
Abstract
Objective: Neuroblastoma is a common neuroendocrine (NE) tumor that presents in early childhood, with a high incidence of malignancy and recurrence. The glycogen synthase kinase-3 (GSK-3) pathway is a potential therapeutic target, as this pathway has been shown to be crucial in the management of other NE tumors. However, it is not known which isoform is necessary for growth inhibition. In this study, we investigated the effect of the GSK-3 inhibitor AR-A014418 on the different GSK-3 isoforms in neuroblastoma.
Methods: NGP and SH-5Y-SY cells were treated with 0–20 μM of AR-A014418 and cell viability was measured by MTT assay. Expression levels of NE markers CgA and ASCL1, GSK-3 isoforms, and apoptotic markers were analyzed by western blot.
Results: Neuroblastoma cells treated with AR-A014418 had a significant reduction in growth at all doses and time points (P < 0.001). A reduction in growth was noted in cell lines on day 6, with 10 μM (NGP-53% vs. 0% and SH-5Y-SY-38% vs. 0%, P < 0.001) treatment compared to control, corresponding with a noticeable reduction in tumor marker ASCL1 and CgA expression.
Conclusion: Treatment of neuroblastoma cell lines with AR-A014418 reduced the level of GSK-3α phosphorylation at Tyr279 compared to GSK-3β phosphorylation at Tyr216, and attenuated growth via the maintenance of apoptosis. This study supports further investigation to elucidate the mechanism(s) by which GSK-3α inhibition downregulates the expression of NE tumor markers and growth of neuroblastoma.
Keywords: :
Introduction
Neuroblastoma is a pediatric malignancy that typically occurs in younger children. Arising from the developing sympathetic nervous system, it accounts for 8% of childhood cancers.Citation1,Citation2 At diagnosis, the tumors can be localized in the adrenal medulla or paraspinal sympathetic ganglia, or be widely metastatic. Neuroblastoma is the most common extracranial solid tumor in childhood, and it is responsible for 15% of pediatric cancer deaths.Citation3
Despite recent significant advances in understanding the genetic basis of tumor initiation and progression, neuroblastoma continues to be responsible for a disproportionate amount of childhood morbidity and mortality. Hence, tumors that present in children over 18 months of age can be lethal at the time of diagnosis, irrespective of aggressive multimodality therapy.Citation4,Citation5
Several intracellular signaling pathways have been demonstrated to play a key role in embryonal tumor biology, including growth factors controlling tumor proliferation, survival, differentiation, and metastasis.Citation6-Citation10 The phosphoinositide 3-kinase (PI3K) pathway has also been shown to play a crucial role in controlling cell proliferation, survival and motility/metastasis downstream of growth factor receptors and Ras.Citation11-Citation13 Glycogen synthase kinase 3 beta (GSK-3β), a ubiquitously expressed multifunctional serine/threonine kinase, is known to regulate a range of cellular functions, including differentiation, growth, proliferation, cell cycle progression, and apoptosis.Citation14-Citation16 GSK-3 is of interest in cancer, as it has been shown to promote apoptotic cell death in various cancers. There are two isoforms, GSK-3α and GSK-3β, with more than 90% similarity in sequence. Recent studies suggest a potential role for GSK3β inhibition in the treatment of neuroblastoma. In vitro studies using the B65 cell line showed SB415286-induced cell cycle arrest with kinase inhibition.Citation17 In Neuro-2A cells SB415286 caused decreased cell proliferation, G2/M cell cycle arrest, and induction of apoptosis.Citation17,Citation18 Furthermore, GSK-3 has been shown to promote DNA damage-induced apoptosis in neuroblastoma cells expressing wild-type p53.Citation19 However it is not known which isoform of GSK-3 regulates cancer cell proliferation. To date there are conflicting and contradictory reports of the role of GSK-3 isoforms in modulation of cell growth.Citation20,Citation21
Selective phosphorylation regulates the activity of both GSK-3 isoforms. GSK-3 is normally active in cells and predominantly regulated through the inhibition of its activity. Activation of GSK-3α and β is dependent upon the phosphorylation of residues Tyr279 and Tyr216 respectively. However, there is still a lack of evidence of the effects on growth, both in vitro and in vivo, by these isoforms.Citation20,Citation21 To further investigate the roles of GSK-3α and β inhibition as possible therapeutic avenues in the treatment of neuroblastoma, we studied the thiazole AR-A014418 (N-[4-methoxybenzyl]-N'-[5-nitro-1,3-thiazol-2-yl]urea) in neuroblastoma cell lines, to evaluate specificity and diversity. In this study, we show that AR-A014418 specifically inhibits phosphorylation of GSK-3α without inhibiting GSK-3β. Furthermore, we show that significant growth reduction is achieved by treatment with AR-A014418, without affecting GSK-3β phosphorylation, indicating that inactivation of GSK-3α is sufficient to inhibit neuroblastoma cell growth.
