
Abstract
The gene encoding the cytoskeletal regulator DIAPH3 is lost at high frequency in metastatic prostate cancer, and DIAPH3 silencing evokes a transition to an amoeboid tumor phenotype in multiple cell backgrounds. This amoeboid transformation is accompanied by increased tumor cell migration, invasion, and metastasis. DIAPH3 silencing also promotes the formation of atypically large (>1 μm) membrane blebs that can be shed as extracellular vesicles (EV) containing bioactive cargo. Whether loss of DIAPH3 also stimulates the release of nano-sized EV (e.g., exosomes) is not established. Here we examined the mechanism of release and potential biological functions of EV shed from DIAPH3-silenced and other prostate cancer cells. We observed that stimulation of LNCaP cells with the prostate stroma-derived growth factor heparin-binding EGF-like growth factor (HB-EGF), combined with p38MAPK inhibition caused EV shedding, a process mediated by ERK1/2 hyperactivation. DIAPH3 silencing in DU145 cells also increased rates of EV production. EV isolated from DIAPH3-silenced cells activated AKT1 and androgen signaling, increased proliferation of recipient tumor cells, and suppressed proliferation of human macrophages and peripheral blood mononuclear cells. DU145 EV contained miR-125a, which suppressed AKT1 expression and proliferation in recipient human peripheral blood mononuclear cells and macrophages. Our findings suggest that EV produced as a result of DIAPH3 loss or growth factor stimulation may condition the tumor microenvironment through multiple mechanisms, including the proliferation of cancer cells and suppression of tumor-infiltrating immune cells.
Introduction
The acquisition of motile behaviors facilitates tumor cell dissemination, a rate-limiting step during the transition to metastatic disease. There has been extensive study of tumor cell migration via a mesenchymal mode (epithelial–mesenchymal transition), characterized by the secretion of matrix metalloproteases, reduced intercellular adhesion, reduced cell polarity, and increased invasiveness.Citation1 However, it has recently become appreciated that certain tumor cells can transition to an alternate mechanism of tumor cell dissemination termed amoeboid motility.Citation2,Citation3 Amoeboid cells are less dependent on pericellular proteolysis, exhibit loss of cell polarity, readily deform their shape through cytoskeletal remodeling, and can migrate rapidly through complex matrices.Citation4,Citation5
Due to heightened actomyosin contractility, amoeboid cells display rounded morphologies and prominent bleb-like protrusions.Citation3,Citation6-Citation9 Our laboratory previously demonstrated that certain amoeboid cells shed large extracellular vesicles (EV), in the size range of 0.5–10 μm in diameter, into the extracellular space.Citation10 Like nano-sized exosomes (generally <100 nm), these large EV, termed “large oncosomes”, harbor abundant biomolecules and are bioactive.Citation10 Large oncosomes are among several types of EV shed from tumor cells,Citation11 of which exosomes have been the most extensively characterized.Citation12 Tumor-derived EV can be enriched in nucleic acids, including DNA, mRNA, miRNA, and small non-coding RNAs, as well as oncoproteins and metabolites, and can thereby mediate the horizontal transfer of oncogenic information to neighboring cells.Citation12 We and others have demonstrated that the interaction of EV with recipient cells is associated with stromal activation,Citation10 promotion of tumor cell invasion,Citation10 endothelial cell migration and angiogenesis,Citation13,Citation14 modulation of inflammatory responses,Citation15 and enhanced drug resistance.Citation16 EV preserve the bioactivity of their molecular cargo and can be readily isolated from multiple biological fluids (e.g., urine, serum, plasma, pleural effusion, and saliva), suggesting that these particles may constitute a potential source of biomarkers for real-time assessments of cancer progression.Citation17
Our laboratory recently identified Diaphanous-related formin-3 (DIAPH3) as a non-canonical metastasis suppressor.Citation18 DIAPH3 belongs to a family of formin orthologs that share tandem FH1 and FH2 domains and regulate cytoskeletal dynamics.Citation19 Formins nucleate, elongate, and bundle linear actin filaments and stabilize microtubules, and have been implicated in endocytosis, cell division, and migration.Citation20-Citation22 We observed that genomic loss at the DIAPH3 locus is strongly associated with metastatic disease in human prostate cancer, breast cancer, and hepatocellular carcinoma.Citation18 Furthermore, silencing of DIAPH3 by RNAi induced a morphological transition to an amoeboid phenotype in cultured prostate and breast cancer cells, a phenotypic switch mediated by cytoskeletal disruption, defective endocytic trafficking, and aberrant signaling through the EGFR/MEK/ERK1/2 axis.Citation18 DIAPH3 silencing increased invasion in vitro and metastasis formation in vivo. Reduced DIAPH3 expression also promoted the genesis and shedding of large oncosomes in some cell backgrounds,Citation23 suggesting that loss or disruption of DIAPH3 may affect cancer progression by modifying the tumor microenvironment.
In this report we demonstrate that shedding of exosome-sized EV is promoted by DIAPH3 loss. ERK1/2-induced shedding of these particles activated oncogenic signal transduction pathways and promoted the proliferation of recipient tumor cells. EV derived from DU145 cells carried miRNAs that suppressed immune cell proliferation. Our findings suggest that a transition to an amoeboid phenotype may alter the tumor microenvironment as a result of enhanced EV secretion and shedding, and that these effects involve direct action on tumor cells and on tumor infiltrating immune cells.
Results
EV shedding from LNCaP cells is enhanced by ERK1/2 activation
We previously reported that heparin-binding EGF-like growth factor (HB-EGF), a product of smooth muscle cells in the prostate stroma, plays a role as a paracrine regulator of prostate tumor cells.Citation24 HB-EGF activates EGFR and ERK1/2 signaling,Citation25 alters proliferation and apoptosis induced by H2O2 or etoposide treatment,Citation26 and promotes an aggressive, neuroendocrine phenotype in prostate cancer cells.Citation25 We also observed that HB-EGF enhances shedding of EV in the size range of large oncosomes.Citation23 To test whether HB-EGF might also increase shedding of exosome-sized (<100 nm) EV, LNCaP cells, which exhibit low basal EV formation,Citation23 were transfected with a constitutively secreted HB-EGF construct (sHB-EGF) or control vector. Immunoblotting confirmed HB-EGF secretion into the conditioned medium (CM), as detected by immunoprecipitation with heparin-conjugated sepharose (). In order to determine whether forced expression of sHB-EGF affects the shedding of exosomes, we purified EV by ultracentrifugation followed by quantitative nanoparticle tracking analysis using the NanoSight system (http://www.nanosight.com/nta). Interestingly, exosome-sized EV from the CM from LNCaP/sHB-EGF cells were ~2-fold more abundant than those from LNCaP/Vector cells (). These findings suggest that HB-EGF stimulation promotes not only the shedding of large oncosomes but also of nanosized particles, and identify HB-EGF as a regulator of EV shedding in prostate cancer cells.
