
Abstract
Use of the inhibitor of ALK fusion onco-protein, crizotinib (PF02341066), has achieved impressive clinical efficacy in patients with ALK-positive non-small cell lung cancer. Nevertheless, acquired resistance to this drug occurs inevitably in approximately a year, limiting the therapeutic benefits of this novel targeted therapy. In this study, we found that autophagy was induced in crizonitib-resistant lung cancer cells and contributed to drug resistance. We observed that ALK was downregulated in the crizotinib-resistant lung cancer cell line, H3122CR-1, and this was causally associated with autophagy induction. The degree of crizotinib resistance correlated with autophagic activity. Activation of autophagy in crizotinib-resistant H3122CR-1 cells involved alteration of the Akt/mTOR signaling pathway. Furthermore, we demonstrated that chloroquine, an inhibitor of autophagy, could restore sensitivity of H3122CR-1 to crizotinib and enhance its efficacy against drug-resistant lung cancer. Thus, modulating autophagy may be worth exploring as a new strategy to overcome acquired crizonitib resistance in ALK-positive lung cancer.
Keywords: :
Introduction
Lung cancer is the most common lethal malignant disease; among all of lung cancer, non-small cell lung cancer (NSCLC) accounts for more than 85% with a majority of them diagnosed at an advanced stage. Targeted therapy represents an important advance in the treatment of lung cancer. Crizotinib (PF02341066) is the first anaplastic lymphoma kinase (ALK)-targeted drug evaluated clinically, and was recently granted accelerated approval for treating patients with NSCLC harboring the echinoderm microtubule-associated protein 4 (EML4)-ALK fusion. Targeting this oncogenic fusion protein not only has achieved impressive clinical efficacy but also enhanced the life quality of patients because of fewer side effects.Citation1-Citation4 Indeed, many patients respond well to initial treatment with crizotinib; nevertheless, ALK+ lung cancer invariably develops resistance to this drug later.Citation5 The median duration of clinical benefit is approximately 10~11 mo. Thus, occurrence of drug resistance limits lung cancer patients from further benefiting from this treatment. It has been known that acquired resistance to crizotinib may result from secondary mutations within the ALK kinase domain, amplification of the ELM4-ALK fusion gene, from activation of the paracrine receptor such as the epidermal growth factor receptor (EGFR)-mediated activation of HER family signaling.Citation4-Citation11 Here, we report a previously unrecognized mechanism of crizotinib resistance: induction of autophagy, a conserved response to various stress and a process of self-digestion of cytoplasm and organelles through which cellular components are recycled for energy utilization, contributes to resistance to crizotinib therapy in ALK+ lung cancer. We found that autophagy was activated in crizotinib-resistant lung cancer cells, and downregulation of ALK protein was causally associated with induction of cyto-protective autophagy. Furthermore, we demonstrated that inhibiting autophagy with chloroquine (CQ) could restore sensitivity of drug-resistant cells to crizotinib and enhance its anticancer efficacy against drug-resistant lung cancer. These observations provide a rationale for targeting autophagy as an approach to modulating crizotinib resistance in lung cancer treatment.