Results
AR-A014418 treatment suppresses neuroblastoma cell growth
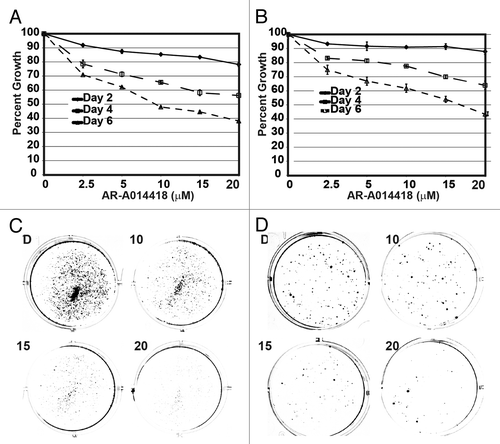
To determine if GSK-3 inhibition affected neuroblastoma cell proliferation, MTT assays were performed. As shown in , suppression of cellular growth in a dose- and time-dependent manner began on day 2 in both NGP () and SH-5Y-SY () neuroblastoma cells. The most significant inhibition was seen with a dose of 10 μM of AR-A014418. After 6 days of treatment, cells exposed to 5 μM of AR-A014418 or greater were all inhibited by ≥25% when compared to DMSO control treated cells. To confirm the growth suppression by AR-A014418, we carried out colony formation assay. As shown in , there is significant reduction in colony formation with increasing concentrations of AR-A014418.
Figure 1. Growth inhibition by GSK-3 inhibitor AR-A014418 in neuroblastoma cells. (A) NGP, (B) SH-5Y-SY cells were incubated with AR-A014418 for up to 6 days at various concentrations and cell viability was measured by both MTT assay and colony formation assay. Significant reduction in both number and size of the colonies were seen in AR-A014418 treatment (C and D).
AR-A014418-induced reduction of GSK-3α phosphorylation correlates with attenuated neuroendocrine marker expression
Neuroblastoma cells express high levels of neuroendocrine tumor markers, including achaete-scute complex-like1 (ASCL1) and chromogranin A (CgA), as a result of their origin from neural crest cells located in the adrenal medulla and sympathetic ganglia. Expression of CgA and ASCL1, a basic helix-loop-helix transcription factor, represents the neuroendocrine phenotype.Citation22
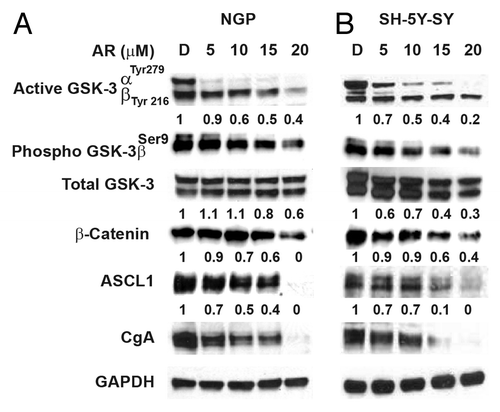
One of the targets for GSK-3 is β-catenin. GSK-3β destabilizes β-catenin. AR-A014418 treatment reduced β-catenin in NB cell lines (). In addition, we have observed a reduction in phosphorylated GSK-3β at ser9th position in both cell lines (). Earlier, we have shown that inactivation of GSK-3 by treatment with either GSK-3 inhibitor or by siRNA against GSK-3 resulted in a significant reduction in neuroendocrine tumor markers, ASCL1 and CgA.Citation23,Citation24 To demonstrate suppression of these neuroendocrine tumor markers by GSK-3 inhibitor, western blot analysis was performed. As seen in , neuroblastoma cells express high levels of both ASCL1 and CgA at baseline. Treatment with AR-A014418 resulted in a decrease in expression of both tumor markers in both cell lines, on day 2, with noticeable reduction starting at 10 μM (). This decreased expression correlated/associated with decreased phosphorylation of GSK-3α, with no change in total GSK-3 expression. The cell lines used in this study have different genetic backgrounds: NGP (gain of chromosome 17q, deletion of chromosome 1p, and amplification of N-myc), and SH-SY5Y (gain of 17q only). Advanced types of NB cancers all reportedly fall into these major genetic groups.Citation25
