
Abstract
Male germ cell tumors (GCTs) are a model for a curable solid tumor. GCTs can differentiate into mature teratomas. Embryonal carcinomas (ECs) represent the stem cell compartment of GCTs and are the malignant counterpart to embryonic stem (ES) cells. GCTs and EC cells are useful to investigate differentiation therapy and chemotherapy response. This study explored mechanistic interactions between all-trans-retinoic acid (RA), which induces differentiation of EC and ES cells, and the Hedgehog (Hh) pathway, a regulator of self-renewal and proliferation. RA was found to induce mRNA and protein expression of Patched 1 (Ptch1), the Hh ligand receptor and negative regulator of this pathway. PTCH1 is also a target gene of Hh signaling through Smoothened (Smo) activation. Yet, this observed RA-mediated Ptch1 induction was independent of Smo. It occurred despite co-treatment with RA and Smo inhibitors. Retinoid induction of Ptch1 also occurred in other RA-responsive cancer cell lines and in normal ES cells. Notably, this enhanced Ptch1 expression was preceded by induction of the homeobox transcription factor Meis1, a direct RA target. Direct interaction between Meis1 and Ptch1 was confirmed using chromatin immunoprecipitation assays. To establish the translational relevance of this work, Ptch1 expression was shown to be deregulated in human ECs relative to mature teratoma and the normal seminiferous tubule. Taken together, these findings reveal a previously unrecognized mechanism through which RA can inhibit the Hh pathway via Ptch1 induction. Engaging this pathway is a new way to repress the Hh pathway that can be translated into the cancer clinic.
Introduction
Male germ cell tumors (GCTs) are models of a curable solid tumor. This is achieved with surgical resection, radiotherapy, platinum-based chemotherapy, or combined regimens of these modalities.Citation1 GCTs are distinct from other cancers because they can undergo in vivo differentiation into mature teratoma, as reviewed.Citation2 Mature teratomas have reduced proliferation and tumorigenicity as compared with the embryonal carcinomas (EC) from which they are derived.Citation2 EC cells are considered to be the stem cell compartment of nonseminomatous GCTs and are viewed as the malignant counterpart to embryonic stem (ES) cells.Citation3,Citation4 EC cells can differentiate into extra-embryonic and embryonic tissues.Citation3 A useful model to study EC induced-differentiation is the multipotent human EC cell line NTERA-2 clone D1 (NT2/D1), which matures along neuronal and other lineages after all-trans-retinoic acid (RA) treatment.Citation5
Retinoids are derivatives of vitamin A, which is essential for fertility, vision, immune function, and cellular homeostasis; retinoids play a key role in normal and neoplastic cell growth.Citation6 Retinoids regulate gene expression via nuclear retinoic acid receptors (RAR) or retinoid X receptors (RXR). RARs and RXRs can heterodimerize and bind to specific retinoic acid response elements (RAREs) in promoter regions, as reviewed.Citation5,Citation7
The hedgehog (Hh) pathway plays a key role in development and adult tissue homeostasis.Citation8 In contrast to nuclear actions of RA, Hh ligands Sonic, Desert, and Indian hedgehog (SHH, DHH, and IHH, respectively) interact with a cell surface receptor, Patched 1 (Ptch1).Citation9 In the absence of ligand, Ptch1 is a constitutive repressor of the Hh pathway, locking the downstream mediator smoothened (SMO) into an inactive conformation. Hh-ligand binding overcomes this inhibition, allowing Smo activation. Target genes of the Hh pathway include GLI1, PTCH1, CCND1 (cyclin D1), and CCNE1 (cyclin E), among others, as reviewed.Citation10
Crosstalk between RA and the Hh pathways is reported during development, where RA can induce SHH expression.Citation11,Citation12 However, the precise mechanisms responsible for interactions between RA and the Hh pathway are not well understood. This study explored this interplay in EC and ES cells. Findings reported here reveal a previously unrecognized link between retinoid signaling and the Hh pathway. This was that RA-treatment augmented Ptch1 expression. This occurred through direct induction of the homeobox transcription factor Meis1. The Meis1 binding site detected within the Ptch1 promoter is evolutionarily conserved. RA-mediated induction of Ptch1 was independent of Smo, since Smo inhibitors did not prevent this. This result highlighted a new mechanism for Hh pathway repression. The mechanism was also engaged after RA treatment of other retinoid-responsive cancer cells and ES cells. Translational relevance of this work was established by determining the expression profile of Ptch1 in human ECs relative to adjacent teratoma or the normal seminiferous tubule. Taken together, the findings presented here provide evidence for a previously unrecognized retinoid-dependent mechanism that can repress Hh signaling.
Results
RA induces Ptch1 and represses Gli1
Gene expression profiling of NT2/D1 cells treated with or without RA revealed upregulation of the Ptch1 gene (data not shown). To independently confirm these results, NT2/D1 cells were treated with RA (10 µM) or vehicle for 5 d, and expression of Ptch1 mRNA was assessed by qPCR assays. This dose of RA was previously optimized for differentiation of NT2/D1 cells into neurons and glia, as reviewed.Citation13 Ptch1 expression was significantly upregulated by day 2 through day 4 (P < 0.01), but was reduced by day 5 (, left panel). Upregulation of Ptch1 protein expression was also confirmed at day 3 (, left panel inset with displayed quantified signal intensity). The RA-resistant NT2/D1-R1Citation14 cells did not augment Ptch1 at the mRNA or protein levels (, right panel and inset).
Figure 1. Ptch1 expression after RA-treatment of human NT2/D1 EC cells. (A) Ptch1 mRNA expression as measured by qPCR assays was displayed independently for NT2/D1 EC cells (left panel; Ptch1 immunoblot with indicated quantification) and RA-resistant NT2/D1-R1 cells (right panel; Ptch1 immunoblot with indicated quantification) treated for 5 d with RA (10 µM) vs. vehicle control. (B) Gli1 mRNA expression is independently displayed for NT2/D1 (left panel) and NT2/D1-R1 cells (right panel). (C) The relative mRNA expression of the indicated Ptch1 isoforms was presented over a 5-d time course of RA (10 µM) treatment vs. vehicle control. Statistical significance is indicated by *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, respectively.