Figure 1. HB-EGF and ERK1/2 activation mediate EV shedding from prostate cancer cells. (A and B) Secreted HB-EGF from LNCaP/sHB-EGF cells stimulated EV shedding. (A) Western blot analysis confirmed HB-EGF secretion. Conditioned medium from LNCaP/sHB-EGF or LNCaP/Vector was precipitated by heparin Sepharose. Western blot was performed using an anti-HB-EGF antibody. (B) Quantitation of EV shed from LNCaP/sHB-EGF or LNCaP/Vector cells by NanoSight optical microscopy. Statistical significance was defined as P < 0.05 (*). (C–E) ERK1/2 activation in DU145 cells in response to p38MAPK inhibition with SB203580 (10 μM) and HB-EGF (100 ng/ml) stimulation increases EV shedding and bioactivity. (C) Western blot analysis indicated ERK1/2 activation by HB-EGF, which was further enhanced when combined with the p38MAPK inhibitor, SB203580. (D) Immunofluorescence staining revealed that formation and shedding of EV were enhanced by ERK1/2 activation. (E) Quantitation of EV shed from tumor cells in response to HB-EGF and SB203580 treatment, as assessed by NanoSight optical microscopy. (F) EV shed from DU145 cells treated with HB-EGF and SB203580 were incubated on FITC-gelatin, in the presence or absence of the MEK1 inhibitor PD98059 (5 μM). The size of cleared spots (regions of gelatinase activity) was measured with Axiovision 4.2 software. A representative image is shown. (G) Inhibition of ERK1/2 phosphorylation by PD98059 was confirmed by western blot.
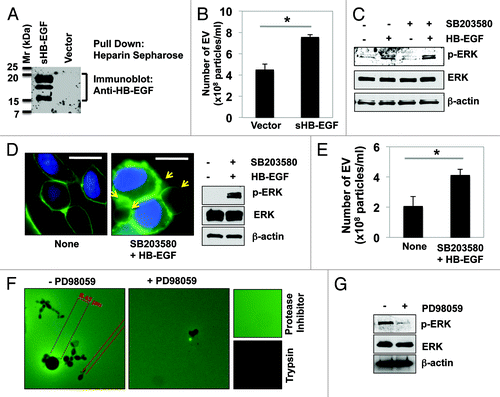
ERK1/2 has been recently implicated in the release of EV from various cell types.Citation27 As this pathway is a downstream effector of HB-EGF signaling, we tested whether ERK1/2 activation promotes EV shedding in DU145 cells. Treatment of serum-starved DU145 cells with a physiological dose of recombinant HB-EGF strongly activated ERK1/2 (); activation was further enhanced in the presence of SB203580, a specific inhibitor of p38MAPK (an ERK1/2 antagonist, ).Citation25,Citation28 Immunofluorescence imaging with CTxB revealed that ERK1/2 activation by HB-EGF enhanced bleb formation, an indicator of large oncosome sheddingCitation10 (, arrows). Nanoparticle tracking analysis showed that CM collected from DU145 treated with HB-EGF and SB203580 contained approximately 2-fold more exosome-sized EV than from control cells (). These results indicate that ERK1/2 activation promotes EV shedding.
We next sought to determine whether HB-EGF-stimulated EV were bioactive. Toward this end, the particles were collected and concentrated by differential centrifugation.Citation23 Incubation of EV derived from HB-EGF-stimulated DU145 cells with FITC-gelatin yielded regions of gelatinase activity (), identifiable as degradation spots of variable size, suggesting marked heterogeneity in the EV population. This result is in line with previous data demonstrating that EV isolated from LNCaP cells stably expressing an activated form of AKT1 (Myr-AKT1), contained matrix metalloproteinase (MMP) activity (e.g., MMP-2 or -9) and degraded FITC-gelatin.Citation10 No such degradation spots were observed in the presence of protease inhibitors or unconditioned medium, while no gelatin fluorescence was observed in the presence of trypsin. Interestingly, inhibition of the upstream activator of ERK1/2, MEK1, with PD98059 markedly inhibited EV gelatinase activity (). The inhibition of ERK1/2 by PD98059 was confirmed by immunoblotting (). These observations suggest that ERK1/2 activation is required for the formation, shedding and bioactivity of EV originating from DU145 cells. Given the demonstrated role of ERK1/2 as an upstream regulator of MMP-2 and MMP-9 in cancer,Citation10,Citation29,Citation30 these results might suggest that the increased aggressiveness of amoeboid cells is potentiated, at least in part, by the shedding of EV containing protease cargo.
DIAPH3 knockdown enhances the shedding of EV, cell invasion, and anchorage-independent growth in DU145 cells
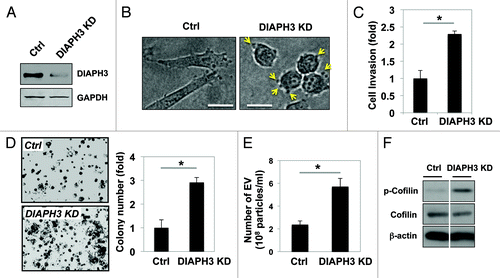
Silencing of DIAPH3 () induced a morphological switch from a spindle-like shape to a more rounded amoeboid phenotype, with the appearance of abundant membrane blebs (, arrows), consistent with our recent demonstration of a transition to an amoeboid phenotype in DIAPH3-silenced cells.Citation18,Citation23 DIAPH3 deficiency enhanced cell invasion () and evoked a 3-fold increase in anchorage-independent growth (). Nanoparticle tracking analysis revealed that silencing of DIAPH3 also enhances the release of exosome-sized particles (). Interestingly, and in agreement with this enhanced shedding of EV, cells deficient for DIAPH3 displayed increased phosphorylation of cofilin (), an inhibitory posttranslational modification that has been implicated in the promotion of EV genesis in cervical carcinoma and breast cancer cells.Citation31 Together, these data implicate DIAPH3 loss as an important mechanism by which prostate cancer cells generate EV.
Figure 2. Loss of DIAPH3 enhances the amoeboid phenotype, cell invasion, anchorage-independent cell proliferation, and EV shedding in DU145 cells. (A) Whole cell lysates from DIAPH3-silenced and control DU145 cells were immunoblotted with an antibody against DIAPH3. (B) Morphological transition to a rounded and amoeboid phenotype as a consequence of DIAPH3 silencing in DU145 cells. (C) Cell invasion through a transwell chamber was measured in DIAPH3 KD and control cells (Ctrl). (D) Anchorage-independent proliferation in soft agar. After staining with MTT solution, colonies were manually counted from 10 fields per condition. (E) EV number was assessed using NanoSight optical microscopy. (F) Western blot analysis demonstrating increased cofilin phosphorylation at Ser3 in DIAPH KD cells as compared with Ctrl.