Figure 2. Phosphorylation of GSK-3α reduction is associated with decreased expression of NE markers. Western analysis showing that decreased phosphorylation of GSK-3 α at Tyr 279 compared to GSK-3β phosphorylation at Tyr216 by AR-A014418. In addition, there is a reduction in GSK-3β phosphorylation at ser9. This decrease in phosphorylation reduced the expression of β-catenin, ASCL1, and CgA in NGP (A) and SH-5Y-SY (B) cells. D, DMSO-treated control. GAPDH was used as a loading control.
AR-A014418 inhibits cellular proliferation via apoptosis
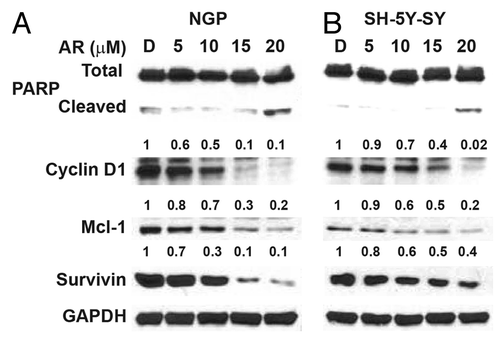
To identify the mechanism by which GSK-3 inhibition attenuates cellular proliferation, western blot analysis was performed. As seen in , NGP and SH-5Y-SY cells treated with AR-A014418 showed marginal expression of cleaved PARP, but decreased expression of cyclin D1, Mcl-1, and survivin. Alteration in apoptotic markers by AR-A014418 treatment suggested induction of apoptosis.
Figure 3. AR-A014418 attenuation of apoptosis inhibitor expression in NGP and SH-5Y-SY cells. Western blot analysis showed there is increase in cleaved PARP, a marker for apoptosis, This was associated with reduction in anti-apoptotic protein Mcl-1 and survivin. D, DMSO-treated control. GAPDH was used as loading control.
Continuous requirement of AR-A014418 treatment for growth suppression
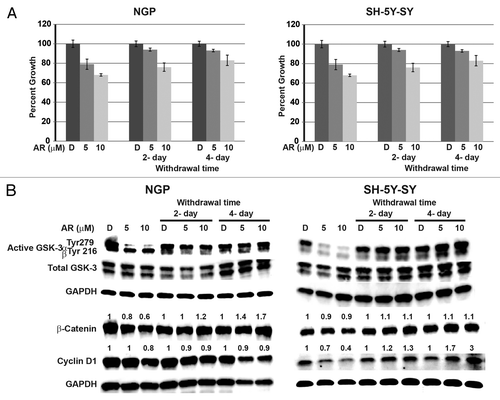
To determine the treatment duration of AR-A014418 in neuroblastoma growth suppression, NGP and SH-5Y-SY cells were treated with AR-A014418 at indicated concentrations for 4-day and then the media was changed to complete medium without AR-A014418 for up to four days. Every two days cellular proliferation was measured by MTT assay as well as cellular extracts were prepared and analyzed for active phosphorylated GSK-3α/β, total GSK-3α/β, and downstream target proteins such as β-catenin, cyclin D1 levels. As shown in , cellular proliferation was reduced in AR-A014418-treated cells in concentration-dependent manner. Interestingly, when the medium was changed to complete medium without AR-A014418, cellular proliferation was increased compared to the treatment control. Importantly, the presence of active phosphorylated GSK-3α at Tyr279 was observed as early as two days after withdrawal of the AR-A014418. Also increase in β-catenin and cyclin D1 were also observed after 2 d of withdrawal in both cell lines (). At present, it is not known whether the increase in β-catenin is due to the change in GSK-3 phosphorylation. The observation of decrease in β-catenin and change in GSK-3 phosphorylation is an associated effect in AR-A014418 treated NB cells. Further investigation is needed. However, it is speculated that inhibition of GSK-3 phosphorylation is required for the growth suppression of NGP and SH-5Y-SY cells. Furthermore, AR-A014418 treatment is required all of the time.