Results
Autophagy is activated in crizotinib-resistant lung cancer cells
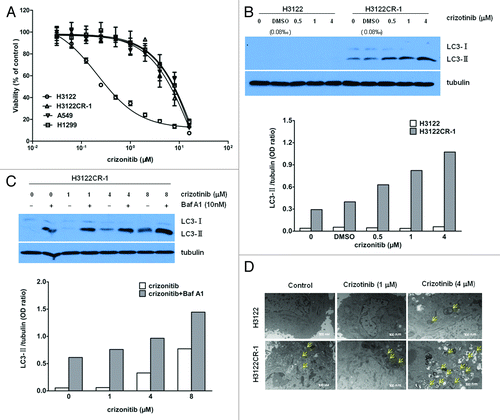
To determine the possible involvement of autophagy in crizotinib resistance, we first compared the autophagy levels in the drug sensitive and drug resistant lung cancer cell lines, H3122 and H3122CR-1, using LC3-II protein as an indicator of autophagy. H3122CR-1 cells were derived from H3122 cells by continuous culture in the crizotinib-containing medium,Citation12 and showed a ~60-fold resistance to the drug as compared with H3122 cells (; IC50: 13.5 µM vs. 0.2 µM). This fold resistance was similar to that seen in the ALK-negative lung cancer cell lines, A549 and H1299 (). shows that in the sensitive cell line H3122, no LC3-II was detected; in contrast, a conspicuous basal level of autophagy was observed in the drug resistant cell line H3122CR-1. Treatment with crizotinib caused a dose-dependent activation of autophagy in H3122CR-1 cells, as evidenced by increases in the amount of LC3-II, but not in H3122 cells (). Activation of autophagy in the crizotinib-resistant cells was verified by autophagic flux assay, which showed a further elevation of LC3-II in H3122CR-1 cells co-treated with crizotinib and bafilomycin A1, an inhibitor of lysosomal protease (), and by an increase in the number of autophagosomes as observed by electron microscopy ().
Figure 1. Autophagy is activated in crizotinib-resistant lung cancer cells. (A) Cells were treated with the indicated concentrations of crizotinib or vehicle for 72 h, and cell viability was assessed using MTT. (B) H3122 and H3122CR-1 cells were treated with the indicated concentrations of crizonitib or vehicle for 24 h; the levels of LC3 were determined by western blot. Tubulin was used as a loading control. (C) H3122CR-1 cells were treated with the indicated concentrations of crizonitib or vehicle for 24 h in the absence or presence of bafilomycin A1 (Baf A1; 10 nM). The levels of LC3 were examined by western blot. (D) Electron microscopic examination of the H3122 and H3122CR-1 cells treated with crizonitib or vehicle for 24 h. The results shown are the representative of three identical experiments.
Downregulation of ALK is associated with induction of autophagy in crizotinib-resistant lung cancer cells
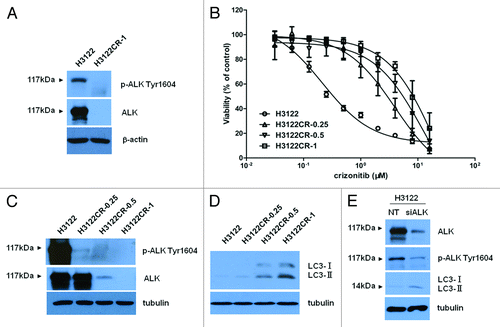
To understand how autophagy is activated in crizotinib-resistant cancer cells, we examined the levels of phospho-ALK and total ALK in the drug-resistant and drug-sensitive cells. shows that H3122 cells were abundant in ALK and phospho-ALK, as determined by western blot; notably, the levels of both phospho-ALK and total ALK were barely detectable in H3122CR-1 cells (). To ascertain that there is an association between the reduction of ALK and crizotinib resistance, we compared the levels of phospho-ALK and total ALK in the H3122 cells with varying degrees of crizotinib resistance. shows that in the drug resistant H3122 cells with ~20- to 60-fold resistance to crizotinib (), the level of ALK was conversely correlated with the degree of crizotinib resistance (). Reduction of phospho-ALK was also observed in those crizotinib-resistant cells, along with an increasingly upregulated autophagy (). To determine whether or not a causal relationship exists between the downregulation of ALK and induction of autophagy, we knocked down the expression of ALK in the drug-sensitive H3122 cells using an ALK-targeted siRNA, followed by an examination of autophagy. shows that in the H3122 cells with knockdown of ALK expression, autophagy was induced. These results suggest that the downregulation of ALK that occurs in the crizotinib-resistant lung cancer cells may account for the initial activation of autophagy.