EV released from DIAPH3-silenced cells stimulate tumor cell proliferation
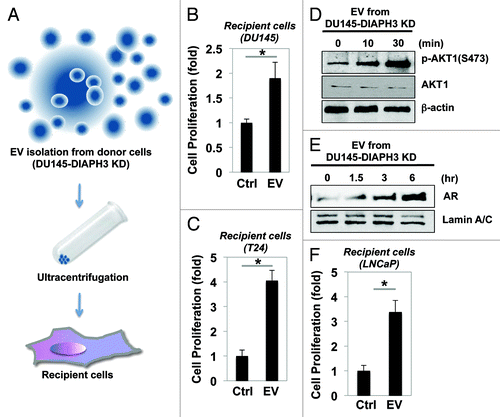
We next assessed whether EV isolated from DIAPH3-deficient cells could alter the biological responses of recipient tumor cells. Toward this end, EV isolated by ultracentrifugation () were added to the medium of recipient cancer cells, and cell proliferation examined after 72 h of incubation. As a negative control, serum-free medium (without cell incubation) was processed in parallel with the isolated EV. We observed that EV derived from DU145-DIAPH3 KD cells enhanced the proliferation of recipient DU145 cells 2-fold (), and those of heterologous cancer cells (T24 bladder cancer cells) 4-fold (). These findings indicate that EV derived from amoeboid tumor cells might induce proliferation in proximal tumor cells, including those with heterogeneous genomic make up.
Figure 3. EV secreted from DIAPH3-silenced DU145 cells enhances the proliferation of recipient cancer cells. (A) Schematic of the EV isolation procedure from the conditioned medium of DU145-DIAPH3 KD cells, by differential centrifugation and subsequent EV addition to recipient cancer cells. (B) DU145 cells were treated with EV from DU145-DIAPH3 KD cells or with cell media (Ctrl) for 72 h. Proliferation was measured by crystal violet analysis. (C) T24 bladder cancer cells were incubated as in (B), and proliferation assessed after 72 h of treatment. Average values derived from 3 independent trials (Student t test, *P < 0.05). (D–F) EV isolated from DU145-DIAPH3 KD cells positively regulated AKT1 phosphorylation and nuclear localization of the androgen receptor, leading to increased cell proliferation of recipient LNCaP cells. (D) LNCaP cells were treated with EV from DU145-DIAPH3 KD cells for the indicated times. Western blotting was performed using antibodies against phospho-AKT1 (S473), AKT1, or β-actin. n = 3 independent experiments. (E) Nuclear extracts were isolated from LNCaP cells treated with DU145-DIAPH3 KD EV for the indicated times. AR levels in the nuclear fraction were determined by western blot analysis. (F) Increased proliferation of LNCaP cells upon exposure to EV from DU145-DIAPH3 KD cells. LNCaP PCa cancer cells were incubated as in (B), and proliferation assessed after 72 h of treatment. Average values derived from 3 independent trials (Student t test, *P < 0.05).
EV from DIAPH3-deficient cells increase AKT1 phosphorylation and the nuclear localization of androgen receptor in LNCaP cells
The increased proliferation of recipient tumor cells by EV suggests that pro-proliferative signals in cancer cells may be stimulated by exogenous treatment with these particles. Because the PI3K/AKT signaling axis and the androgen receptor (AR) both play central roles in prostate cancer carcinogenesis and progression,Citation32 we tested whether exposure to EV from amoeboid cells alters these signaling networks in LNCaP cells. As shown in , EV from DIAPH3-deficient cells increased the phosphorylation of AKT1, but not that of ERK1/2 or of p38MAPK (data not shown). Notably, treatment with EV also increased levels of AR in the nuclear compartment (), in a time-dependent fashion. Consistent with this increased pro-oncogenic signaling, cell proliferation of recipient LNCaP cells was stimulated by exposure to EV (). These findings suggest that EV shed from amoeboid tumor cells alter intracellular signaling networks in trans and might contribute to stimulating more aggressive behavior in neighboring tumor cells.
EV shed from DIAPH3-silenced cells inhibit the proliferation of immune cells through the transfer of miR125a
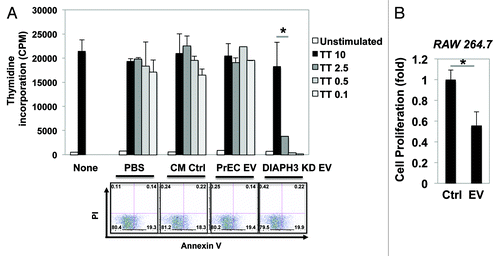
The findings above indicate that EV from amoeboid cells can modulate the behavior of recipient tumor cells. Having previously demonstrated that EV from LNCaP overexpressing the oncogene MyrAKT1 induce aspects of reactive stroma in prostate myofibroblasts,Citation10 we now focused on the influence of EV shed from DIAPH3-deficient cells on immune cell function. EV collected from DIAPH3-silenced DU145 cells were added to the media of cultured human peripheral blood mononuclear cells (PBMC), and a tritiated thymidine (3H-thymidine) incorporation assay was used to assess cell proliferation. As controls, no treatment, PBS only, control conditioned medium (CM, without cell incubation), and EV from normal prostate epithelial cells (PrEC) were employed. As shown in (upper graph), PBMC were dose-dependently treated with tetanus toxoid, a well-known stimulator of PBMC proliferation. The addition of EV significantly suppressed this proliferative response, without induction of cell apoptosis (). Similarly, exposure to EV markedly decreased the proliferation of RAW 264.7 mouse-derived leukemic monocyte/macrophage cells (). These observations imply that amoeboid prostate cell-derived EV affect immune components of the tumor microenvironment.
Figure 4. EV obtained from DU145-DIAPH3 KD cells reduces the proliferation of RAW264.7 (mouse macrophage) and PBMC (human peripheral blood mononuclear) immune cells. (A) Proliferation of PBMC treated with EV from DIAPH3 KD (PBS, CM, or PrEC EV as controls) was measured by 3H-thymidine incorporation (upper graph). Cell apoptosis was measured by flow cytometry analysis after cell staining with propidium iodide (PI) and Annexin V (bottom panels). (B) Proliferation of RAW264.7 cells treated with EV from DU145-DIAPH3 KD (or Ctrl) was measured by crystal violet staining.
