g-5p and let-7i-5p probes were noted to show extremely high specificity through low cross hybridization in a report on the microarray technologyCitation28 we used here. There are examples of miRNAs that show oncogenic effects in one tissue but act as a tumor suppressor in another tissue type e.g., miR-29.Citation26 There are also miRNAs with “opposing activities” in the same normal cell e.g., miR-430, and miRNAs that potentially have rival actions in the same tumor cell, e.g., miR-200, which is known to be involved in inhibition of EMT but also to promote the ability of metastatic breast cancer cells to colonize.Citation26 It will be important to further examine a role for the let-7 miRNAs in the development and progression of ccRCC.
miR-204 is identified as one of the most downregulated miRNAs in ccRCC by several studies.Citation21,Citation22,Citation29 Functional studies in ccRCC cells demonstrated that miR-204 is a VHL-regulated tumor suppressor that acts by inhibiting macroautophagy, with MAP1LC3B as a direct functional target. Of note, higher tumor grade of human ccRCC was correlated with a concomitant decrease in miR-204 expression and an increase in MAP1LC3B levels, indicating that MAP1LC3B-mediated macroautophagy is necessary for RCC progression.Citation30 A study of glioma reported that miR-204 was downregulated and provided evidence that miR-204 targeted the SOX4 transcription factor leading to suppression of the self-renewal capacity of stem cells, and also targeted EphB2 resulting in inhibition of cell migration.Citation31
miR-145-5p is downregulated in general in cancer, is considered a tumor suppressor, and inhibits angiogenesis, growth, and invasion by targeting VEGF.Citation32 miR-145-5p was implicated in prognosis of RCC by Slaby et al.Citation33 miR-30a-3p appears to be downregulated in cancer and to be a tumor suppressor by inhibition of EMT through regulation of Snail1.Citation34 Wang et al. suggest caution in any assessment of miR-30a-3p expression by the probes on the Agilent array.Citation28 Since we verified the downregulation of miR-30a-3p by stem-loop RT-PCR in the same ccRCC, we included the data. The Mir-193b-365 cluster is essential for brown fat differentiationCitation35 which may indicate a role for miR-193b in lipid metabolism. miR-99b-5p was reported to be downregulated in prostate cancer and overexpression by transfection to inhibit growth of prostate tumor cells.Citation36
Analysis by DIANA mirpathCitation37 (http://diana.cslab.ece.ntua.gr/?sec=homeof) identified KEGG pathways significantly (P < 0.05) overrepresented as targets of the 51 dysregulated miRNAs. These pathways included the Wnt, TGF-β, MAPK, Hedgehog, and mTOR signaling pathways, various pathways of metabolism, epithelial to mesenchymal transition (EMT), apoptosis, pathways in cancer, renal cell carcinoma, and other cancers (Sup. Materials).
miRNA expression in ccRCC and normal renal cells
We also examined differences in miRNA expression levels between the 94 ccRCC and 5 NRP since there are only two reports of analysis of ccRCC by the Agilent v2 arrayCitation38,Citation39 As noted by other investigators in studies of different types of cancer,Citation26 we also found more miRNAs to be downregulated than upregulated in ccRCC. Furthermore, the level of downregulation was greater than of upregulation. Our analysis implicated several miRNAs as dysregulated in ccRCC for the first time including upregulation of miR-494 reported to target PTEN,Citation40 142-3p implicated in angiogenesisCitation20 as discussed above, 22-3p involved in the regulation of differentiation of mesenchymal stem cellsCitation41 and also known to be a repressor of PTENCitation42 as well as other miRNAs such as miR-1225-5p and 342-3p of which little or nothing is known of targets and functional role. We also identified downregulation of the let-7 family involved in the inhibition of cell growth and proliferation,Citation17 miR-200a-3p and 429 which are both members of the miR-200 family that target the ZEB1 and ZEB2 transcriptional repressors of E-cadherin and so are implicated in EMT and tumor invasion,Citation17 199a-5p involved in the regulation of the IKKβ/NFκB and PTEN/AKT pathwaysCitation43 and other miRNAs such as miR-30c-5p of unknown function. Analysis by DIANA mirpath on a set of the 20 most upregulated and 30 most downregulated miRNAs identified the PI3K-Akt, MAPK, Wnt, TGF-β, p53, mTOR, Hedgehog, and VEGF signaling pathways, various pathways of metabolism, EMT, apoptosis, pathways in cancer, renal cell carcinoma, and other cancers as over-represented (P < 0.05) (Sup. Materials).