Figure 2. Downregulation of ALK is associated with induction of autophagy in crizotinib-resistant lung cancer cells. (A) H3122 and H3122CR-1 cells were cultured for 24 h, and the levels of total ALK and phospho-ALK were detected by western blot. β-actin was used as a loading control. (B) Cells were treated with the indicated concentrations of crizotinib or vehicle for 72 h, and cell viability was determined by MTT. (C and D) Parental H3122 cell line and the crizotinib-resistant H3122 cell lines, H3122CR-0.25, H3122CR-0.5, and H3122CR-1, were cultured for 24 h and the levels of ALK, phospho-ALK, LC3, and tubulin were determined by western blot. (E) H3122 was transfected with an ALK-targeted siRNA or a non-targeting siRNA. Seventy-two hours later, the levels of ALK, phospho-ALK, LC3, and tubulin were examined by western blot. The results shown are the representative of three identical experiments.
Akt/mTOR pathway is involved in autophagy activation in crizotinib-resistant lung cancer cells
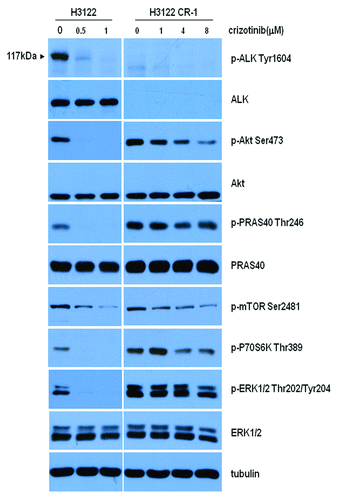
We next wanted to explore the mechanism of autophagy induction in crizotinib-resistant cancer cells. It is known that the aberrant expression of the ALK fusion oncogene leads to activation of several canonical signaling pathways including PI3K/Akt/mTOR and Ras/MEK/ERK cascades,Citation13-Citation17 which were reported to participate in the regulation of autophagy.Citation18-Citation20 Thus, we analyzed and compared the activity of Akt, mTOR, and MEK/ERK pathways in H3122 and H3122CR-1 cells. shows that in the H3122 cells, not only the activity of ALK but also ERK, PRAS40 and Akt/mTOR/S6 kinase signaling pathways were markedly inhibited by crizotinib at concentration of 0.5 μM. Although H3122CR-1 cells showed a dramatic downregulation of ALK and phospho-ALK, the activity of ERK, PRAS40, and Akt/mTOR/S6 kinase pathways were upregulated (). Crizotinib showed inhibitory effects on the activity of PRAS40 and Akt/mTOR/S6 kinase at 4 µM, but had no effects on ERK activity (). As inhibiting Akt is known to induce autophagy, these results suggest that autophagy activation observed in the drug-resistant cancer cells following crizotinib treatment may result from Akt inhibition.
Figure 3. Akt/mTOR pathway is involved in autophagy activation in crizotinib-resistant lung cancer cells. H3122 and H3122CR-1 cells were treated with the indicated concentrations of crizonitib or vehicle for 2 h. The levels of phospho-ALK, phospho-Akt, phospho-PRAS40, phospho-mTOR, phospho-P70S6K, phospho-ERK, ALK, Akt, PRAS40, ERK, and tubulin were determined by western blot.
Inhibiting autophagy increases sensitivity of drug-resistant lung cancer cells to crizotinib
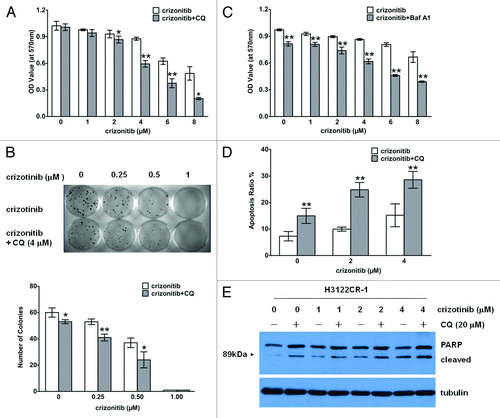
Because induction of autophagy was observed in drug resistant H3122CR-1 cells (), we next wanted to know what role that autophagy may play in altering cellular response to crizotinib treatment. Using chloroquine (CQ), a late-stage autophagy inhibitor, we demonstrated that sensitivity of the drug-resistant H3122CR-1 cells to crizotinib was significantly increased in the presence of CQ, as determined by MTT () and clonogenic assays (). Similarly, bafilomycin A1, another inhibitor of the late stage autophagy, could also sensitize H3122CR-1 cells to crizonitib (). Furthermore, we showed that combined treatment of H3122CR-1 cells with crizotinib and CQ caused greater apoptosis than treatment with crizotinib alone, as evidenced by increases in the percentage of the cells with Annexin V staining () and in the amount of cleaved PARP (). These results suggest that inhibition of autophagy can partially restore the sensitivity of drug-resistant cancer cells to crizotinib.