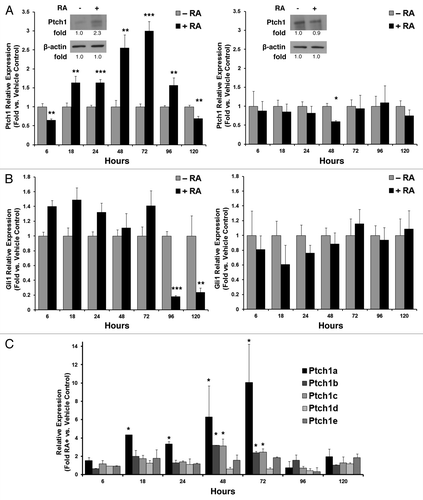
Because Ptch1 is a negative regulator of the Hh pathway, we next asked whether the observed Ptch1 induction affected the expression of Gli1, a known Hh pathway target gene.Citation10 Gli1 mRNA was significantly (P < 0.001) downregulated at days 4 and 5 of RA treatment in NT2/D1 (, left panel), but not in RA-resistant NT2/D1-R1 cells (, right panel). Because Ptch1 mRNA exists as multiple isoforms, isoform-specific qPCR assays were used to assess expression of the 5 family members (Ptch1a, Ptch1b, Ptch1c, Ptch1d, and Ptch1e) over the 5-d treatment regimen. RA treatment of NT2/D1 cells induced Ptch1a, Ptch1b, and Ptch1c isoforms (). Of these isoforms, Ptch1b was the predominant isoform expressed in NT2/D1 EC cells. It was expressed basally at approximately 1000-fold greater mRNA levels than Ptch1a and 50-fold greater levels than Ptch1c mRNA, and these relative differences were closely maintained after RA-treatment despite the prominent induction of Ptch1a (data not shown). It was not possible to differentiate between two closely-related Ptch1a isoforms (a and a’) that encode for protein isoforms of different sizes using qPCR primers.Citation15 This might overestimate the contribution of this isoform to overall Ptch1 expression after RA treatment.
Ptch1 induction by RA was independent of Smo
Ptch1 is a Hh pathway target gene.Citation10 Whether RA-mediated augmentation of Ptch1 expression depended on Smo activation was studied. NT2/D1 cells were independently treated for 3 d with one of two Smo inhibitors: cyclopamine (10 µM)Citation16 or GDC-0449 (1 µM, data not shown, and 10 µM).Citation17 These treatments did not affect Ptch1 induction despite RA treatment (). This established that this observed effect on Ptch1 did not depend on Smo activity. To confirm that Ptch1 would inhibit the Hh pathway in NT2/D1 cells, these cells were transiently transfected with a Gli binding site luciferase reporter construct (Gli-BSLuc) or with a Gli1-unresponsive constitutive TK-luciferase construct, with a renilla luciferase construct to control for transfection efficiency. These dual luciferase assays showed a significant (P < 0.05) and time-dependent decrease in Gli-luciferase activity following RA-treatment ().
Figure 2. Ptch1 upregulation after RA-treatment was independent of Smo activity. (A) Ptch1 mRNA expression as measured by qPCR assays was displayed for NT2/D1 cells following single or combined treatment with RA (10 µM) or a Smo inhibitor (cyclopamine [10 µM] or GDC-0449 [10 µM]), vs. vehicle-treated control. (B) The activity of a Gli-BSLuc Gli-responsive luciferase reporter plasmid relative to a constitutive TK-luciferase reporter plasmid was examined following RA (10 µM) treatment or vehicle treatment over the indicated time period. Each transfection was normalized to renilla luciferase activity. (C) The relative mRNA expression profiles of Ptch1 and Gli1 were compared between AB2.2 murine ES cell embryoid bodies formed in the presence of RA (0.5 µM) vs. vehicle control at the indicated time points, with the symbol * indicating P ≤ 0.05 relative to day 0 baseline expression, set to a value of 1. (D) The relative mRNA expression of Ptch1 following RA-treatment vs. vehicle control was assessed in these differentiation-sensitive cancer cell lines: NB4, Tera-1, and P19. The symbol * indicated P ≤ 0.05 in all panels.
![Figure 2. Ptch1 upregulation after RA-treatment was independent of Smo activity. (A) Ptch1 mRNA expression as measured by qPCR assays was displayed for NT2/D1 cells following single or combined treatment with RA (10 µM) or a Smo inhibitor (cyclopamine [10 µM] or GDC-0449 [10 µM]), vs. vehicle-treated control. (B) The activity of a Gli-BSLuc Gli-responsive luciferase reporter plasmid relative to a constitutive TK-luciferase reporter plasmid was examined following RA (10 µM) treatment or vehicle treatment over the indicated time period. Each transfection was normalized to renilla luciferase activity. (C) The relative mRNA expression profiles of Ptch1 and Gli1 were compared between AB2.2 murine ES cell embryoid bodies formed in the presence of RA (0.5 µM) vs. vehicle control at the indicated time points, with the symbol * indicating P ≤ 0.05 relative to day 0 baseline expression, set to a value of 1. (D) The relative mRNA expression of Ptch1 following RA-treatment vs. vehicle control was assessed in these differentiation-sensitive cancer cell lines: NB4, Tera-1, and P19. The symbol * indicated P ≤ 0.05 in all panels.](/cms/asset/2460f77b-9885-4475-a034-8f71046406e8/kcbt_a_10927821_f0002.gif)
These findings were examined in other cell contexts. The murine ES cell line AB2.2 was plated in embryoid body-forming conditions with RA (0.5 µM) or vehicle as a control. Relative mRNA expression profiles for Ptch1 and Gli1 were assayed by qPCR assays every 2 d. Ptch1 was upregulated through day 8 of RA-treatment followed by its subsequent repression; Gli1 mRNA was significantly (P < 0.05) repressed after day 4 of RA treatment (). Significant Ptch1 mRNA upregulation (P < 0.05) was also observed after 3 d of independent RA treatments of NB4, Tera-1, and P19 cell lines ().