Studies of miRNA expression and prognosis in RCC
There have been only two prior studies of ccRCC using the Agilent v2 miRNA expression array of 723 human miRNAs. Weng et al. studied 3 pairs of benign kidney and ccRCCCitation38 and Wu et al. examined 13 localized and 15 metastatic ccRCC as well as 10 benign kidney specimens.Citation39 Another two studies used the Agilent v1 array with 470 miRNAs. Jung et al. examined 12 ccRCC of mainly higher stage and matched normalCitation29 while Nakada et al.Citation44 looked at 16 mostly low stage ccRCC and 6 normal specimens. Our ccRCC vs. NRP data are in good agreement with all four studies demonstrating a degree of consistency with the same platform technology. The miRNAs identified here by microarray as differentially expressed between NRP and ccRCC also agree well with a recent study by RNA sequencing on 11 ccRCC and 11 patient matched adjacent normal specimens.Citation22
The same RNA sequencing study examined miRNA expression in 22 ccRCC divided into prognostic subgroups.Citation22 Among the miRNAs associated with worse prognosis were upregulation of miR-193b-3p, 221-3p, and 146a-5p as well as downregulation of miR-204-5p and 139-5p: all of which were also significantly dysregulated in the same direction in our study. In another study that used Taqman low density arrays of 754 miRNAs to examine prognosis, Slaby et al. found downregulation of miR-145-5pCitation33 as we did also. From a comparison of 13 localized and 15 metastatic ccRCC, Wu et al. validated a panel of 4 miRNAsCitation39 one of which, miR-139-5p, was also identified in Osanto et al.Citation22 and in our study here. White et al. used microparaflo microfluidic technology to assess 875 miRNAs in an initial 18 primary and 10 unmatched metastatic fresh-frozen ccRCC.Citation45 The miRNAs identified in this study as downregulated in metastatic ccRCC correspond well to our findings but the upregulated miRNAs do not. Recently, TCGA reported that unsupervised clustering of RNA sequencing data identified upregulation of miR-21 and downregulation of miR-204 as distinguishing a group of ccRCC with poor overall survival from other ccRCC.Citation14 These two miRNAs showed the most dysregulated expression in advanced ccRCC in our study also. Disparities between the set of miRNAs identified in our data with other studies are likely due to differences in: the histopathology of tumor specimens examined, technology platform used, changes in nomenclature, filtering of probes, and statistical analysis.
Caveats to our study
Since our study began, more human miRNAs than the 723 on the Agilent v2 microarray have been discovered (http://www.mirbase.org/) and will need to be examined for a more comprehensive survey of miRNA expression in ccRCC. We observed relatively modest fold changes in miRNA expression between groups of ccRCC of different stage, grade or biological behavior. It seems likely that fold changes between one clear cell tumor to another might be less pronounced than between normal and tumor or between different histological cell subtypes of RCC. A related point concerns the tumor cell content of a primary ccRCC specimen. It should be noted that while RCC is encapsulated so that contamination by surrounding normal renal parenchyma is not an issue, all ccRCC contain lymphocytes, endothelial cells, and connective tissue cells that will dilute the tumor cell miRNA expression profile. In our experience, a cut-off of ≥70% tumor cell content allows the inclusion of any ccRCC thereby avoiding bias in tumor selection. The use of laser capture microdissection could increase the tumor cell content but invariably will facilitate RNA degradation to a greater or lesser degree.Citation46
Summary