Figure 4. Inhibiting autophagy increases sensitivity of drug-resistant lung cancer cells to crizotinib in vitro. (A) H3122CR-1 cells were treated with crizonitib at the indicated concentrations or vehicle in the absence or presence of 20 μM of chloroquine (CQ) for 72 h. At the end of treatments, the cell viability was measured using MTT assay. (B) H3122CR-1 cells were plated in 35-mm tissue culture dishes (100 cells per dish) and treated with vehicle or crizonitib at the indicated concentration in the presence or absence of 4 μM of CQ for 10 d. At the end of treatment, cells were fixed in 25% methanol and stained with 1% methylene blue in 50% methanol. Colonies were counted following staining. (C) H3122CR-1 cells were treated with vehicle or crizonitib at the indicated concentrations in the absence or presence of 10 nM of Baf A1 for 72 h. At the end of treatment, the cell viability was measured by MTT assay. (D and E) H3122CR-1 cells were treated with vehicle or crizonitib at the indicated concentrations in the absence or presence of 20 μM of chloroquine (CQ) for 48 h. At the end of treatment, the proportion of apoptotic cells was measured by flow cytometric analysis of Annexin V and 7-aminoactinomycin D staining (D); the levels of cleaved PARP were examined by western blot. Tubulin was used as a loading control (E). Bars are mean ± SD from three identical experiments. *P < 0.05, **P < 0.01.
In vivo sensitizing effect of chloroquine on crizotinib-resistant lung cancer
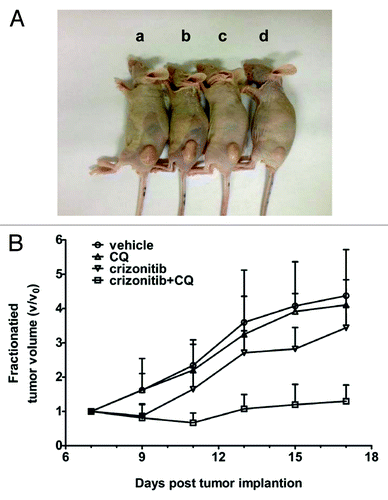
The in vitro sensitizing effect of CQ on crizotinib-resistant cancer cells could be recapitulated in vivo. In a mouse xenograft model in which the drug resistant H3122CR-1 cells were injected into in the hind flanks of the animals, we observed that crizotinib (100 mg/kg, oral gavage, qd) alone did not effectively inhibit tumor growth; however, combination of crizotinib with CQ (50 mg/kg, i.p., qd) significantly suppressed the growth of the drug-resistant tumors, as compared with the vehicle treatment or crizotinib alone (). These in vivo observations provide further support for the role of autophagy in cancer cell resistance to crizotinib.