Differentiation effects
The consequences of Ptch1 overexpression were analyzed in NT2/D1 cells. Engineered Ptch1 overexpression was confirmed by qPCR assays (Fig. S1, left panel). This Ptch1 overexpression relative to control transfectants repressed Gli1 expression at the mRNA level in these cells (Fig. S1, right panel), confirming that this transfected Ptch1 species was functional in NT2/D1 cells.
To assess the consequences of Ptch1 upregulation following RA-treatment, NT2/D1 cells were stably engineered with independent shRNA constructs targeting Ptch1, or a control vector. Knockdown efficiency was confirmed after puromycin selection (, left panel). Transfectants were independently treated with RA for 5 d vs. a vehicle control. Flow cytometry expression analyses of the immature marker SSEA3 and of the neuronal differentiation marker A2B5 were performed. Ptch1 knockdown decreased the decline of SSEA3 expressing NT2/D1 cells following RA-treatment, but did not reduce the accumulation of the A2B5 expressing cells (, middle and right panels).
Figure 3. The Hh pathway contributes to induced NT2/D1 cell differentiation. (A) NT2/D1 cells were individually stably transduced with two independent shRNAs targeting Ptch1 or an insertless control vector, and Ptch1 mRNA knockdown was confirmed (left panel). The consequences of stable Ptch1 knockdown on differentiation marker expression following 5 d of RA treatment vs. controls are shown (SSEA3, middle panel; A2B5, right panel). Analysis of expression of the (B) SSEA3 undifferentiated marker, (C) A2B5 neuronal marker, and (D) Vinis53 non-neuronal marker in NT2/D1 cells by flow cytometry after 5 d treatment with RA, cyclopamine, or GDC-0449, alone or in the indicated combinations are displayed. The symbol * depicts P ≤ 0.05 vs. control in panel (A). In (B–D), the symbol * depicts P ≤ 0.05 vs. vehicle-treated control; the symbol # depicts P ≤ 0.05 for the indicated combination treatments vs. each of the respective single treatments.
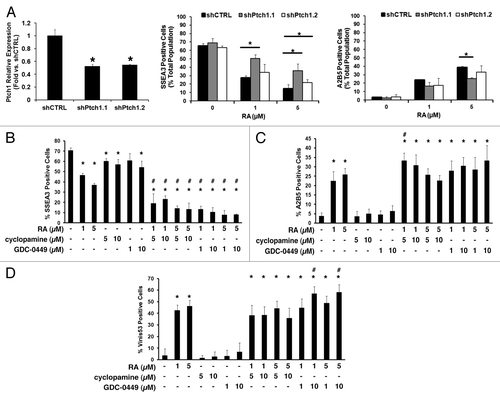
To assess effects of Hh pathway inhibition on EC cell differentiation, independent immunophenotypic analyses of NT2/D1 cells were performed using flow cytometry after 5 d with individual vehicle, RA, cyclopamine, or GDC-0449 treatments, and with combinations of a Smo inhibitor and RA. Dosages of RA were chosen to uncover cooperative effects of these agents. Treatments with RA (1 µM or 5 µM) decreased expression of the SSEA3 marker (), increased expression of the A2B5 marker (), and increased expression of the non-neuronal differentiation marker Vinis53 (). Cyclopamine and GDC-0449 each had modest effects on immunophenotypic marker profiles (). However, combinations of Smo inhibitors and RA had at least additive effects in decreasing expression of SSEA3 (), and the selective Smo inhibitor, GDC-0449, had at least additive effects on augmenting Vinis53 expression at higher doses ().
RA augmented Ptch1 through Meis1
The kinetics of Ptch1 upregulation after RA-treatment implied that this effect was indirect. Bioinformatic analysis did not identify RAREs in the Ptch1 promoter (data not shown). Potential mediators of this Ptch1 induction were next sought. Analysis of the Ptch1 gene in MultiTF (available from http://multitf.dcode.org/) revealed the presence of an intronic Meis1 binding site that was conserved in all of the examined species (; ). Meis1 is a homeobox transcription factor that regulates self-renewal.Citation18,Citation19 MEIS1 is a recognized RA-target gene in the salamander, where it coordinates RA-dependent cell fate specification in regenerating limbs.Citation20 Meis1 induction was also reported in P19 EC cells after RA-treatment.Citation21 RA induced Meis1 expression in NT2/D1 cells was studied by qPCR assays over 5 d vs. vehicle controls. Meis1 mRNA and protein expression profiles were rapidly and prominently augmented in a timeline consistent with a direct RA target gene (, left panel). The RA-resistant NT2/D1-R1 cell line did not exhibit these effects (, right panel).