Our study has examined genome-wide miRNA expression in a large number of ccRCC, broadly representative of the disease as it presents by stage and grade,Citation47 from a single institution, and re-reviewed by a single uropathologist for cell type and grade. miRNA expression was determined from fresh-frozen specimens, with a tumor cell content assessed as ≥70%, by the Agilent microarray platform that utilizes direct labeling less prone to amplification bias and that is both sequence and size selective and so specific for the mature form of miRNAs,Citation28 and is reported to have performed well in comparative studies of genome-wide miRNA expression technologies.Citation48,Citation49 That, after statistical analysis, several of the miRNAs with significant differential expression were represented by more than one probe for an individual miRNA strengthens confidence, as does the verification of a subset of the most dysregulated miRNAs by a quantitative independent technology, and that the miRNAs identified as most significantly dysregulated in our study agree well with recent RNA sequencing studies.Citation14,Citation22
We found a signature of miRNA expression that was consistently present in ccRCC of high stage, or of high grade, or with progression as well as signatures that associated with each of these parameters of more advanced disease. The signatures include many miRNAs novel to ccRCC or to progression in ccRCC that target pathways of clear biological relevance to this disease. Further study of the mRNA targets of the miRNAs we have identified may provide additional insight into the pathogenesis and biology of RCC. The individual miRNAs or signatures may have translational relevance to early detection in body fluids. The miRNA expression signature could also be used to predict prognosis by analysis of body fluids or needle biopsy of patients considered for active surveillance or, more readily, in the tumors from patients having undergone surgical resection. Lastly, the dysregulated miRNAs are candidate targets for therapy and also implicate pathways, such as of metabolism, as therapeutic targets in ccRCC.
Materials and Methods
Specimens
Specimens were collected under a Fox Chase Cancer Center (FCCC) Institutional Review Board (IRB) and all patients provided written consent. Snap-frozen RCC specimens from patients who underwent partial or radical nephrectomy for RCC between 2001 and 2009 were obtained from the FCCC Biospecimen Repository. These tissues were re-examined for histological cell type and Fuhrman nuclear gradeCitation50 by a single pathologist experienced in RCC, Dr Al-Saleem. Ninety-five ccRCC were selected for this study. Clinicopathological data are given in . Grades 1 and 2 were categorized as low grade carcinoma (LG). Grades 3 and 4 were grouped as high grade carcinoma (HG).Citation50 Clinical stages 1 and 2 were categorized as low stage (LS) and stages 3 and 4 as high stage (HS) carcinoma.Citation51 Normal renal parenchyma specimens were obtained from five FCCC patients (3 male, 2 female) with unifocal ccRCC and of similar age (mean 62.8 y) to RCC patients at diagnosis (median 64 y) (http://seer.cancer.gov/statfacts/html/kidrp.html).
Sample preparation
Frozen tumor tissue sections were stained with H&E and examined by the pathologist, Dr Al-Saleem, for selection of an area of ≥70% tumor cell content to be used for RNA extraction. The chosen area of tissue was cut into smaller pieces and disrupted using Kimble–Kontes disposable pestles (Sigma-Aldrich Z359947) followed by homogenization with Qiashredder (Qiagen 79656) columns. Total RNA was extracted using Trizol (Invitrogen 15596-026). The RNA quality was measured for all tissues in this study using a Nanodrop (Thermo-Scientific) and an Agilent Bioanalyzer 2100 (Agilent Technologies) to obtain the optical density 260/280 nm and 260/230 nm ratios. We included samples that had a 260/280 nm ratio between 1.8 and 2.0 and a 260/230 nm ratio between 1.8 and 2.2 as well as a discrete peak for the miRNA population as measured by the Small RNA assay.Citation52
miRNA expression microarray
One hundred nanograms of total RNA was labeled with Cy3 and hybridized with a miRNA labeling and hybridization kit (Agilent Technologies 5190-0456) according to the manufacturer’s instructions. We used Human miRNA microarray Version 2.0 (Agilent Technologies G4112F), which contains probes for 723 human (and 76 human viral) miRNAs based on Sanger miRBase release 10.1. The arrays were washed then scanned with a laser confocal scanner (Agilent Technologies, G2505B). The fluorescence intensity was calculated by Feature Extraction Software (Agilent Technologies). The raw miRNA microarray data set is available at Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/). miRNAs are annotated according to miRBase release 18 (http://www.mirbase.org/).