Figure 5. Combination treatment with crizotinib and CQ effectively inhibits the growth of H3122CR-1 tumor in vivo. (A) CD-1 nude mice (6–7 wk old) were inoculated with H3122CR-1 cells (4 × 106 cells/mouse) subcutaneously. When tumors reached an average volume of ~120 mm3 (7 d after tumor implantation), the mice were randomly divided in groups (5 mice per group) and treated for 10 d with: (a) vehicle (DMSO, oral gavage, qd); (b) chloroquine (50mg/kg/day, intra-peritoneal injection, qd); (c) crizotinib (100 mg/kg/day, oral gavage, qd); (d) combination of chloroquine and crizotinib. Picture was taken on day 17. (B) Tumor sizes were measured every two days with calipers and the tumor volumes (mm3) were calculated using the following formula: length × width2 × 0.5. Each point represents mean tumor volume (V) normalized to the initial volume (V0); bars, SD. By-day comparisons show that: (1) crizotinib + CQ vs. vehicle control or CQ from day 11 on, P < 0.01; (2) crizotinib + CQ vs. crizotinib from day 11 to day 15, P < 0.05, and on day 17, P < 0.01. There is no statistic difference between crizotinib alone and CQ or vehicle control.
Discussion
Acquired drug resistance constitutes a major hurdle that limits the therapeutic benefits of crizotinib, the first available ALK inhibitor in the clinic, for patients with ALK positive NSCLC. Several mechanisms of crizotinib resistance have been reported, including second mutation or amplification of the ALK fusion geneCitation5-Citation7,Citation9 and activation of compensatory signaling pathways such as activation of EGFR.Citation5,Citation7,Citation8 Therefore, approaches to circumventing crizotinib resistance are of immensely importance. Combination of crizotinib with gefitinib, a small molecule inhibitor of EGFR tyrosine kinase, was tested and showed significant growth-inhibitory effects against those drug-resistant tumor cells only containing enhanced ERBB signalings without a secondary ALK mutation or ALK gene amplification; however, this combined treatment failed to completely reverse crizotinib resistance,Citation5 suggesting that there might exist other mechanisms that are possibly involved in cellular resistance to this drug. To better understand how cancer cells alter sensitivity to crizotinib, we explored the role of autophagy, a critical cellular process that determines cell death or survival. We found that autophagy level is upregulated in crizotinib-resistant lung cancer cells (), and activation of autophagy contributes to acquired drug resistance. Inhibitors of autophagy such as CQ can restore crizotinib sensitivity to the drug resistant cells both in vitro and in vivo ( and ).
The crizotinib-resistant lung cancer cell line, H3122CR-1, shows a downregulation of ALK and phospho-ALK with activation of autophagy (). Although we demonstrate a causal relationship between downregulation of ALK and upregulation of autophagy, the precise mechanisms by which ALK affects autophagy remains unclear. It is likely that the Akt/mTOR pathway links ALK activity to autophagy induction, as altered expression of ALK is known to activate several signaling pathways including PI3K/Akt/mTOR. Loss or downregulation of ALK has also been observed in other crizotinib-resistant cancer cell lines.Citation5,Citation12 The mechanism by which ALK is lost or downregulated is not clear. In addition, although crizotinib-induced inhibition of Akt was observed in both of H3122 and H3122CR-1 cells (), activation of autophagy was only observed in the drug-resistant cells (). This may be because that the sensitive cancer cells were dying when subjected to crizotinib-induced inhibition of Akt but the resistant cancer cells, which have high expression and activity of Akt, remain alive and activate autophagy as a survival tactic.
The role of autophagy in cellular resistance to a variety of therapeutic interventions has been appreciated in recent years, and targeting autophagy has been considered a new approach to overcoming therapeutic resistance.Citation21 However, inhibiting autophagy at different stages may yield discrepant results. For example, it was shown that inhibiting autophagy at an early stage using 3-methyladenine or small interfering RNA against Atg5 decreases the imatinib-induced cytotoxicity in human malignant glioma cells while suppressing autophagy at a late stage with bafilomycin A1 or RTA 203 enhanced imatinib-induced cytotoxicity.Citation22 Also, pretreatment with 3-methyladenine suppresses the cell death-promoting effect of CQ in PC3-shAKT123 cells.Citation23 Consistent with those observations, we found that silencing of Atg5 expression does not increase crizotinib-induced cytotoxicity in H3122CR-1 cells (data not shown); however, CQ, which inhibits autophagy at a late stage, can sensitize drug resistant cells to crizotinib (). We realize that in addition to inhibition of autophagy, CQ has other pharmacologic effect. Thus, to confirm that inhibiting autophagy at late stage can enhance the cytotoxicity of crizotinib, we also used bafilomycin A1, another agent that inhibits autophagy at a late stage. Bafilomycin A1 shows similar sensitizing effect to CQ in crizotinib-resistant H3122CR-1 cells (). Promotion of apoptosis in drug resistant cells may account for the sensitizing effect caused by autophagy inhibition, as shown in .