Figure 4. Continuous treatment of AR-A014418 is required for growth suppression of NGP and SH-5Y-SY cells. Cells were treated with AR-A014418 for 4 d and then media was changed to complete media without AR-A014418 for up to 4 d. Cell viability was measured by MTT assay (A) and western blot analysis for the levels of GSK-3 phosphorylation, β-catenin, and cyclin D1 (B). Significant reduction in cellular proliferation was observed with AR-A014418 treatment whereas cellular growth is increased compared to the treatment when the medium was changed to regular medium without AR-A014418 (A) in both cell lines. Importantly, reversal of phosphorylation of GSK-3α protein and increase in β-catenin and cyclin D1 protein was seen in withdrawal condition (B).
Discussion
Neuroblastoma is one of the most common pediatric malignancies, and is responsible for a significant portion of pediatric cancer deaths. Surgical resection is a major aspect of the therapeutic regimen, especially for the indolent, low risk and intermediate risk tumors, which occur in adolescents and adults. For high-risk, metastatic and recurrent tumors, multimodality therapy with high intensity chemotherapy, surgical resection, and radiation prove ineffective. In recent years, research has focused on the GSK-3 signaling pathway, which has been noted to play a role in other neurodegenerative diseases, namely Alzheimer disease.Citation26 Previous studies have examined the potential benefits from either the inhibition or activation of this molecular target.
Targeting GSK-3 provides a more specific inhibition than targeting its upstream precursor, Akt, taking into consideration the multiple downstream effects of Akt.Citation22 GSK-3 is constitutively active and phosphorylated at a single tyrosine residue (Tyr279 in the α-isoform and Tyr216 in the β-isoform).Citation27 Previous studies have commonly used GSK-3 inhibitors as a method of GSK-3β inhibition; however, their nonspecific nature and extensive side effects warrant investigation of other therapeutic options.Citation27
In this study, we show both dose- and time-dependent attenuation of growth in two neuroblastoma cell lines, NGP and SH-5Y-SY, with an associated decreased expression of neuroendocrine tumor markers in these cell lines. At present, the mechanism of reduction in ASCL1 after GSK-3α reduction is not clear. However, we have observed that siRNA against ASCL1 in medullary thyroid cancer cells resulted in reduction in CgA.Citation28 One can speculate that down regulation of β-catenin may cause reduction in the level of ASCL1, since ASCL1 is a possible downstream target of Wnt/β-catenin signaling. On the other hand, GSK-3α reduction may alter HES-1 activity which may downregulate ASCL1. Importantly, AR-A014418 decreased the phosphorylation of the GSK-3α subunit at the Tyr-279 position; attenuating the activation of GSK-3. We also observed that specific GSK-3α inhibition is associated with reduction in the neuroendocrine markers. This study is novel in demonstrating a possible anti-cancer role for the inhibition of GSK-3α.
Materials and Methods
Cell culture
Maintenance of the human NGP and SH-5Y-SY neuroblastoma cell lines, was accomplished by growth of the cells in RPMI1640 medium (Gibco-BRL), supplemented with 10% fetal bovine serum (Sigma Cell Culture), 100 IU/mL penicillin, and 100 μg/mL streptomycin (Gibco-BRL) in a humidified atmosphere of 5% CO2 in air at 37 °C. AR-A014418 was dissolved in dimethyl sulfoxide (DMSO) (Sigma Cell Culture) to prepare stock solutions of 50 μM. An equivalent volume of DMSO alone served as a negative control.