Quantitative RT-PCR verification
Total RNA samples were reverse transcribed using a TaqMan MicroRNA Reverse Transcription kit (Applied Biosystems 4366596) following the manufacturer’s protocol. Real-time PCR was performed on an Applied Biosystems 7500 Real Time PCR system for 40 cycles using predesigned TaqMan miRNA Assays (Applied Biosystems). Small nucleolar RNA RNU44 (Applied Biosystems 4427975) was used as a control in qRT-PCR assays. The expression levels of a miRNA relative to RNU44 were measured using the comparative threshold cycle (Ct) method. ΔCt values were obtained from Ct values of the miRNA probes and that of RNU44 for each sample (in duplicate) and 2−ΔCt values were calculated.
Statistical analysis
The microarray raw data for all probes was assessed by pairwise Pearson correlation among all samples. One sample showed poor correlation against all other samples (Fig. S1) and was removed from further analysis. The raw data were background corrected and normalized using the quantile normalization method. Probes with a coefficient of variation of less than 0.1 were excluded. Probes with maximum expression <100 on a linear scale, i.e., uniformly low expression, were marked as low expression and not considered for significance. Differential expression analyses were done using Limma.Citation53 Statistical significance was measured by P values controlled for the false discovery rate (FDR) using the Benjamini–Hochberg method to account for multiple testing. Probes with a FDR < 0.2 were considered to be statistically significant. Biological significance was measured by a log fold change and then converted to a linear scale with ≥1.5-fold upregulation or downregulation of expression considered as significant.
Pathways targeted by dysregulated miRNA expression were identified using DIANA miRPath v.2.0 that utilizes the data of the Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=mirpath/reverse). The score cutoff used for target prediction was 0.8. Pathways were filtered based on a one-sided Fisher exact test P value (<0.05) adjusted for multiple testing using FDR.Citation37
Additional material
Download Zip (1.8 MB)Disclosure of Potential Conflicts of Interest
The authors declare that there are no potential conflicts of interest.
Acknowledgments
This publication was supported in part by the EDRN through grant number 3U01CA111242-05, by P30 CA006927 from the National Cancer Institute, and by Action to Cure Kidney Cancer. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Additional funds were provided by Fox Chase Cancer Center via institutional support of the Kidney Cancer Keystone Program.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11 - 30; http://dx.doi.org/10.3322/caac.21166; PMID: 23335087
- Motzer RJ, Bacik J, Mariani T, Russo P, Mazumdar M, Reuter V. Treatment outcome and survival associated with metastatic renal cell carcinoma of non-clear-cell histology. J Clin Oncol 2002; 20:2376 - 81; http://dx.doi.org/10.1200/JCO.2002.11.123; PMID: 11981011
- Banumathy G, Cairns P. Signaling pathways in renal cell carcinoma. Cancer Biol Ther 2010; 10:658 - 64; http://dx.doi.org/10.4161/cbt.10.7.13247; PMID: 20814228
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990; 61:759 - 67; http://dx.doi.org/10.1016/0092-8674(90)90186-I; PMID: 2188735
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100:57 - 70; http://dx.doi.org/10.1016/S0092-8674(00)81683-9; PMID: 10647931
- Cairns P. Renal cell carcinoma. Cancer Biomark 2010; 9:461 - 73; PMID: 22112490