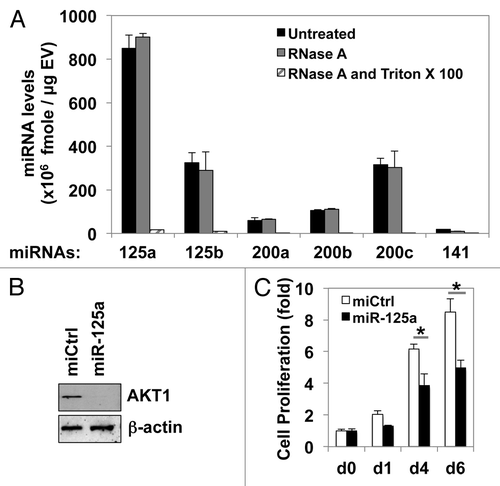
The effects on disparate cell types observed above support the emerging notion that EV are important mediators of cell–cell communication.Citation12,Citation17 We hypothesized that this is facilitated in part by cargo microRNAs (miRNAs), non-coding RNA molecules that negatively regulate gene expression and thereby play important roles in tumor progression and metastasis.Citation33-Citation35 We examined the expression of the miR-200 family (miR-200a, 200b, 200c, and 141) and the miR-125 family (miR-125a and 125b), which are involved in prostate tumor cell proliferation, invasion, and disease progression,Citation36-Citation38 in EV derived from DU145 cells. This analysis revealed that miR-125a is a highly abundant miRNA in EV shed from DU145 cells. Experiments using RNase A and/or Triton X-100 indicated that both miR125a and the other detected miRNAs were protected within EV (). The miR-125 family is known to repress the ERK1/2 and AKT1 pathways. In our experimental setting, overexpression of miR-125a in RAW264.7 cells reduced AKT1 expression (). In line with the pro-proliferative role of AKT1, and with the suppression of proliferation of RAW264.7 cells by EV (), miR-125a overexpression significantly attenuated cell proliferation (). These data implicate the horizontal transfer of miR-125a by EV in the suppression of macrophage proliferation.
Figure 5. The shed EV from DU145 contain abundant miR-125a, which targets AKT1 and suppresses the proliferation of RAW264.7 cells. (A) Expression of microRNAs in EV shed from DU145-DIAPH3 KD cells was measured by qRT-PCR analysis, in the presence or absence of RNase A or Triton X-100. (B) RAW264.7 cells were transfected with miR-125a or control miR (miR-Ctrl). Western blot was performed to assess AKT1 or β-actin expression. (C) Overexpression of miR-125a, but not a control miRNA, in RAW264.7 cells reduced cell proliferation, as assessed by crystal violet analysis (Student t test, *P < 0.05).
Discussion
In this report we demonstrate enhanced shedding of exosome-sized EV from amoeboid prostate cancer cells and demonstrate several novel biological effects of the shed vesicles. EV formation and shedding in DU145 cells was promoted by stimulation with HB-EGF, a product of the prostate stroma that was previously identified as a potent pro-proliferative growth factor in prostate cancer,Citation39 and by DIAPH3 silencing, an event that can trigger a transition to an amoeboid phenotype in prostate and breast tumor cells.Citation18 EV were bioactive, displaying gelatinase activity, a process facilitated by ERK1/2 hyperactivation. EV shed from DIAPH3-silenced cells enhanced cell proliferation in both autologous and heterologous tumor cells, and significantly reduced immune cell proliferation. Our data suggest that the latter was mediated, at least in part, by EV-resident miR-125a.
In the past, EV were generally considered to be a means for egress and/or exclusion of molecules from cells.Citation40 It is now clear they are critical delivery vehicles of oncogenic and other types of molecular cargoCitation12,Citation27,Citation41-Citation44 and are increasingly appreciated as mediators of communication within the tumor microenvironment.Citation17,Citation45,Citation46 EV modulate signaling in neighboring cells,Citation47 contribute to priming of the metastatic niche, and facilitate tumor cell homing.Citation48 Since EV can exist in biofluids such as urine, blood, pleural effusions, spinal fluids, saliva, or ascites, analysis of these particles potentially represents a non- or minimally-invasive clinical resource for diagnosis and prognosis.
Our observation that DIAPH3 silencing promotes the secretion of small as well as large EV extends our previous findings demonstrating that DIAPH3 loss enhances the formation of large oncosomes.Citation10,Citation23 Overexpression of the formins FHOD1 (FH1/FH2 domain-containing protein 1),Citation49 Dia1,Citation50 formin-like 2,Citation51 or DIP (mDia2 regulator diaphanous interacting protein)Citation28,Citation52 has been shown to promote membrane bleb formation and in some cases amoeboid motility in various cell types. Our findings on DIAPH3 are the first demonstration that loss of a formin protein evokes EV shedding.
Our quantitative miRNA analysis of EV shed from prostate cancer cells detected multiple miRNA in these particles. The miRNA-200 family has been implicated in the regulation of EMT (miR-200a)Citation53 and mesenchymal-amoeboid transition (miR-200c),Citation54 raising the possibility that EV contribute to the transformation of recipient cells into more aggressive tumor cell variants. We also detected miR-125a in EV shed from DU145, a miRNA found to be upregulated in prostate tumors relative to normal tissue.Citation55 Interestingly, we now show that miR-125a, which is shed in prostate cancer cell-derived EV, can suppress the proliferation of cultured macrophages. These findings add to emerging evidence that EV contain abundant biochemical information that have the capacity to alter the tumor microenvironment.
The insensitivity of miRNAs detected in EV to RNase digestion indicates that EV protect their molecular cargo from environmental insults that induce degradation.Citation47 These findings are in line with reports that EV may serve as novel drug delivery vehicles for autologous therapy.Citation56 Indeed, the delivery of siRNAs, anti-inflammatory agents, and pharmacologic inhibitors to various sites in vivo has been recently achieved using cell-derived microvesicles.Citation57,Citation58 Given their derivation from cells, EV represent a more physiological route of delivery than liposomes, the most common lipid-based delivery mechanism employed clinically.Citation59 Liposomes suffer from a short half-life and leakage of encapsulated drugs, issues potentially surmountable through the greater biostability of EV.Citation59,Citation60 Given that EV can act as decoys for certain immunotherapies, engineering of these particles has the potential to significantly improve chemotherapeutic efficacy.