In conclusion, we have identified the activation of cyto-protective autophagy as a contributor to acquired crizonitib-resistance in ALK-positive lung cancer, along with the possible signaling pathway involved. Our results demonstrate that suppressing autophagy may be exploited as a means to sensitize drug resistant cancer to crizotinib, and autophagy inhibitors such as CQ have potential to be utilized as clinical modulators of crizotinib resistance.
Materials and Methods
Cell lines and culture
The human lung cancer cell lines, A549 and H1299, were obtained from American Type Culture Collection. H3122 and its crizotinib-resistant counterpart, H3122CR1, were a kindly provided by David Proia (Synta Pharmaceuticals Corp.). All cell lines were cultured in RPMI-1640 supplemented with 10% FBS, 100 units/mL of penicillin, and 100 mg/mL of streptomycin. All cells were cultured in a humidified atmosphere containing 95% O2/5% CO2 at 37 °C.
Antibodies and reagents
Antibody to ALK, phospho-ALK (Tyr1604), Akt, phospho-Akt (Ser473), PRAS40, phospho-PRAS40 (Thr246), ERK, phospho-ERK1/2 (Thr202/Tyr204), phospho-mTOR (Ser2481), phospho-p70S6K (Thr389), PRAP, α-tubulin, β-actin, were purchased from Cell Signaling Technology. Antibody to LC3B was purchased from Novus Biologicals. Atg-5 antibody was purchased from MBL International Corporation. Crizotinib were purchased from LC Laboratories. Chloroquine diphosphate salt (CQ), bafilomycin A1 and MTT were purchased from Sigma-Aldrich. Atg-5-targeted siRNA, ALK targeted siRNA and non-targeted siRNA were synthesized by Santa Cruz Biotechnology. Lipofectamine RNAi-max was purchased from Invitrogen. Fetal bovine serum was purchased from Invitrogen. RPMI-1640 was purchased from Thermo Scientific. Western blot reagents were obtained from Pierce Biotechnology.
Western blotting
Cells were lysed in the Solulyse-M mammalian protein extraction reagent (Genlantis) supplemented with a protease inhibitor cocktail (Roche) at room temperature for 5 min. Cell lysates were centrifuged at 14 000 × g for 10 min and the supernatants were collected. Proteins (10–40 µg) were resolved by SDS-PAGE and then transferred to polyvinylidenedifluoride (PVDF) membranes (Bio-Rad). The PVDF membranes were probed with primary antibodies in 3% BSA/TBST at 4 °C overnight, followed by incubation with secondary antibodies at room temperature for 1 h. The protein signals were detected by ECL method.
Electron microscopy
Cells were harvested by trypsinization, fixed in 2.5% glutaraldehyde/4% para-formaldehyde in 0.1 M cacodylate buffer, and post-fixed in 1% osmium tetroxide buffer. After dehydration in a graded series of acetone, the cells were embedded in spur resin. Ninety nm thin sections were cut on a Reichert Ultracut E microtome. Sectioned grids were stained with saturated solution of uranyl acetate and lead citrate. Sections were examined at 80 kV with a JEOL 1200EX transmission electron microscope.