- Lam JS, Klatte T, Kim HL, Patard JJ, Breda A, Zisman A, Pantuck AJ, Figlin RA. Prognostic factors and selection for clinical studies of patients with kidney cancer. Crit Rev Oncol Hematol 2008; 65:235 - 62; http://dx.doi.org/10.1016/j.critrevonc.2007.08.003; PMID: 17931881
- Kapur P, Peña-Llopis S, Christie A, Zhrebker L, Pavía-Jiménez A, Rathmell WK, Xie XJ, Brugarolas J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol 2013; 14:159 - 67; http://dx.doi.org/10.1016/S1470-2045(12)70584-3; PMID: 23333114
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 2009; 19:92 - 105; http://dx.doi.org/10.1101/gr.082701.108; PMID: 18955434
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer 2006; 6:857 - 66; http://dx.doi.org/10.1038/nrc1997; PMID: 17060945
- Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer 2006; 6:259 - 69; http://dx.doi.org/10.1038/nrc1840; PMID: 16557279
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005; 33:e179; http://dx.doi.org/10.1093/nar/gni178; PMID: 16314309
- Crispen PL, Boorjian SA, Lohse CM, Leibovich BC, Kwon ED. Predicting disease progression after nephrectomy for localized renal cell carcinoma: the utility of prognostic models and molecular biomarkers. Cancer 2008; 113:450 - 60; http://dx.doi.org/10.1002/cncr.23566; PMID: 18523999
- Atlas CG, Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499:43 - 9; http://dx.doi.org/10.1038/nature12222; PMID: 23792563
- Beroukhim R, Brunet JP, Di Napoli A, Mertz KD, Seeley A, Pires MM, Linhart D, Worrell RA, Moch H, Rubin MA, et al. Patterns of gene expression and copy-number alterations in von-hippel lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res 2009; 69:4674 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-09-0146; PMID: 19470766
- Faragalla H, Youssef YM, Scorilas A, Khalil B, White NM, Mejia-Guerrero S, Khella H, Jewett MA, Evans A, Lichner Z, et al. The clinical utility of miR-21 as a diagnostic and prognostic marker for renal cell carcinoma. J Mol Diagn 2012; 14:385 - 92; http://dx.doi.org/10.1016/j.jmoldx.2012.02.003; PMID: 22580180
- Spizzo R, Nicoloso MS, Croce CM, Calin GA. SnapShot: MicroRNAs in Cancer. Cell 2009; 137:586 - e1; http://dx.doi.org/10.1016/j.cell.2009.04.040; PMID: 19410551
- Talotta F, Cimmino A, Matarazzo MR, Casalino L, De Vita G, D’Esposito M, Di Lauro R, Verde P. An autoregulatory loop mediated by miR-21 and PDCD4 controls the AP-1 activity in RAS transformation. Oncogene 2009; 28:73 - 84; http://dx.doi.org/10.1038/onc.2008.370; PMID: 18850008
- Papagiannakopoulos T, Shapiro A, Kosik KS. MicroRNA-21 targets a network of key tumor-suppressive pathways in glioblastoma cells. Cancer Res 2008; 68:8164 - 72; http://dx.doi.org/10.1158/0008-5472.CAN-08-1305; PMID: 18829576
- Olson P, Lu J, Zhang H, Shai A, Chun MG, Wang Y, Libutti SK, Nakakura EK, Golub TR, Hanahan D. MicroRNA dynamics in the stages of tumorigenesis correlate with hallmark capabilities of cancer. Genes Dev 2009; 23:2152 - 65; http://dx.doi.org/10.1101/gad.1820109; PMID: 19759263
- Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C, Ganesan S, Bhanot G, Liou LS. Identification of a microRNA panel for clear-cell kidney cancer. Urology 2010; 75:835 - 41; http://dx.doi.org/10.1016/j.urology.2009.10.033; PMID: 20035975
- Osanto S, Qin Y, Buermans HP, Berkers J, Lerut E, Goeman JJ, van Poppel H. Genome-wide microRNA expression analysis of clear cell renal cell carcinoma by next generation deep sequencing. PLoS One 2012; 7:e38298; http://dx.doi.org/10.1371/journal.pone.0038298; PMID: 22745662
- Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, Hong M, Jiang T, Jiang Q, Lu J, et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia 2012; 26:769 - 77; http://dx.doi.org/10.1038/leu.2011.273; PMID: 21979877