Figure 4. Meis1 binds to the PTCH1 genomic site and contributes to retinoid induction of Ptch1 mRNA. (A) Bioinformatic analysis of the PTCH1 gene revealed an evolutionarily conserved intronic Meis1 binding site, as shown in this schema. (B) Meis1 mRNA expression profile as measured by qPCR assays was displayed for NT2/D1 cells (left panel, immunoblot analysis with quantification and arrow depicting Meis1) or NT2/D1-R1 RA-resistant EC cells (right panel, immunoblot analysis with quantification and arrow depicting Meis1) treated for 5 d with RA (10 µM, +RA) vs. vehicle as a control (−RA). (C) NT2/D1 cells were transiently transfected with two independent siRNAs targeting Meis1 or a scrambled control siRNA. Knockdown was confirmed (left panel Meis1 immunoblot analysis with quantification). Relative levels of Meis1 induction after 3 d of treatment with RA (10 µM) or vehicle as a control for the Meis1 knockdown is displayed (center panel). Relative levels of Ptch1 induction after 3 d of treatment with RA (10 µM) or vehicle as a control in NT2/D1 cells are shown after Meis1 knockdown (right panel). (D) ChIP assays identified a PTCH1 DNA complex by use of a Meis1-specific antibody following 3 d of treatment with RA (10 µM) vs. vehicle as a control (−RA). The symbols indicated *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.001.
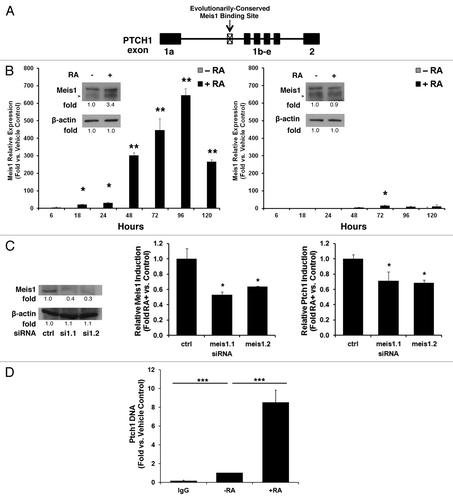
Table 1. The Meis1 binding site in the PTCH1 gene is evolutionarily conserved.
To determine if Meis1 was directly responsible for Ptch1 induction, two different siRNAs targeting Meis1 or a scrambled control siRNA were independently transfected into NT2/D1 cells. Knockdown was confirmed, as shown (, left panel). Meis1 knockdown was maintained over the course of this experiment (, center panel). Meis1 knockdown significantly (P < 0.05) reduced Ptch1 induction after RA-treatment, vs. controls (, right panel). ChIP assays were next used to establish a direct link between Ptch1 and Meis1. Substantially more Ptch1 than GAPDH (which lacks Meis1 binding sites) chromatin was pulled down from NT2/D1 cells with a Meis1/2-specific antibody as compared with a nonspecific IgG; this interaction was enhanced by 3 d of RA (10 µM) treatment vs. controls ().
Ptch1 expression in GCTs, teratomas, and seminiferous tubules
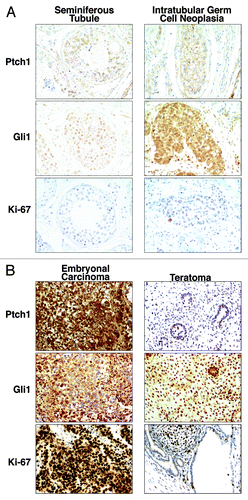
Seven GCT orchiectomy specimens were examined immunohistochemically for Gli1, Ptch1, and Ki-67 expression. These tissues contained both EC and teratoma components. Some displayed histologically normal seminiferous tubules and adjacent intratubular germ cell neoplasia (ITGCN), a GCT precursor lesion. Low levels of Gli1, Ptch1, and Ki-67 expression were observed in the seminiferous tubule, but levels were increased in ITGCN (). EC elements expressed the highest levels of Gli1, Ptch1, and Ki-67 (). Notably, the adjacent teratomas expressed much lower levels of Gli1, Ptch1, and Ki-67 immunostaining as compared with the EC components ().
Figure 5. Hedgehog pathway member immunohistochemical expression profiles in embryonal carcinoma, teratoma, and testis. Gli1, Ptch1, and Ki-67 (a proliferation marker) immunohistochemical expression patterns are displayed in (A) a representative histopathologically normal seminiferous tubule vs. intratubular germ cell neoplasia and (B) a representative embryonal carcinoma vs. adjacent teratoma. All panels are shown as 100× magnification.
Testes of Ptch1+/LacZ heterozygotic mice were next examined. This was done to examine Ptch1 and Gli1 expression in a setting without testicular pathologic abnormalities. Ptch1 was highly expressed in the Leydig cells. There was staining of occasional endothelial cells, but no staining was detected in Sertoli cells, spermatogonia, spermatocytes, and spermatids (Fig. S2, top panels). Gli1 expression was observed in seminiferous tubules, in agreement with prior reports in non-transgenic adult mice (Fig. S2, bottom panels).Citation22 Most tubules displayed nuclear and cytoplasmic Gli1 staining in the Sertoli cells, spermatogonia, and spermatocytes, and immunostaining only in the cytoplasm was seen in a subset of Leydig cells. Reduced staining was observed in spermatids. Variant staining patterns were observed in some seminiferous tubules; these included absent staining (data not shown), preferential staining in late spermatocytes and spermatids (Fig. S3A), or punctuate staining of spermatids (Fig. S3B). Abundant Gli1 staining was also observed in the murine epididymus (Fig. S3C).
Discussion
A role for Gli1 and Ptch1 in cell cycle regulation and proliferation of NT2/D1 cells was previously proposed.Citation23 Yet, this prior study focused on changes in Gli1 and Ptch1 mRNA levels at later time points of RA-treatment of NT/D1 cells than did this study. The current study conducted a detailed kinetic analysis and uncovered an RA-mediated induction of Ptch1, as shown in . The results presented here establish that the previously-observed Ptch1 and Gli1 repressionCitation23 was preceded by an induction of Ptch1 after RA-treatment of NT2/D1 cells. Notably, Ptch1 induction itself was functional in these cells because it conferred Gli1 repression (Fig. S1). The inability of Smo inhibitors to prevent Ptch1 induction after RA-treatment, as shown in , indicates that this is a previously unrecognized mechanism through which RA can repress Hh signaling. Ptch1 induction by RA-treatment was also observed in ES cells and other RA-responsive cancer cell lines. This indicated that this mechanism was active in diverse cell contexts. The Ptch1 knockdown results combined with the RA and Smo inhibitor differentiation marker studies in implicated a role for the Hh pathway in regulating EC cell differentiation, especially through a non-neuronal lineage.