The mechanisms through which tumors evade immune surveillance are still poorly understood. Our results suggest, for the first time, that EV from aggressive prostate cancer cells suppress macrophage proliferation through the action of miR-125a. This observation highlights the potential of EV to act as immuno-modulators within the tumor microenvironment through non-coding RNA. It would be of interest to determine whether other miRNAs are similarly involved and what the downstream transcript targets are. Given the diverse effects of EV described here, our results also point to the context-dependence of the EV source for the resulting biological activities in recipient cells, and the need to identify EV signatures predictive of such responses.Citation61
In conclusion, we have shown here that bioactive EV shed from amoeboid prostate cancer cells promote the proliferation of recipient tumor cells, while inhibiting the proliferation of immune cells. Our findings indicate that EV may condition the tumor microenvironment, in part through the horizontal transfer of genetic information such as miRNAs. Many questions regarding EV biology remain to be answered. For example, what is the minimum quantity of EV-encapsulated bioactive material (e.g., miRNAs or proteins) required to elicit effects in neighboring cells? What are the mechanisms by which cargo are selectively packaged into EV and absorbed by neighboring cells? Lastly, what signaling cascades are modulated by EV exposure, and how can these be regulated pharmacologically? Elucidating some of these facets of EV biology would improve possibilities for pharmacologic intervention in cancer.
Materials and Methods
Reagents
Recombinant human HB-EGF was purchased from R&D Biotechnology, and SB203580 from Biochem. Antibodies: phospho-ERK1/2, ERK1/2, phospho-AKT1 (S473), AKT1, cofilin, phospho-cofilin (S3) and AR were obtained from Cell Signaling Technology; Lamin A/C and β-actin were from Santa Cruz Biotechnology; DIAPH3 was kindly provided by Dr Henry Higgs, Dartmouth Medical School.Citation18 FITC-CTxB was obtained from Sigma. Real-time PCR kit was from SABiosciences. ECL detection kit was from BioRad and New England Nuclear. Matrigel was purchased from BD Biosciences.
Cell culture and transfection
LNCaP, DU145, T24, and RAW264.7 cells were purchased from and cultured as instructed by ATCC. Media were supplemented with 10% fetal bovine serum, 2% glutamine, and 1% antibiotics (Invitrogen) under a humidified atmosphere of 5% CO2 at 37 °C. The cells were subcultured at a density of 6 × 103 cells/cm2. For cell transfections of synthetic miRNA oligonucleotides, RAW264.7 cells were grown to 80% confluence in 60-mm culture plates, at which time they were transiently transfected with the 150 pmol miRNAs (hsa-miR-125a and miR negative control, Thermo Scientific) using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Immunofluorescence microscopic analysis
Cell membranes were labeled with FITC-conjugated CTxB (green), and imaged using an Axioplan 2 microscope (Zeiss) as previously described.Citation23
Isolation of EV from culture medium
Shed EV were collected from conditioned medium and purified as previously described.Citation23 Alternatively, where indicated, EV shed from DU145 were collected from conditioned medium from cells stimulated with SB203580 (10 μM) and HB-EGF (100 ng/ml) for 16 h, in the presence or absence of a 30 min pretreatment with PD98059 (10 μM). Briefly, conditioned medium was subjected to two successive, rapid centrifugations at 300× g and 800× g to eliminate cells and debris, respectively. The resulting supernatants were subjected to ultracentrifugation for 2 h at 100 000× g to pellet the EV. Recovered material was resuspended in 100 μl of ice-cold phosphate-buffered saline (PBS) per tube. Recipient cells (LNCaP, DU145, T24, or RAW264.7) were serum-starved for 16 h and then treated with 20 μg of EV for the indicated times.
Measurement of EV using nanoparticle tracking analysis
Cells were washed twice with PBS, and then incubated in serum-free medium for an additional 24 h. Conditioned medium was harvested, and cell debris were removed by centrifugation at 300× g for 15 min. Supernatants were applied to nanoparticle tracking analysis using the NanoSight system.
In vitro gelatin degradation assay
Shed EV were isolated from DU145 stimulated with SB203580 (10 μM) and HB-EGF (100 ng/ml), in the presence or absence of pretreatment of PD98059 (10 μM), as described above. The gelatin degradation assay was performed as previously described.Citation10 In brief, EV were seeded onto FITC-labeled gelatin-coated coverslips, and incubated for 1 h at 37 °C. Trypsin and protease inhibitor were used as positive and negative controls, respectively.
Western blotting analysis
Cells were lysed in RIPA buffer (50 mM TRIS-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate) supplemented with 1% SDS, boiled at 95 °C with constant shaking for 5 min, frozen at −20 °C, homogenized through a 26G1/2 needle (BD Biosciences), frozen again at −80 °C, and upon thawing debris were removed by centrifugation. Equivalent protein amounts (25 μg) were resolved by SDS-PAGE, transferred to a nitrocellulose membrane, and specific antibodies used to determine protein expression.
Imaging of DU145 morphology
Control or DIAPH3-silenced cells (18 750 cells per well) were seeded into 24-well plates with No 1.5 glass-bottom coverslips (MatTek). Plates were precoated with 1.6 mg/ml collagen I dissolved in 0.1% acetic acid; after overnight dehydration in a laminar flow hood, wells were washed once with serum-free DMEM and collagen I rehydrated in DMEM + 10% FBS for 3 h at 37 °C. Brightfield images of cells were collected with a 20× objective on an Ultravox Spinning Disc Confocal microscope at the Intellectual and Developmental Disabilities Research Core facility (IDDRC, NIH-P30-HD-18655). Still images from frames acquired every 15 min are shown.
Invasion assay
Thick matrigel-coated inserts were prepared 1 d before experiment. DU145 Control or DIAPH3-silenced cells were serum-starved for 4 h, and each cell suspension (3 × 105 cells/ml, 300 μl total) was seeded on the upper surface of each insert. FBS-containing normal culture medium were added in each well. After incubation for an additional 12 h at 37 °C, 5% CO2, non-invasive cells were removed. Invasive cells, which moved to the bottom surface of the inserts, were gently stained with cell staining solutionCitation62 for 20 min. After washing twice to remove background staining, 300 μl of extraction solutionCitation62 containing 10% acetic acid was used for extraction. Absorbance was read at 560 nm using a FLUOstar plate reader.
Cell proliferation assay
Recipient cells were seeded onto 6-well culture plates 1 d before the experiment (1 × 103 cells/well). After washing twice with PBS and 16 h-serum starvation, 20 μg of isolated EV from donor cells were added. Cell proliferation of recipient cells was quantified by measuring cell mass, using crystal violet analysis. Briefly, cells were stained with crystal violet solution, extracted in 10% acetic acid solution and absorbance measured at 570 nm.
Anchorage-independent growth assay
DIAPH3 loss and control DU145 PCa cells were seeded at 1 × 104 in 3 ml of 0.35% agar in DMEM/10% FBS, overlaid on 2 ml of 0.7% agar in DMEM/FBS, in 6-well plates. Plates were incubated for 7 d and cells fed every 2 d. At the end of the assay, colonies were visualized by staining with MTT reagent and images captured using a Zeiss Axioplan 2 microscope. Positively stained metabolically active cells in 10 independent images were scored.