- Ghosh G, Subramanian IV, Adhikari N, Zhang X, Joshi HP, Basi D, Chandrashekhar YS, Hall JL, Roy S, Zeng Y, et al. Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J Clin Invest 2010; 120:4141 - 54; http://dx.doi.org/10.1172/JCI42980; PMID: 20972335
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer 2010; 17:F19 - 36; http://dx.doi.org/10.1677/ERC-09-0184; PMID: 19779035
- Lujambio A, Lowe SW. The microcosmos of cancer. Nature 2012; 482:347 - 55; http://dx.doi.org/10.1038/nature10888; PMID: 22337054
- Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res 2007; 101:59 - 68; http://dx.doi.org/10.1161/CIRCRESAHA.107.153916; PMID: 17540974
- Wang H, Ach RA, Curry B. Direct and sensitive miRNA profiling from low-input total RNA. RNA 2007; 13:151 - 9; http://dx.doi.org/10.1261/rna.234507; PMID: 17105992
- Jung M, Mollenkopf HJ, Grimm C, Wagner I, Albrecht M, Waller T, Pilarsky C, Johannsen M, Stephan C, Lehrach H, et al. MicroRNA profiling of clear cell renal cell cancer identifies a robust signature to define renal malignancy. J Cell Mol Med 2009; 13:9B 3918 - 28; http://dx.doi.org/10.1111/j.1582-4934.2009.00705.x; PMID: 19228262
- Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, Gallo CA, Plas DR, Biesiada J, Meller J, et al. VHL-regulated MiR-204 suppresses tumor growth through inhibition of LC3B-mediated autophagy in renal clear cell carcinoma. Cancer Cell 2012; 21:532 - 46; http://dx.doi.org/10.1016/j.ccr.2012.02.019; PMID: 22516261
- Ying Z, Li Y, Wu J, Zhu X, Yang Y, Tian H, Li W, Hu B, Cheng SY, Li M. Loss of miR-204 expression enhances glioma migration and stem cell-like phenotype. Cancer Res 2013; 73:990 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-12-2895; PMID: 23204229
- Zou C, Xu Q, Mao F, Li D, Bian C, Liu LZ, Jiang Y, Chen X, Qi Y, Zhang X, et al. MiR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle 2012; 11:2137 - 45; http://dx.doi.org/10.4161/cc.20598; PMID: 22592534
- Slaby O, Redova M, Poprach A, Nekvindova J, Iliev R, Radova L, Lakomy R, Svoboda M, Vyzula R. Identification of MicroRNAs associated with early relapse after nephrectomy in renal cell carcinoma patients. Genes Chromosomes Cancer 2012; 51:707 - 16; http://dx.doi.org/10.1002/gcc.21957; PMID: 22492545
- Kumarswamy R, Mudduluru G, Ceppi P, Muppala S, Kozlowski M, Niklinski J, Papotti M, Allgayer H. MicroRNA-30a inhibits epithelial-to-mesenchymal transition by targeting Snai1 and is downregulated in non-small cell lung cancer. Int J Cancer 2012; 130:2044 - 53; http://dx.doi.org/10.1002/ijc.26218; PMID: 21633953
- Sun L, Xie H, Mori MA, Alexander R, Yuan B, Hattangadi SM, Liu Q, Kahn CR, Lodish HF. Mir193b-365 is essential for brown fat differentiation. Nat Cell Biol 2011; 13:958 - 65; http://dx.doi.org/10.1038/ncb2286; PMID: 21743466
- Sun D, Lee YS, Malhotra A, Kim HK, Matecic M, Evans C, Jensen RV, Moskaluk CA, Dutta A. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res 2011; 71:1313 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-10-1031; PMID: 21212412
- Vlachos IS, Kostoulas N, Vergoulis T, Georgakilas G, Reczko M, Maragkakis M, Paraskevopoulou MD, Prionidis K, Dalamagas T, Hatzigeorgiou AG. DIANA miRPath v.2.0: investigating the combinatorial effect of microRNAs in pathways. Nucleic Acids Res 2012; 40:W498-504; http://dx.doi.org/10.1093/nar/gks494; PMID: 22649059
- Weng L, Wu X, Gao H, Mu B, Li X, Wang JH, Guo C, Jin JM, Chen Z, Covarrubias M, et al. MicroRNA profiling of clear cell renal cell carcinoma by whole-genome small RNA deep sequencing of paired frozen and formalin-fixed, paraffin-embedded tissue specimens. J Pathol 2010; 222:41 - 51; PMID: 20593407