The kinetics of Ptch1 induction implied that Ptch1 was not a direct RA target gene. Indeed, evolutionary conservation of the Meis1 binding site in the Ptch1 promoter suggested a role for this transcription factor in this Ptch1 regulation. This hypothesis was confirmed by both Meis1 knockdown and ChIP assays, displayed in . Hence, a direct role for Meis1 in the retinoid induction of Ptch1 was found.
Prior work indicated the importance of the Hh pathway in development of the mouse testis,Citation24-Citation26 and in the adult testis.Citation22,Citation27-Citation29 The findings presented here regarding Ptch1 and Gli1 expression in the human testis are consistent with this work, as shown in . Those findings were extended by analysis of both Ptch1 and Gli1 expression profiles in the Ptch+/LacZ mouse, as displayed in Figures S2 and S3. Ptch+/LacZ studies confirmed Ptch1 expression was detected in the adult murine testis and primarily in Leydig cells. Gli1 staining was observed in spermatogonia and spermatocytes within seminiferous tubules. This indicated that basal Hh signaling was observed in these cells despite restricted Ptch1 expression.
Our findings from clinical GCT specimens revealed that the Hh pathway components Gli1 and Ptch1 were abundantly expressed in the highly proliferative undifferentiated EC regions that exhibit prominent Ki-67 immunostaining. In marked contrast, reduced expression profiles of the same markers was observed in the adjacent teratoma elements in the same GCT, as shown in . We hypothesize from this result that the activation of the Hh signaling in EC promotes deregulated growth of GCTs, while the activity of the same pathway is reduced in the differentiated teratoma present in the same GCT.
The Hh inhibitor GDC-0449 (Vismodegib) has shown clinical activity in Gorlin syndrome-associated invasive basal cell carcinomas and medulloblastoma.Citation30-Citation33 This inhibitor targets Smo. While Gorlin syndrome-associated cancers are driven by inactivating mutations of the PTCH1 gene, there are many other cancers that overexpress Hh pathway members, including lung, breast, pancreatic, and other cancers, as reviewed.Citation34 An important implication of the current work is that cooperative treatment with an optimal retinoid and an Hh pathway inhibitor might have additive effects in repressing Hh signaling, and perhaps in reducing the onset of mutations that confer resistance to each of these agents. There is precedent for crosstalk between RA and other antineoplastics. An example of this is reported between retinoids and estrogen receptor signaling in breast cancer.Citation35 Clinical crosstalk is also found between an RXR agonist (rexinoid) and an epidermal growth factor receptor tyrosine kinase inhibitor.Citation36,Citation37
In conclusion, this study has shown that RA can inhibit the Hh pathway in EC cells through a previously unrecognized mechanism. This mechanism is one that induces expression of Ptch1 through a Smo-independent pathway. Ptch1 augmentation was detected in other cell contexts, including retinoid-responsive cancer cells and ES cells. Ptch1 induction was determined to be due to the retinoid-dependent upregulation of the transcription factor Meis1. The translational relevance of this finding was established by finding differential Ptch1 expression in human EC vs. teratomas and the normal testis. Whether pharmacologically engaging this pathway can be translated into the cancer clinic is the subject of future work.
Materials and Methods
Chemicals
The Smo inhibitors cyclopamine (LC Laboratories) and GDC-0449 (StemRD) were purchased, as was RA (Sigma-Aldrich). Stock solutions in ethanol or dimethyl sulfoxide (DMSO; Sigma-Aldrich), were prepared and stored at −80 °C.
Cell culture
The multipotent human EC NT2/D1 and its RA-resistant subclone NT2/D1-R1,Citation14 human EC Tera-1, human 293T embryonic kidney, and murine EC P19 cell lines (ATCC) were each cultured in high glucose DMEM (Life Technologies) medium supplemented with 2 mM l-glutamine (Corning) and 10% fetal bovine serum (FBS; Thermo Fisher Scientific). Human NB4 acute promyelocytic leukemia (APL) cells were cultured in RPMI 1640 medium (Corning) supplemented with 10% FBS. Murine AB2.2 ES cells (ATCC) were cultured in Mouse ES Cell Basal Medium (ATCC) supplemented with 0.1 mM 2-mercaptoethanol (Sigma-Aldrich), 1000 U/mL leukemia inhibitory factor (LIF; EMD-Millipore), and 10% ES-cell qualified FBS (ATCC). Cells were cultured at 37 °C with 5% CO2 and in a humidified incubator.
Semiquantitative real time RT-PCR (qPCR) assays
Total mRNA was isolated using the RNeasy kit with on-column DNase digestion (Qiagen). Reverse transcription (RT) was performed with the High Capacity cDNA RT Kit (Applied Biosystems) and a Peltier Thermal Cycler (MJ Research). The qPCR assays were performed using iTaq Fast SYBR Green Supermix with ROX (Bio-Rad Laboratories) and a 7500 Fast Real-Time PCR System (Applied Biosystems). Assays were performed in triplicate with at least two replicates. Primer sequences are displayed in Supplemental File 4.
Immunoblot analysis
Immunoblotting was performed as before.Citation38 Antibodies and dilutions were: Ptch1 1:200 (sc-6148; Santa Cruz Biotechnology), Meis1/2 1:500 (sc-10599; Santa Cruz Biotechnology), and Actin C-11 1:5000 (sc-1615; Santa Cruz Biotechnology). Species-specific horseradish peroxidase (HRP)-linked secondary antibodies were used with the Lumina Forte electrochemiluminescent detection reagent (EMD-Millipore). Signal intensities were determined with NIH ImageJ software (available from http://rsbweb.nih.gov/ij/).