Nuclear fractionation for detection of AR
Cytoplasmic and nuclear extracts were prepared as described.Citation63,Citation64
PBMC preparation and proliferation assay
PBMC were isolated from human blood by density-gradient separation using Histopaque-1077 as described in previous literature.Citation65 For proliferation assay in PBMC, 2 × 105 cells were cultured with or without tetanus toxoid (TT, 0, 0.1, 0.5, 2.5, or 10.0 μg/ml). Proliferation of PBMC was measured as a 3H-thymidine incorporation assay during the last 16 h of the 5-d culture. Assays were performed in triplicate to obtain statistical significance.
qRT-PCR analysis
Total RNA was isolated from DU145-shed EV, untreated or treated with 0.1 mg/ml RNase A (Sigma) and 0.1% Triton X-100 (Sigma), using Trizol LS (Invitrogen) according to the manufacturer’s instructions. The levels of selected miRNAs were quantified using a Taqman® miRNA assay (Life Technologies) according to manufacturer’s instructions. The absolute amounts of each miRNA per microgram of EV were determined based on a standard curve of threshold values obtained at different concentrations of synthetic miR-200c.
Statistical analysis
The mean of 3 or more replicates was used as average. Data were represented as mean ± SD. The P values were calculated using a standard unpaired Student t test for simple comparisons, and statistical significance is displayed as P < 0.05 (*) or P < 0.005 (**).
| Abbreviations: | ||
| AR | = | androgen receptor |
| CTxB | = | cholera toxin B |
| CM | = | conditioned medium |
| EV | = | extracellular vesicles |
| EMT | = | epithelial-to-mesenchymal transition |
| HB-EGF | = | heparin-binding EGF-like growth factor |
| MAT | = | mesenchymal-to-amoeboid transition |
| MMP | = | matrix metalloproteinase |
| PBMC | = | peripheral blood mononuclear cells |
| PBS | = | phosphate-buffered saline |
| PCa | = | prostate cancer |
| PrEC | = | normal prostate epithelial cells |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr Anthony Hill of the Boston Children’s Hospital IDDRC Imaging Core for technical assistance with confocal imaging. This study was supported by NIH 1R01CA143777, 1R01CA112303, and US Department of Defense PC093459 (to M.R.F), NIH 5R00CA131472 (to D.D.V.), a Ladies Auxiliary VFW Cancer Research Postdoctoral Fellowship (to S.M.) and the Fishbein Family IC Research Foundation/Interstitial Cystitis Association (ICA), Pilot Research Program/ICA, New York Academy of Medicine, and Boston Children’s Hospital Faculty Development (to J.K.). The research described was also supported by NIH/National Center for Advancing Translational Science (NCATS) UCLA CTSI Grant Number UL1TR000124. J.K. is an American Urological Association Foundation Research Scholar and a Harvard Medical School Eleanor and Miles Shore Scholar. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
J.K. and M.R.F. conceived of the study, designed experiments, evaluated data, and wrote the paper. J.K., S.M., M.L., and D.B. performed experiments. D.T.U. provided expertise and supervised data interpretation. S.M. and D.D.V. contributed conceptually and intellectually and to the writing of the manuscript.
References
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest 2009; 119:1420 - 8; http://dx.doi.org/10.1172/JCI39104; PMID: 19487818
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Bröcker EB, Friedl P. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 2003; 160:267 - 77; http://dx.doi.org/10.1083/jcb.200209006; PMID: 12527751
- Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 2003; 5:711 - 9; http://dx.doi.org/10.1038/ncb1019; PMID: 12844144
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol 2010; 188:11 - 9; http://dx.doi.org/10.1083/jcb.200909003; PMID: 19951899
- Belletti B, Nicoloso MS, Schiappacassi M, Berton S, Lovat F, Wolf K, Canzonieri V, D’Andrea S, Zucchetto A, Friedl P, et al. Stathmin activity influences sarcoma cell shape, motility, and metastatic potential. Mol Biol Cell 2008; 19:2003 - 13; http://dx.doi.org/10.1091/mbc.E07-09-0894; PMID: 18305103
- Shea KF, Wells CM, Garner AP, Jones GE. ROCK1 and LIMK2 interact in spread but not blebbing cancer cells. PLoS One 2008; 3:e3398; http://dx.doi.org/10.1371/journal.pone.0003398; PMID: 18852895
- Yoshida K, Soldati T. Dissection of amoeboid movement into two mechanically distinct modes. J Cell Sci 2006; 119:3833 - 44; http://dx.doi.org/10.1242/jcs.03152; PMID: 16926192
- Eisenmann KM, Harris ES, Kitchen SM, Holman HA, Higgs HN, Alberts AS. Dia-interacting protein modulates formin-mediated actin assembly at the cell cortex. Curr Biol 2007; 17:579 - 91; http://dx.doi.org/10.1016/j.cub.2007.03.024; PMID: 17398099
- Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol 2008; 181:879 - 84; http://dx.doi.org/10.1083/jcb.200802081; PMID: 18541702
- Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, Morley S, Mulholland D, Rotinen M, Hager MH, Insabato L, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol 2012; 181:1573 - 84; http://dx.doi.org/10.1016/j.ajpath.2012.07.030; PMID: 23022210
- Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet 2012; 21:R1 R125 - 34; http://dx.doi.org/10.1093/hmg/dds317; PMID: 22872698
- Muralidharan-Chari V, Clancy JW, Sedgwick A, D’Souza-Schorey C. Microvesicles: mediators of extracellular communication during cancer progression. J Cell Sci 2010; 123:1603 - 11; http://dx.doi.org/10.1242/jcs.064386; PMID: 20445011
- Zhuang G, Wu X, Jiang Z, Kasman I, Yao J, Guan Y, Oeh J, Modrusan Z, Bais C, Sampath D, et al. Tumour-secreted miR-9 promotes endothelial cell migration and angiogenesis by activating the JAK-STAT pathway. EMBO J 2012; 31:3513 - 23; http://dx.doi.org/10.1038/emboj.2012.183; PMID: 22773185
- Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, Bruno S, Bussolati B, Camussi G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood 2007; 110:2440 - 8; http://dx.doi.org/10.1182/blood-2007-03-078709; PMID: 17536014
- Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, Lovat F, Fadda P, Mao C, Nuovo GJ, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci U S A 2012; 109:E2110 - 6; http://dx.doi.org/10.1073/pnas.1209414109; PMID: 22753494
- Bebawy M, Combes V, Lee E, Jaiswal R, Gong J, Bonhoure A, Grau GE. Membrane microparticles mediate transfer of P-glycoprotein to drug sensitive cancer cells. Leukemia 2009; 23:1643 - 9; http://dx.doi.org/10.1038/leu.2009.76; PMID: 19369960
- Rak J, Guha A. Extracellular vesicles--vehicles that spread cancer genes. Bioessays 2012; 34:489 - 97; http://dx.doi.org/10.1002/bies.201100169; PMID: 22442051
- Hager MH, Morley S, Bielenberg DR, Gao S, Morello M, Holcomb IN, Liu W, Mouneimne G, Demichelis F, Kim J, et al. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med 2012; 4:743 - 60; http://dx.doi.org/10.1002/emmm.201200242; PMID: 22593025
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem 2007; 76:593 - 627; http://dx.doi.org/10.1146/annurev.biochem.75.103004.142647; PMID: 17373907
- Breitsprecher D, Goode BL. Formins at a glance. J Cell Sci 2013; 126:1 - 7; http://dx.doi.org/10.1242/jcs.107250; PMID: 23516326
- Thurston SF, Kulacz WA, Shaikh S, Lee JM, Copeland JW. The ability to induce microtubule acetylation is a general feature of formin proteins. PLoS One 2012; 7:e48041; http://dx.doi.org/10.1371/journal.pone.0048041; PMID: 23110170
- Bartolini F, Gundersen GG. Formins and microtubules. Biochim Biophys Acta 2010; 1803:164 - 73; http://dx.doi.org/10.1016/j.bbamcr.2009.07.006; PMID: 19631698
- Di Vizio D, Kim J, Hager MH, Morello M, Yang W, Lafargue CJ, True LD, Rubin MA, Adam RM, Beroukhim R, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res 2009; 69:5601 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-3860; PMID: 19549916
- Freeman MR, Paul S, Kaefer M, Ishikawa M, Adam RM, Renshaw AA, Elenius K, Klagsbrun M. Heparin-binding EGF-like growth factor in the human prostate: synthesis predominantly by interstitial and vascular smooth muscle cells and action as a carcinoma cell mitogen. J Cell Biochem 1998; 68:328 - 38; http://dx.doi.org/10.1002/(SICI)1097-4644(19980301)68:3<328::AID-JCB4>3.0.CO;2-W; PMID: 9518259
- Kim J, Adam RM, Freeman MR. Activation of the Erk mitogen-activated protein kinase pathway stimulates neuroendocrine differentiation in LNCaP cells independently of cell cycle withdrawal and STAT3 phosphorylation. Cancer Res 2002; 62:1549 - 54; PMID: 11888934
- Lin J, Adam RM, Santiestevan E, Freeman MR. The phosphatidylinositol 3′-kinase pathway is a dominant growth factor-activated cell survival pathway in LNCaP human prostate carcinoma cells. Cancer Res 1999; 59:2891 - 7; PMID: 10383151
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, D’Souza-Schorey C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr Biol 2009; 19:1875 - 85; http://dx.doi.org/10.1016/j.cub.2009.09.059; PMID: 19896381
- Kim J, Keay SK, Freeman MR. Heparin-binding epidermal growth factor-like growth factor functionally antagonizes interstitial cystitis antiproliferative factor via mitogen-activated protein kinase pathway activation. BJU Int 2009; 103:541 - 6; http://dx.doi.org/10.1111/j.1464-410X.2008.08097.x; PMID: 18990151
- Dong QZ, Wang Y, Tang ZP, Fu L, Li QC, Wang ED, Wang EH. Derlin-1 is overexpressed in non-small cell lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated up-regulation of MMP-2 and MMP-9. Am J Pathol 2013; 182:954 - 64; http://dx.doi.org/10.1016/j.ajpath.2012.11.019; PMID: 23306155
- Xiao LJ, Lin P, Lin F, Liu X, Qin W, Zou HF, Guo L, Liu W, Wang SJ, Yu XG. ADAM17 targets MMP-2 and MMP-9 via EGFR-MEK-ERK pathway activation to promote prostate cancer cell invasion. Int J Oncol 2012; 40:1714 - 24; PMID: 22200661
- Li B, Antonyak MA, Zhang J, Cerione RA. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012; 31:4740 - 9; http://dx.doi.org/10.1038/onc.2011.636; PMID: 22266864
- Wegiel B, Evans S, Hellsten R, Otterbein LE, Bjartell A, Persson JL. Molecular pathways in the progression of hormone-independent and metastatic prostate cancer. Curr Cancer Drug Targets 2010; 10:392 - 401; http://dx.doi.org/10.2174/156800910791208562; PMID: 20384583
- Zhang N, Wang X, Huo Q, Sun M, Cai C, Liu Z, Hu G, Yang Q. MicroRNA-30a suppresses breast tumor growth and metastasis by targeting metadherin. Oncogene 2013; Forthcoming http://dx.doi.org/10.1038/onc.2013.286; PMID: 23851509
- Valastyan S. Roles of microRNAs and other non-coding RNAs in breast cancer metastasis. J Mammary Gland Biol Neoplasia 2012; 17:23 - 32; http://dx.doi.org/10.1007/s10911-012-9241-9; PMID: 22293951
- Inui M, Martello G, Piccolo S. MicroRNA control of signal transduction. Nat Rev Mol Cell Biol 2010; 11:252 - 63; http://dx.doi.org/10.1038/nrm2868; PMID: 20216554
- Kong D, Li Y, Wang Z, Banerjee S, Ahmad A, Kim HR, Sarkar FH. miR-200 regulates PDGF-D-mediated epithelial-mesenchymal transition, adhesion, and invasion of prostate cancer cells. Stem Cells 2009; 27:1712 - 21; http://dx.doi.org/10.1002/stem.101; PMID: 19544444
- Shi XB, Xue L, Yang J, Ma AH, Zhao J, Xu M, Tepper CG, Evans CP, Kung HJ, deVere White RW. An androgen-regulated miRNA suppresses Bak1 expression and induces androgen-independent growth of prostate cancer cells. Proc Natl Acad Sci U S A 2007; 104:19983 - 8; http://dx.doi.org/10.1073/pnas.0706641104; PMID: 18056640
- Shi XB, Xue L, Ma AH, Tepper CG, Kung HJ, White RW. miR-125b promotes growth of prostate cancer xenograft tumor through targeting pro-apoptotic genes. Prostate 2011; 71:538 - 49; http://dx.doi.org/10.1002/pros.21270; PMID: 20886540
- Duque JL, Adam RM, Mullen JS, Lin J, Richie JP, Freeman MR. Heparin-binding epidermal growth factor-like growth factor is an autocrine mediator of human prostate stromal cell growth in vitro. J Urol 2001; 165:284 - 8; http://dx.doi.org/10.1097/00005392-200101000-00080; PMID: 11125426