Cellular viability assay
Cell viability was measured by MTT assay. Briefly, cells were plated in 96-well tissue culture plates at 6 × 103 cells per well and subjected to different treatments for 72 h. At the end of treatments, MTT was added to each well and incubated for another 4 h. The formazan product was dissolved in dimethyl sulfoxide (DMSO) and read at 570 nm on a Victor3 Multi-Label plate reader (PerkinElmer).
Clonogenic assay
Cells were seeded into 10-mm tissue culture dishes (100 cells per dish) and incubated in a humidified atmosphere containing 95% O2/5% CO2 at 37 °C. Twelve hours later, the cells were subjected to treatment with either crizonitib alone or combination of crizotinib and CQ at the indicated concentration and incubated for 10–14 d. At the end of the incubation period, cells were stained with 1% methylene blue in 50% methanol for 30 min, washed with water, and colonies counted.
Apoptosis assay
Apoptosis was measured by flow cytometric analysis of Annexin V and 7-aminoactinomycin D staining. Briefly, 100 μl of Guava Nexin reagent (Millipore) was added to 2 × 104~1 × 105 cells in 100 μl. After incubation with the reagent for 20 min at room temperature in the dark, the cells were analyzed by a Guava Easy-CyteTM Plus Flow Cytometry System (Millipore).
Animal experiments
Six to seven weeks old female CD-1 nude mice (ordered from Charles River Laboratories) were used for animal experiments. Briefly, crizotinib-resistant H3122CR-1 cells (4 × 106 cells/mouse) were subcutaneously implanted into the lower flank of each mouse. When tumors reached an average volume of ~120 mm3, tumor-bearing mice were randomly divided into four groups (5 mice per group): (1) vehicle control (100% DMSO, oral gavage, qd), (2) CQ (50mg/kg, i.p. injection. qd), (3) crizotinib (100mg/kg, oral gavage, qd), and (4) CQ plus crizotinib. The animals received the treatment for 10 d, and the tumor sizes were measured every 2 d. Tumor volumes (V: mm3) were calculated using the following formula: V = length × width2 × 0.5. The animal study was approved by the Institutional Animal Care and Use Committee of The Pennsylvania State University College of Medicine.
Statistical analysis
Quantitative data were presented as mean ± SD. IC50 values, which indicate the concentration of an agent that gives half-maximal inhibition, were calculated by fitting the experimental data with nonlinear regression using GraphPad Prism (GraphPad Software, Inc., San Diego, CA). Results of the clonogenic assay and apoptosis assay were analyzed by unpaired two-tailed Student t test. The tumor volume V was calculated (length × width2 × 0.5) and normalized to V0 (the volume at the onset of treatment). Statistical evaluations of tumor growth were performed by daily comparisons of the volumes and were analyzed by one-way ANOVA, followed by S-N-K test, using SAS version 9.2 software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We wish to thank Dr David A Proia (Synta Pharmaceuticals Corp) for providing us H3122 and H3122CR-1 cell lines.