Ptch1 knockdown
Two independent Ptch1 short hairpin RNA (shRNA) constructs and a non-targeting scrambled control were purchased (Thermo Fisher Scientific) and are listed in Figure S4. Lentiviruses were generated with an optimized systemCitation39 consisting of packaging plasmids pCMV-dR8.2 (plasmid 8455; Addgene) and pMD2.G (plasmid 12259; Addgene). Vectors were transfected into 293T cells using Fugene 6 transfection reagent (Roche) according to the manufacturer’s protocol. Lentiviral titers supplemented with 1% bovine serum albumin (BSA; Sigma-Aldrich) were harvested and used to infect NT2/D1 cells in the presence of 4 ug/mL polybrene (Sigma-Aldrich). Transduced cells were selected with 0.5 µg/mL puromycin (Sigma-Aldrich).
Ptch1 transient overexpression and luciferase assays
The Ptch1b cDNA insert in a pCI-Ptch1b-Flag vector (a gift of Dr Rune Toftgårds, Karolinska Institutet) was removed from the vector using restriction endonucleases Nhe1 and Not1 (New England Biolabs) and ligated into the respective sites in the multiple cloning region of the pCI-neo G418-selectable mammalian expression backbone (Promega) to generate a pCI-neo-Ptch1b-flag vector, which was sequence-confirmed. The empty vector served as a negative control. NT2/D1 cells were transfected with pCI-neo-Ptch1b-flag or the empty vector with TransIT-LT1 transfection reagent (Mirus Bio). Short-term selection was with G418 (250 µg/mL; Corning) for 5 d.
Hh reporter assays were performed following co-transfection of the GliBS-Luc or TK-luciferase control plasmids with a renilla-expression construct (RL-TK) into NT2/D1 cells with Fugene 6, as before.Citation40 Luciferase activity was assayed at indicated time points using the Dual Luciferase Reporter Assay System (Promega).
Differentiation assays
Fluorescence activated cell sorting (FACS) analyses of expression of differentiation markers in NT2-D1 cells were performed as before.Citation14 NT2/D1 cells treated with the indicated agents or vehicle control for 5 d were harvested and independently analyzed with antibodies that recognized A2B5 (R&D Systems), SSEA3 (R&D Systems), or Vinis-53 (Developmental Studies Hybridoma Bank, University of Iowa) along with appropriate secondary antibodies (Life Technologies). Cells were analyzed by FACScan (BD Biosciences) and MACSQuant VYB (Miltenyi Biotec).
Silencing with siRNAs
Independent small interfering RNAs (siRNAs) that targeted human Meis1 transcripts and a RISC-free control siRNA (Dharmacon) were used, as shown in Supplemental File 4. NT2/D1 (2 × 105) cells were plated in 6-well tissue culture plates and transfected with these siRNAs using Lipofectamine 2000 (Life Technologies).
Chromatin immunoprecipitation (ChIP) assays
NT2/D1 cells (5 × 106) were plated in 15cm tissue culture plates (Corning) and treated 24 h later with RA (10 µM) or DMSO as a vehicle. After 3 d of treatment, cells were harvested and chromatin binding assessed via an optimized ChIP protocol.Citation41 DNA–protein complexes were pulled down with nonspecific IgG or Meis1/2 antibody (10 µg) complexed to protein-G magnetic Dynabeads (Life Technologies). The qPCR primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH; negative control) and Ptch1 loci are displayed in Supplemental File 4. Data were normalized to 10% input samples.
In vivo analysis of Ptch1 and Gli1 expression
Animal protocols were reviewed and approved by the relevant Institutional Animal Care and Use Committee (IACUC). Analysis of human tissues was approved by Dartmouth’s Institutional Review Board (IRB). Seven GCTs that contained both EC and mature teratoma in the same specimen, and/or histopathologically normal seminiferous tubules were processed for immunohistochemistry (IHC), as before.Citation42 Antibodies independently recognized Ptch1 (sc-6149; Santa Cruz Biotechnology), Gli1 (sc-20867; Santa Cruz Biotechnology), and Ki-67 (clone MIB1; Dako).
Testes from Ptch+/LacZ transgenic mice were harvested at 7 wk of age. These tissues were fixed in 4% formaldehyde on ice for an hour, cryoprotected overnight in 30% sucrose in phosphate buffered saline (PBS), and embedded in Tissue-Tek Optimal Cutting Temperature Medium (Ted Pella Inc.). Slides were immunostained for Gli1 via IHCCitation43 or X-Gal (Life Technologies) to detect β-galactosidase activity.Citation44
Statistical analyses
Two-tailed t tests were used. Results were displayed as means ± SD. Statistical significance was set at P ≤ 0.05.
Additional material
Download Zip (1.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Flow cytometry analyses were performed in the DartLab: Immunoassay and Flow Cytometry Shared Resource at the Geisel School of Medicine at Dartmouth. A.B. was supported by a National Research Service Award from the National Institutes of Health (NIH, T32-CA009658) and through a fellowship from the Albert J. Ryan Foundation. E.D. is an American Cancer Society Clinical Research Professor supported by a generous gift from the F.M. Kirby Foundation. This work was supported in part by an American Cancer Society Institutional Grant, Prouty Multi-Investigator Award, and a grant from Uniting Against Lung Cancer with Mary Jo’s Fund to Fight Cancer (S. Freemantle) and by NIH and National Cancer Institute (NCI) grants R01-CA111422 and R01-CA062275 and by a Samuel Waxman Cancer Research Foundation Award (E. D.). This work was supported in part from the NCI core grant 5 P30 CA023108 that supports the Norris Cotton Cancer Center at Dartmouth.