Cell proliferation assay
Neuroblastoma cell proliferation was measured using a 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay according to manufacturer instructions. Cells were plated in quadruplicate at a concentration of 25 × 104 cells/mL in 24-well plates and incubated overnight. The following day (day 0), the cells were treated with AR-A014418 (0–20 μM) and incubated for up to 6 d, with the medium being changed every 2 d. Cell growth was assessed on days 2, 4, and 6. The assay was repeated three times, and one representative plot is shown.
Colony formation assay
A colony formation assay was performed to confirm the growth inhibition by AR-A014418 treatment. Cells were plated into 6-well plates and treated with various concentrations of AR-A014418 for 4 d. Then media were replaced with regular media without drug for 1 wk. After 1 wk the cells were fixed with crystal violet staining and photographs were taken using Molecular ImagerTM ChemiDoc XRS+ imager with image lab software (Bio-Rad).
Western analysis
Cells were plated on 10 cm tissue culture plates (BD Biosciences) at a concentration of 1 × 106 cells/mL. After attachment, the cells were treated with 0–20 μM doses of AR-A014418. On day 2, the cells were washed with 1× phosphate-buffered saline (PBS) (Invitrogen), and then harvested the cells by centrifugation at 2000 rpm for 10 minutes at 4 °C. The pellets were lysed according to a previously described protocol.Citation29 Total cellular protein concentrations were determined by a 660 nM protein assay (Pierce). Denatured cellular extracts (20 μg) were boiled with equal volumes (1:1) of loading dye (2% sodium dodecyl sulfate, 20% glycerol, 0.1mol/L Tris 5, 13-mercapto-ethanol, 0.04% bromophenol blue) for 10 min and electrophoresed through 7.5%, 10%, and/or 12% Mini-Protean TGX gels (Bio-Rad Laboratories). The proteins were then transferred onto nitrocellulose membranes (Bio-Rad Laboratories), blocked in milk solution (5% dry skim milk and 0.05% Tween-20 in PBS) for 1 h, and then primary antibodies were applied.
The antibody dilutions were as follows: 1:1000 for active phosphorylated GSK-3αY279/βY216, phosphorylated GSK-3βser9, cyclin D1 (Abcam), Mcl1 (Cell Signaling) and chromogranin A (Zymed Laboratories); 1:2000 for ASCL1 (BD Pharmingen), total regular GSK-3α/β (Santa Cruz Biotechnologies), and PARP (Cell Signaling); 1:10 000 for GAPDH and 1:500 for β-catenin (Santa Cruz Biotechnologies); and 1:2000 for survivin and cyclin D1 (Cell Signaling). Membranes were incubated overnight at 4 °C, and then washed in wash buffer (0.05% Tween-20 in PBS) the following day. HRP-conjugated goat anti-rabbit IgG antibody was then applied in a dilution of 1:3000 for CgA, GAPDH, active GSK-3α/β, and MCL1. A dilution of 1:5000 goat anti-mouse IgG linked with HRP secondary antibody was used for ASCL1 and PARP, and 1:3000 for total GSK-3α/β. After incubating for 1 h on a shaker at room temperature, the membranes were washed 3–5 times with wash buffer. The proteins were then visualized using SuperSignal West Femto (Pierce) Immun-Star (Bio-Rad) or Dura (Pierce) kits according to manufacturer instructions.
Densitometry analysis
After visualizing proteins bands on the membrane, the band intensities were quantified using ImageJ software (National Institutes of Health). The relative number for the band intensities were calculated by normalized against GAPDH.
| Abbreviations: | ||
| GSK | = | glycogen synthase kinase |
| NB | = | neuroblastoma |
| NE | = | neuroendocrine |
| ASCL1 | = | achaete scute complex-like1 |
| CgA | = | chromogranin A |
| MTT | = | 3-(4,5-Dimethylthiazole-2-yl)-2,5-diphenl tetrazolium bromide |
| PARP | = | poly(ADP) ribose polymerase |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
This research was supported in part by National Institutes of Health Grant R01 CA121115 (H.C.), the American Cancer Society MEN Research Scholars Grant (H.C.), American Cancer Society MEN2 Professorship (H.C.), National Institutes of Health/National Cancer Institute Supplemental grant R01-CA12115-02S1 (H.C., Y.C.), and funding from “The Medical College of Wisconsin Dean’s Program Development Funding”.