Reference
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Jänne PA, Costa DB, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med 2010; 363:1693 - 703; http://dx.doi.org/10.1056/NEJMoa1006448; PMID: 20979469
- Ou SH, Bazhenova L, Camidge DR, Solomon BJ, Herman J, Kain T, Bang YJ, Kwak EL, Shaw AT, Salgia R, et al. Rapid and dramatic radiographic and clinical response to an ALK inhibitor (crizotinib, PF02341066) in an ALK translocation-positive patient with non-small cell lung cancer. J Thorac Oncol 2010; 5:2044 - 6; http://dx.doi.org/10.1097/JTO.0b013e318200f9ff; PMID: 21102269
- Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol 2011; 12:1004 - 12; http://dx.doi.org/10.1016/S1470-2045(11)70232-7; PMID: 21933749
- Shaw AT, Engelman JA. ALK in lung cancer: past, present, and future. J Clin Oncol 2013; 31:1105 - 11; http://dx.doi.org/10.1200/JCO.2012.44.5353; PMID: 23401436
- Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med 2012; 4:20ra17; http://dx.doi.org/10.1126/scitranslmed.3003316; PMID: 22277784
- Choi YL, Soda M, Yamashita Y, Ueno T, Takashima J, Nakajima T, Yatabe Y, Takeuchi K, Hamada T, Haruta H, et al, ALK Lung Cancer Study Group. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med 2010; 363:1734 - 9; http://dx.doi.org/10.1056/NEJMoa1007478; PMID: 20979473
- Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, Lathan C, Marcoux JP, Du J, Okuda K, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res 2011; 71:6051 - 60; http://dx.doi.org/10.1158/0008-5472.CAN-11-1340; PMID: 21791641
- Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, Kaneda H, Takezawa K, Kuwata K, Yamaguchi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res 2012; 18:6219 - 26; http://dx.doi.org/10.1158/1078-0432.CCR-12-0392; PMID: 22843788
- Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res 2012; 18:1472 - 82; http://dx.doi.org/10.1158/1078-0432.CCR-11-2906; PMID: 22235099
- Katayama R, Khan TM, Benes C, Lifshits E, Ebi H, Rivera VM, Shakespeare WC, Iafrate AJ, Engelman JA, Shaw AT. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A 2011; 108:7535 - 40; http://dx.doi.org/10.1073/pnas.1019559108; PMID: 21502504
- Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, Nakamura T, Matsumoto K, Soda M, Mano H, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res 2012; 18:3592 - 602; http://dx.doi.org/10.1158/1078-0432.CCR-11-2972; PMID: 22553343
- Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, Jiang Q, Xue L, Lovly CM, Jimenez JP, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov 2013; 3:430 - 43; http://dx.doi.org/10.1158/2159-8290.CD-12-0440; PMID: 23533265
- Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008; 14:4275 - 83; http://dx.doi.org/10.1158/1078-0432.CCR-08-0168; PMID: 18594010
- Takezawa K, Okamoto I, Nishio K, Jänne PA, Nakagawa K. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res 2011; 17:2140 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-10-2798; PMID: 21415216
- McDermott U, Iafrate AJ, Gray NS, Shioda T, Classon M, Maheswaran S, Zhou W, Choi HG, Smith SL, Dowell L, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res 2008; 68:3389 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-07-6186; PMID: 18451166
- Marzec M, Kasprzycka M, Liu X, El-Salem M, Halasa K, Raghunath PN, Bucki R, Wlodarski P, Wasik MA. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene 2007; 26:5606 - 14; http://dx.doi.org/10.1038/sj.onc.1210346; PMID: 17353907
- Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer 2013; 13:685 - 700; http://dx.doi.org/10.1038/nrc3580; PMID: 24060861
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528 - 39; http://dx.doi.org/10.1038/nrclinonc.2011.71; PMID: 21587219
- Cheng Y, Qiu F, Tashiro S, Onodera S, Ikejima T. ERK and JNK mediate TNFalpha-induced p53 activation in apoptotic and autophagic L929 cell death. Biochem Biophys Res Commun 2008; 376:483 - 8; http://dx.doi.org/10.1016/j.bbrc.2008.09.018; PMID: 18796294
- Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J 2010; 277:2 - 21; http://dx.doi.org/10.1111/j.1742-4658.2009.07366.x; PMID: 19843174
- Cheng Y, Ren X, Hait WN, Yang JM. Therapeutic targeting of autophagy in disease: biology and pharmacology. Pharmacol Rev 2013; 65:1162 - 97; http://dx.doi.org/10.1124/pr.112.007120; PMID: 23943849
- Shingu T, Fujiwara K, Bögler O, Akiyama Y, Moritake K, Shinojima N, Tamada Y, Yokoyama T, Kondo S. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity in human malignant glioma cells. Int J Cancer 2009; 124:1060 - 71; http://dx.doi.org/10.1002/ijc.24030; PMID: 19048625
- Degtyarev M, De Mazière A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol 2008; 183:101 - 16; http://dx.doi.org/10.1083/jcb.200801099; PMID: 18838554