References
- Verhoeven RHA, Gondos A, Janssen-Heijnen MLG, Saum KU, Brewster DH, Holleczek B, Crocetti E, Rosso S, Hakulinen T, Aareleid T, et al, EUNICE Survival Working Group. Testicular cancer in Europe and the USA: survival still rising among older patients. Ann Oncol 2013; 24:508 - 13; http://dx.doi.org/10.1093/annonc/mds460; PMID: 23110807
- Spinella MJ, Kerley JS, White KA, Curtin JC. Retinoid target gene activation during induced tumor cell differentiation: human embryonal carcinoma as a model. J Nutr 2003; 133:273S - 6S; PMID: 12514308
- Andrews PW. Teratocarcinomas and human embryology: pluripotent human EC cell lines. Review article. APMIS 1998; 106:158 - 67, discussion 167-8; http://dx.doi.org/10.1111/j.1699-0463.1998.tb01331.x; PMID: 9524574
- Andrews PW. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev Biol 1984; 103:285 - 93; http://dx.doi.org/10.1016/0012-1606(84)90316-6; PMID: 6144603
- Freemantle SJ, Spinella MJ, Dmitrovsky E. Retinoids in cancer therapy and chemoprevention: promise meets resistance. Oncogene 2003; 22:7305 - 15; http://dx.doi.org/10.1038/sj.onc.1206936; PMID: 14576840
- Duester G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008; 134:921 - 31; http://dx.doi.org/10.1016/j.cell.2008.09.002; PMID: 18805086
- Bastien J, Rochette-Egly C. Nuclear retinoid receptors and the transcription of retinoid-target genes. Gene 2004; 328:1 - 16; http://dx.doi.org/10.1016/j.gene.2003.12.005; PMID: 15019979
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev 2001; 15:3059 - 87; http://dx.doi.org/10.1101/gad.938601; PMID: 11731473
- Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993; 75:1417 - 30; http://dx.doi.org/10.1016/0092-8674(93)90627-3; PMID: 7916661
- Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 2009; 9:873 - 86; http://dx.doi.org/10.2174/156652409789105570; PMID: 19860666
- Ribes V, Wang Z, Dollé P, Niederreither K. Retinaldehyde dehydrogenase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development 2006; 133:351 - 61; http://dx.doi.org/10.1242/dev.02204; PMID: 16368932
- Niederreither K, Dollé P. Retinoic acid in development: towards an integrated view. Nat Rev Genet 2008; 9:541 - 53; http://dx.doi.org/10.1038/nrg2340; PMID: 18542081
- Langlois A, Duval D. Differentiation of the human NT2 cells into neurons and glia. Methods Cell Sci 1997; 19:213 - 9; http://dx.doi.org/10.1023/A:1009731707443
- Moasser MM, Khoo K-S, Maerz WJ, Zelenetz A, Dmitrovsky E. Derivation and characterization of retinoid-resistant human embryonal carcinoma cells. Differentiation 1996; 60:251 - 7; http://dx.doi.org/10.1046/j.1432-0436.1996.6040251.x; PMID: 8765055
- Kogerman P, Krause D, Rahnama F, Kogerman L, Undén AB, Zaphiropoulos PG, Toftgård R. Alternative first exons of PTCH1 are differentially regulated in vivo and may confer different functions to the PTCH1 protein. Oncogene 2002; 21:6007 - 16; http://dx.doi.org/10.1038/sj.onc.1205865; PMID: 12203113
- Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev 2002; 16:2743 - 8; http://dx.doi.org/10.1101/gad.1025302; PMID: 12414725
- Robarge KD, Brunton SA, Castanedo GM, Cui Y, Dina MS, Goldsmith R, Gould SE, Guichert O, Gunzner JL, Halladay J, et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg Med Chem Lett 2009; 19:5576 - 81; http://dx.doi.org/10.1016/j.bmcl.2009.08.049; PMID: 19716296
- Yamada T, Urano-Tashiro Y, Tanaka S, Akiyama H, Tashiro F. Involvement of crosstalk between Oct4 and Meis1a in neural cell fate decision. PLoS One 2013; 8:e56997; http://dx.doi.org/10.1371/journal.pone.0056997; PMID: 23451132
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013; 497:249 - 53; http://dx.doi.org/10.1038/nature12054; PMID: 23594737
- Mercader N, Tanaka EM, Torres M. Proximodistal identity during vertebrate limb regeneration is regulated by Meis homeodomain proteins. Development 2005; 132:4131 - 42; http://dx.doi.org/10.1242/dev.01976; PMID: 16107473
- Oulad-Abdelghani M, Chazaud C, Bouillet P, Sapin V, Chambon P, Dollé P. Meis2, a novel mouse Pbx-related homeobox gene induced by retinoic acid during differentiation of P19 embryonal carcinoma cells. Dev Dyn 1997; 210:173 - 83; http://dx.doi.org/10.1002/(SICI)1097-0177(199710)210:2<173::AID-AJA9>3.0.CO;2-D; PMID: 9337137
- Kroft TL, Patterson J, Won Yoon J, Doglio L, Walterhouse DO, Iannaccone PM, Goldberg E. GLI1 localization in the germinal epithelial cells alternates between cytoplasm and nucleus: upregulation in transgenic mice blocks spermatogenesis in pachytene. Biol Reprod 2001; 65:1663 - 71; http://dx.doi.org/10.1095/biolreprod65.6.1663; PMID: 11717126
- Vestergaard J, Lind-Thomsen A, Pedersen MW, Jarmer HØ, Bak M, Hasholt L, Tommerup N, Tümer Z, Larsen LA. GLI1 is involved in cell cycle regulation and proliferation of NT2 embryonal carcinoma stem cells. DNA Cell Biol 2008; 27:251 - 6; http://dx.doi.org/10.1089/dna.2007.0625; PMID: 18241033