References
- Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010; 362:2202 - 11; http://dx.doi.org/10.1056/NEJMra0804577; PMID: 20558371
- Zage PE, Nolo R, Fang W, Stewart J, Garcia-Manero G, Zweidler-McKay PA. Notch pathway activation induces neuroblastoma tumor cell growth arrest. Pediatr Blood Cancer 2012; 58:682 - 9; http://dx.doi.org/10.1002/pbc.23202; PMID: 21744479
- Wojtalla A, Salm F, Christiansen DG, Cremona T, Cwiek P, Shalaby T, Gross N, Grotzer MA, Arcaro A. Novel agents targeting the IGF-1R/PI3K pathway impair cell proliferation and survival in subsets of medulloblastoma and neuroblastoma. PLoS One 2012; 7:e47109; http://dx.doi.org/10.1371/journal.pone.0047109; PMID: 23056595
- Franks LM, Bollen A, Seeger RC, Stram DO, Matthay KK. Neuroblastoma in adults and adolescents: an indolent course with poor survival. Cancer 1997; 79:2028 - 35; http://dx.doi.org/10.1002/(SICI)1097-0142(19970515)79:10<2028::AID-CNCR26>3.0.CO;2-V; PMID: 9149032
- Kushner BH, Kramer K, LaQuaglia MP, Modak S, Cheung NK. Neuroblastoma in adolescents and adults: the Memorial Sloan-Kettering experience. Med Pediatr Oncol 2003; 41:508 - 15; http://dx.doi.org/10.1002/mpo.10273; PMID: 14595707
- Cianfarani S, Rossi P. Neuroblastoma and insulin-like growth factor system. New insights and clinical perspectives. Eur J Pediatr 1997; 156:256 - 61; http://dx.doi.org/10.1007/s004310050595; PMID: 9128806
- Gilbertson R. Paediatric embryonic brain tumours. biological and clinical relevance of molecular genetic abnormalities. Eur J Cancer 2002; 38:675 - 85; http://dx.doi.org/10.1016/S0959-8049(01)00315-X; PMID: 11916550
- Ho R, Eggert A, Hishiki T, Minturn JE, Ikegaki N, Foster P, Camoratto AM, Evans AE, Brodeur GM. Resistance to chemotherapy mediated by TrkB in neuroblastomas. Cancer Res 2002; 62:6462 - 6; PMID: 12438236
- Rikhof B, de Jong S, Suurmeijer AJ, Meijer C, van der Graaf WT. The insulin-like growth factor system and sarcomas. J Pathol 2009; 217:469 - 82; http://dx.doi.org/10.1002/path.2499; PMID: 19148905
- Wechsler-Reya R, Scott MP. The developmental biology of brain tumors. Annu Rev Neurosci 2001; 24:385 - 428; http://dx.doi.org/10.1146/annurev.neuro.24.1.385; PMID: 11283316
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD. Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol 2001; 17:615 - 75; http://dx.doi.org/10.1146/annurev.cellbio.17.1.615; PMID: 11687500
- Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol 2010; 11:329 - 41; http://dx.doi.org/10.1038/nrm2882; PMID: 20379207
- Vanhaesebroeck B, Vogt PK, Rommel C. PI3K: from the bench to the clinic and back. Curr Top Microbiol Immunol 2010; 347:1 - 19; http://dx.doi.org/10.1007/82_2010_65; PMID: 20549473
- Carter Y, Jaskula-Sztul R, Chen H, Mazeh H. Signaling pathways as specific pharmacologic targets for neuroendocrine tumor therapy: RET, PI3K, MEK, growth factors, and Notch. Neuroendocrinology 2013; 97:57 - 66; http://dx.doi.org/10.1159/000335136; PMID: 22343668
- Luo J. Glycogen synthase kinase 3beta (GSK3beta) in tumorigenesis and cancer chemotherapy. Cancer Lett 2009; 273:194 - 200; http://dx.doi.org/10.1016/j.canlet.2008.05.045; PMID: 18606491
- Martinez A. Preclinical efficacy on GSK-3 inhibitors: towards a future generation of powerful drugs. Med Res Rev 2008; 28:773 - 96; http://dx.doi.org/10.1002/med.20119; PMID: 18271054