- Lee TH, D’Asti E, Magnus N, Al-Nedawi K, Meehan B, Rak J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin Immunopathol 2011; 33:455 - 67; http://dx.doi.org/10.1007/s00281-011-0250-3; PMID: 21318413
- Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, Guha A, Rak J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol 2008; 10:619 - 24; http://dx.doi.org/10.1038/ncb1725; PMID: 18425114
- Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci U S A 2009; 106:3794 - 9; http://dx.doi.org/10.1073/pnas.0804543106; PMID: 19234131
- Balaj L, Lessard R, Dai L, Cho YJ, Pomeroy SL, Breakefield XO, Skog J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat Commun 2011; 2:180; http://dx.doi.org/10.1038/ncomms1180; PMID: 21285958
- Noerholm M, Balaj L, Limperg T, Salehi A, Zhu LD, Hochberg FH, Breakefield XO, Carter BS, Skog J. RNA expression patterns in serum microvesicles from patients with glioblastoma multiforme and controls. BMC Cancer 2012; 12:22; http://dx.doi.org/10.1186/1471-2407-12-22; PMID: 22251860
- Camussi G, Deregibus MC, Tetta C. Tumor-derived microvesicles and the cancer microenvironment. Curr Mol Med 2013; 13:58 - 67; http://dx.doi.org/10.2174/156652413804486304; PMID: 22834836
- D’Souza-Schorey C, Clancy JW. Tumor-derived microvesicles: shedding light on novel microenvironment modulators and prospective cancer biomarkers. Genes Dev 2012; 26:1287 - 99; http://dx.doi.org/10.1101/gad.192351.112; PMID: 22713869
- Filipazzi P, Bürdek M, Villa A, Rivoltini L, Huber V. Recent advances on the role of tumor exosomes in immunosuppression and disease progression. Semin Cancer Biol 2012; 22:342 - 9; http://dx.doi.org/10.1016/j.semcancer.2012.02.005; PMID: 22369922
- Peinado H, Alečković M, Lavotshkin S, Matei I, Costa-Silva B, Moreno-Bueno G, Hergueta-Redondo M, Williams C, García-Santos G, Ghajar C, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med 2012; 18:883 - 91; http://dx.doi.org/10.1038/nm.2753; PMID: 22635005
- Hannemann S, Madrid R, Stastna J, Kitzing T, Gasteier J, Schönichen A, Bouchet J, Jimenez A, Geyer M, Grosse R, et al. The Diaphanous-related Formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J Biol Chem 2008; 283:27891 - 903; http://dx.doi.org/10.1074/jbc.M801800200; PMID: 18694941
- Kitzing TM, Sahadevan AS, Brandt DT, Knieling H, Hannemann S, Fackler OT, Grosshans J, Grosse R. Positive feedback between Dia1, LARG, and RhoA regulates cell morphology and invasion. Genes Dev 2007; 21:1478 - 83; http://dx.doi.org/10.1101/gad.424807; PMID: 17575049
- Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R. Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 2010; 29:2441 - 8; http://dx.doi.org/10.1038/onc.2009.515; PMID: 20101212
- Wyse MM, Lei J, Nestor-Kalinoski AL, Eisenmann KM. Dia-interacting protein (DIP) imposes migratory plasticity in mDia2-dependent tumor cells in three-dimensional matrices. PLoS One 2012; 7:e45085; http://dx.doi.org/10.1371/journal.pone.0045085; PMID: 23024796
- Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10:593 - 601; http://dx.doi.org/10.1038/ncb1722; PMID: 18376396
- Elson-Schwab I, Lorentzen A, Marshall CJ. MicroRNA-200 family members differentially regulate morphological plasticity and mode of melanoma cell invasion. PLoS One 2010; 5:e13176; http://dx.doi.org/10.1371/journal.pone.0013176; PMID: 20957176
- Walter BA, Valera VA, Pinto PA, Merino MJ. Comprehensive microRNA Profiling of Prostate Cancer. J Cancer 2013; 4:350 - 7; http://dx.doi.org/10.7150/jca.6394; PMID: 23781281
- Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C, Barnes S, Grizzle W, Miller D, Zhang HG. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther 2010; 18:1606 - 14; http://dx.doi.org/10.1038/mt.2010.105; PMID: 20571541
- van Dommelen SM, Vader P, Lakhal S, Kooijmans SA, van Solinge WW, Wood MJ, Schiffelers RM. Microvesicles and exosomes: opportunities for cell-derived membrane vesicles in drug delivery. J Control Release 2012; 161:635 - 44; http://dx.doi.org/10.1016/j.jconrel.2011.11.021; PMID: 22138068
- EL Andaloussi S, Mäger I, Breakefield XO, Wood MJ, S ELA. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov 2013; 12:347 - 57; http://dx.doi.org/10.1038/nrd3978; PMID: 23584393
- Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine 2006; 1:297 - 315; PMID: 17717971
- Kooijmans SA, Vader P, van Dommelen SM, van Solinge WW, Schiffelers RM. Exosome mimetics: a novel class of drug delivery systems. Int J Nanomedicine 2012; 7:1525 - 41; PMID: 22619510
- Whiteside TL. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans 2013; 41:245 - 51; http://dx.doi.org/10.1042/BST20120265; PMID: 23356291
- Kim J, Kim WJ, Liu Z, Loda M, Freeman MR. The ubiquitin-specific protease USP2a enhances tumor progression by targeting cyclin A1 in bladder cancer. Cell Cycle 2012; 11:1123 - 30; http://dx.doi.org/10.4161/cc.11.6.19550; PMID: 22370483
- Mukhopadhyay NK, Kim J, Cinar B, Ramachandran A, Hager MH, Di Vizio D, Adam RM, Rubin MA, Raychaudhuri P, De Benedetti A, et al. Heterogeneous nuclear ribonucleoprotein K is a novel regulator of androgen receptor translation. Cancer Res 2009; 69:2210 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-08-2308; PMID: 19258514
- Kim J, Jahng WJ, Di Vizio D, Lee JS, Jhaveri R, Rubin MA, Shisheva A, Freeman MR. The phosphoinositide kinase PIKfyve mediates epidermal growth factor receptor trafficking to the nucleus. Cancer Res 2007; 67:9229 - 37; http://dx.doi.org/10.1158/0008-5472.CAN-07-1333; PMID: 17909029
- Bedoret D, Singh AK, Shaw V, Hoyte EG, Hamilton R, DeKruyff RH, Schneider LC, Nadeau KC, Umetsu DT. Changes in antigen-specific T-cell number and function during oral desensitization in cow’s milk allergy enabled with omalizumab. Mucosal Immunol 2012; 5:267 - 76; http://dx.doi.org/10.1038/mi.2012.5; PMID: 22318492