- Yao HH-C, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev 2002; 16:1433 - 40; http://dx.doi.org/10.1101/gad.981202; PMID: 12050120
- Barsoum I, Yao HHC. Redundant and differential roles of transcription factors Gli1 and Gli2 in the development of mouse fetal Leydig cells. Biol Reprod 2011; 84:894 - 9; http://dx.doi.org/10.1095/biolreprod.110.088997; PMID: 21209421
- Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol 1996; 6:298 - 304; http://dx.doi.org/10.1016/S0960-9822(02)00480-3; PMID: 8805249
- Clark AM, Garland KK, Russell LD. Desert hedgehog (Dhh) gene is required in the mouse testis for formation of adult-type Leydig cells and normal development of peritubular cells and seminiferous tubules. Biol Reprod 2000; 63:1825 - 38; http://dx.doi.org/10.1095/biolreprod63.6.1825; PMID: 11090455
- Szczepny A, Hime GR, Loveland KL. Expression of hedgehog signalling components in adult mouse testis. Dev Dyn 2006; 235:3063 - 70; http://dx.doi.org/10.1002/dvdy.20931; PMID: 16958114
- Morales CR, Fox A, El-Alfy M, Ni X, Argraves WS. Expression of Patched-1 and Smoothened in testicular meiotic and post-meiotic cells. Microsc Res Tech 2009; 72:809 - 15; http://dx.doi.org/10.1002/jemt.20733; PMID: 19484749
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 2009; 361:1173 - 8; http://dx.doi.org/10.1056/NEJMoa0902903; PMID: 19726761
- Caro I, Low JA. The role of the hedgehog signaling pathway in the development of basal cell carcinoma and opportunities for treatment. Clin Cancer Res 2010; 16:3335 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-09-2570; PMID: 20439455
- Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med 2009; 361:1164 - 72; http://dx.doi.org/10.1056/NEJMoa0905360; PMID: 19726763
- Low JA, de Sauvage FJ. Clinical experience with Hedgehog pathway inhibitors. J Clin Oncol 2010; 28:5321 - 6; http://dx.doi.org/10.1200/JCO.2010.27.9943; PMID: 21041712
- Pasca di Magliano M, Hebrok M. Hedgehog signalling in cancer formation and maintenance. Nat Rev Cancer 2003; 3:903 - 11; http://dx.doi.org/10.1038/nrc1229; PMID: 14737121
- White KA, Yore MM, Deng D, Spinella MJ. Limiting effects of RIP140 in estrogen signaling: potential mediation of anti-estrogenic effects of retinoic acid. J Biol Chem 2005; 280:7829 - 35; http://dx.doi.org/10.1074/jbc.M412707200; PMID: 15632153
- Dragnev KH, Ma T, Cyrus J, Galimberti F, Memoli V, Busch AM, Tsongalis GJ, Seltzer M, Johnstone D, Erkmen CP, et al. Bexarotene plus erlotinib suppress lung carcinogenesis independent of KRAS mutations in two clinical trials and transgenic models. Cancer Prev Res (Phila) 2011; 4:818 - 28; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0376; PMID: 21636548
- Dragnev KH, Petty WJ, Shah S, Biddle A, Desai NB, Memoli V, Rigas JR, Dmitrovsky E. Bexarotene and erlotinib for aerodigestive tract cancer. J Clin Oncol 2005; 23:8757 - 64; http://dx.doi.org/10.1200/JCO.2005.01.9521; PMID: 16314636
- Snow GE, Kasper AC, Busch AM, Schwarz E, Ewings KE, Bee T, Spinella MJ, Dmitrovsky E, Freemantle SJ. Wnt pathway reprogramming during human embryonal carcinoma differentiation and potential for therapeutic targeting. BMC Cancer 2009; 9:383; http://dx.doi.org/10.1186/1471-2407-9-383; PMID: 19874621
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9:493 - 501; http://dx.doi.org/10.1261/rna.2192803; PMID: 12649500
- Galimberti F, Busch AM, Chinyengetere F, Ma T, Sekula D, Memoli VA, Dragnev KH, Liu F, Johnson KC, Guo Y, et al. Response to inhibition of smoothened in diverse epithelial cancer cells that lack smoothened or patched 1 mutations. Int J Oncol 2012; 41:1751 - 61; PMID: 22923130
- Mao P, Hever MP, Niemaszyk LM, Haghkerdar JM, Yanco EG, Desai D, Beyrouthy MJ, Kerley-Hamilton JS, Freemantle SJ, Spinella MJ. Serine/threonine kinase 17A is a novel p53 target gene and modulator of cisplatin toxicity and reactive oxygen species in testicular cancer cells. J Biol Chem 2011; 286:19381 - 91; http://dx.doi.org/10.1074/jbc.M111.218040; PMID: 21489989
- Petty WJ, Dragnev KH, Memoli VA, Ma Y, Desai NB, Biddle A, Davis TH, Nugent WC, Memoli N, Hamilton M, et al. Epidermal growth factor receptor tyrosine kinase inhibition represses cyclin D1 in aerodigestive tract cancers. Clin Cancer Res 2004; 10:7547 - 54; http://dx.doi.org/10.1158/1078-0432.CCR-04-1169; PMID: 15569985
- Ma Y, Fiering S, Black C, Liu X, Yuan Z, Memoli VA, Robbins DJ, Bentley HA, Tsongalis GJ, Demidenko E, et al. Transgenic cyclin E triggers dysplasia and multiple pulmonary adenocarcinomas. Proc Natl Acad Sci U S A 2007; 104:4089 - 94; http://dx.doi.org/10.1073/pnas.0606537104; PMID: 17360482
- Náray-Fejes-Tóth A, Fejes-Tóth G. Novel mouse strain with Cre recombinase in 11β-hydroxysteroid dehydrogenase-2-expressing cells. Am J Physiol Renal Physiol 2007; 292:F486 - 94; http://dx.doi.org/10.1152/ajprenal.00188.2006; PMID: 16896181