- Dickey A, Schleicher S, Leahy K, Hu R, Hallahan D, Thotala DK. GSK-3β inhibition promotes cell death, apoptosis, and in vivo tumor growth delay in neuroblastoma Neuro-2A cell line. J Neurooncol 2011; 104:145 - 53; http://dx.doi.org/10.1007/s11060-010-0491-3; PMID: 21161565
- Pizarro JG, Folch J, Esparza JL, Jordan J, Pallàs M, Camins A. A molecular study of pathways involved in the inhibition of cell proliferation in neuroblastoma B65 cells by the GSK-3 inhibitors lithium and SB-415286. J Cell Mol Med 2009; 13:9B 3906 - 17; http://dx.doi.org/10.1111/j.1582-4934.2008.00389.x; PMID: 18624766
- Ngok-Ngam P, Watcharasit P, Thiantanawat A, Satayavivad J. Pharmacological inhibition of GSK3 attenuates DNA damage-induced apoptosis via reduction of p53 mitochondrial translocation and Bax oligomerization in neuroblastoma SH-SY5Y cells. Cell Mol Biol Lett 2013; 18:58 - 74; http://dx.doi.org/10.2478/s11658-012-0039-y; PMID: 23161404
- Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res 2005; 65:2076 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-04-3642; PMID: 15781615
- Wilson W 3rd, Baldwin AS. Maintenance of constitutive IkappaB kinase activity by glycogen synthase kinase-3alpha/beta in pancreatic cancer. Cancer Res 2008; 68:8156 - 63; http://dx.doi.org/10.1158/0008-5472.CAN-08-1061; PMID: 18829575
- Pinchot SN, Adler JT, Luo Y, Ju J, Li W, Shen B, Kunnimalaiyaan M, Chen H. Tautomycin suppresses growth and neuroendocrine hormone markers in carcinoid cells through activation of the Raf-1 pathway. Am J Surg 2009; 197:313 - 9; http://dx.doi.org/10.1016/j.amjsurg.2008.10.007; PMID: 19245907
- Adler JT, Cook M, Luo Y, Pitt SC, Ju J, Li W, Shen B, Kunnimalaiyaan M, Chen H. Tautomycetin and tautomycin suppress the growth of medullary thyroid cancer cells via inhibition of glycogen synthase kinase-3beta. Mol Cancer Ther 2009; 8:914 - 20; http://dx.doi.org/10.1158/1535-7163.MCT-08-0712; PMID: 19372564
- Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Inactivation of glycogen synthase kinase-3beta, a downstream target of the raf-1 pathway, is associated with growth suppression in medullary thyroid cancer cells. Mol Cancer Ther 2007; 6:1151 - 8; http://dx.doi.org/10.1158/1535-7163.MCT-06-0665; PMID: 17363508
- Lastowska M, Cullinane C, Variend S, Cotterill S, Bown N, O’Neill S, Mazzocco K, Roberts P, Nicholson J, Ellershaw C, et al, United Kingdom Children Cancer Study Group and the United Kingdom Cancer Cytogenetics Group. Comprehensive genetic and histopathologic study reveals three types of neuroblastoma tumors. J Clin Oncol 2001; 19:3080 - 90; PMID: 11408505
- Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 2004; 3:479 - 87; http://dx.doi.org/10.1038/nrd1415; PMID: 15173837
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J 1993; 12:803 - 8; PMID: 8382613
- Kunnimalaiyaan M, Vaccaro AM, Ndiaye MA, Chen H. Overexpression of the NOTCH1 intracellular domain inhibits cell proliferation and alters the neuroendocrine phenotype of medullary thyroid cancer cells. J Biol Chem 2006; 281:39819 - 30; http://dx.doi.org/10.1074/jbc.M603578200; PMID: 17090547
- Greenblatt DY, Cayo MA, Adler JT, Ning L, Haymart MR, Kunnimalaiyaan M, Chen H. Valproic acid activates Notch1 signaling and induces apoptosis in medullary thyroid cancer cells. Ann Surg 2008; 247:1036 - 40; http://dx.doi.org/10.1097/SLA.0b013e3181758d0e; PMID: 18520232