- Wu X, Weng L, Li X, Guo C, Pal SK, Jin JM, Li Y, Nelson RA, Mu B, Onami SH, et al. Identification of a 4-microRNA signature for clear cell renal cell carcinoma metastasis and prognosis. PLoS One 2012; 7:e35661; http://dx.doi.org/10.1371/journal.pone.0035661; PMID: 22623952
- Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, Wang X, Wang J, Yu H, Cao X, et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol 2012; 188:5500 - 10; http://dx.doi.org/10.4049/jimmunol.1103505; PMID: 22544933
- Huang S, Wang S, Bian C, Yang Z, Zhou H, Zeng Y, Li H, Han Q, Zhao RC. Upregulation of miR-22 promotes osteogenic differentiation and inhibits adipogenic differentiation of human adipose tissue-derived mesenchymal stem cells by repressing HDAC6 protein expression. Stem Cells Dev 2012; 21:2531 - 40; http://dx.doi.org/10.1089/scd.2012.0014; PMID: 22375943
- Bar N, Dikstein R. miR-22 forms a regulatory loop in PTEN/AKT pathway and modulates signaling kinetics. PLoS One 2010; 5:e10859; http://dx.doi.org/10.1371/journal.pone.0010859; PMID: 20523723
- Yin G, Alvero AB, Craveiro V, Holmberg JC, Fu HH, Montagna MK, Yang Y, Chefetz-Menaker I, Nuti S, Rossi M, et al. Constitutive proteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene 2013; 32:39 - 49; http://dx.doi.org/10.1038/onc.2012.33; PMID: 22349827
- Nakada C, Matsuura K, Tsukamoto Y, Tanigawa M, Yoshimoto T, Narimatsu T, Nguyen LT, Hijiya N, Uchida T, Sato F, et al. Genome-wide microRNA expression profiling in renal cell carcinoma: significant down-regulation of miR-141 and miR-200c. J Pathol 2008; 216:418 - 27; http://dx.doi.org/10.1002/path.2437; PMID: 18925646
- White NM, Khella HW, Grigull J, Adzovic S, Youssef YM, Honey RJ, Stewart R, Pace KT, Bjarnason GA, Jewett MA, et al. miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br J Cancer 2011; 105:1741 - 9; http://dx.doi.org/10.1038/bjc.2011.401; PMID: 22033272
- Morrogh M, Olvera N, Bogomolniy F, Borgen PI, King TA. Tissue preparation for laser capture microdissection and RNA extraction from fresh frozen breast tissue. Biotechniques 2007; 43:41 - 2, 44, 46 passim; http://dx.doi.org/10.2144/000112497; PMID: 17695251
- Hock LM, Lynch J, Balaji KC. Increasing incidence of all stages of kidney cancer in the last 2 decades in the United States: an analysis of surveillance, epidemiology and end results program data. J Urol 2002; 167:57 - 60; http://dx.doi.org/10.1016/S0022-5347(05)65382-7; PMID: 11743275
- Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, Hadfield J, Bertone P, Caldas C. Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA 2010; 16:991 - 1006; http://dx.doi.org/10.1261/rna.1947110; PMID: 20360395
- Pradervand S, Weber J, Lemoine F, Consales F, Paillusson A, Dupasquier M, Thomas J, Richter H, Kaessmann H, Beaudoing E, et al. Concordance among digital gene expression, microarrays, and qPCR when measuring differential expression of microRNAs. Biotechniques 2010; 48:219 - 22; http://dx.doi.org/10.2144/000113367; PMID: 20359303
- Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 1982; 6:655 - 63; http://dx.doi.org/10.1097/00000478-198210000-00007; PMID: 7180965
- Edge SB, Byrd DR, Compton CC, Fritz AG, Greene FL, Trotti A III. Chapter 43 Kidney. AJCC Cancer Staging Manual Seventh Edition 2009:479-90
- Tissot C. Analysis of miRNA content in total RNA preparations using the Agilent 2100 bioanalyzer. Agilent Technologies Application Note 2008; Publication Number 5989-7870EN. Available from: http://www.chem.agilent.com/Library/applications/5989-7870EN.pdf
- Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004; 3:e3; http://dx.doi.org/10.2202/1544-6115.1027; PMID: 16646809