
Abstract
Triple negative breast cancer (TNBC) accounts for 15–20% of breast carcinomas and represents one of the most aggressive forms of this disease. Basal and claudin-low are the two main molecular subtypes among TNBCs. We previously reported that deletion of Lfng in mouse mammary gland caused deregulated Notch activation and induced basal-like and claudin-low tumors with co-selection for Met amplification. In human breast cancers, the vast majority of basal tumors and a subset of claudin-low tumors show reduced Lfng expression. Elevated Met expression and activation is associated with basal as well as claudin-low subtypes. To examine roles of Met and Notch in TNBC cells, we established two cell lines that harbor Met amplification as well as Lfng deletion, and possess features of basal and claudin-low breast cancer subtypes. Pharmacological inhibition of Met not only suppressed cell growth, tumorsphere and colony formation, but also reversed epithelial-to-mesenchymal transition and inhibited cell migration in both cell lines. In contrast, inhibition of Notch signaling using a γ-secretase inhibitor (GSI) only suppressed colony formation. Interestingly, GSI had no effect as single agent, but exerted a synergistic effect with Met inhibitor, on cell growth in 2D culture. We found that inhibition of Met resulted in downregulation of Dll ligands and upregulation of Jagged ligands, leading to differential modulation of Notch signaling. Our results suggest that combination targeting of Met and Notch may prove beneficial for TNBC patients with Met overexpression and Notch hyperactivation.
Introduction
Triple negative breast cancer (TNBC) is a heterogeneous group of diseases that are defined on the basis of being negative for estrogen receptor α (ERα), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2).Citation1-Citation4 TNBC accounts for approximately 15–20% of breast carcinomas. It preferentially affects younger patients, is more prevalent among African-American women, and is often more aggressive than other types of breast cancer. At present no targeted treatment exists for TNBC and standard chemotherapy remains the only therapeutic modality. In recent years transcriptional profiling has been used to classify breast cancers into at least five molecular subtypes (basal, claudin-low, luminal A, luminal B, and HER2-enriched). Among these, the basal and claudin-low subtypes constitute the majority of TNBCs.Citation5 Basal subtype breast cancers express markers of myoepithelial/basal cells, and are thought to originate from mammary bi-potent or luminal progenitor cells, with which they share features.Citation6,Citation7 In contrast, the claudin-low subtype share more features with mammary stem cells and cells that have undergone epithelial-to-mesenchymal transition (EMT).Citation8-Citation11 These tumors may therefore originate from mammary stem cells.
The Notch signaling pathway has been shown to control stem cell self-renewal, cell fate specification and differentiation in the mammary gland.Citation12-Citation14 Notch activation has also been shown to regulate EMT in both developmental and pathological contexts.Citation15 Thus, aberrant Notch activation is most likely to play an important role in triple negative tumors, which exhibit characteristics of stem/progenitor cells and EMT. Indeed, Notch1 was shown to be highly expressed in breast cancer compared with normal tissue, and was segregated with basal-like breast cancer.Citation16-Citation18 Recent studies suggested a Jagged1-Notch1-CyclinD1 axis playing a key role in maintaining proliferation of TNBC, as opposed to other types of breast cancer.Citation19,Citation20 Notch3 signaling controls survival of hypoxic TNBC cells,Citation21 and Notch4 is involved in self-renewal of breast cancer stem cells.Citation22 A surgical pathology series of TNBC demonstrated Notch1 and Notch4 expression in tumor cells with predominantly nuclear and cytoplasmic location.Citation23 In addition, chromosomal rearrangements producing constitutively active versions of Notch1 or Notch2 were detected almost exclusively in TNBC cell lines and tumors, further supporting the notion that Notch pathway is pathogenetically relevant in TNBC.Citation24 As a result, Notch has emerged as a potential drug target for poor prognosis TNBC.
The Notch extracellular domain is heavily glycosylated. One of two main forms of glycosylation in this context, involves fucose linkage to Notch by Pofut-1 fucosyltransferase followed by extension of these sites through Fringe protein-mediated addition of Glc-NAc.Citation25 The degree of glycosylation by Fringe modulates the relative affinity of Notch receptors for its two main ligands: Delta and Jagged/Serrate.Citation25-Citation27 We previously reported that Lunatic Fringe (Lfng) exhibits decreased expression in the vast majority of TNBC, and deletion of this gene in mouse mammary gland enhances Notch receptor activation/signaling, increases proliferation, and induces TNBC in cooperation with amplification of the Met/Caveolin gene locus.Citation28 Met, a tyrosine kinase receptor involved in EMT, is frequently expressed at high levels in aggressive human breast cancer with EMT features, and expression of oncogenic Met (together with p53 loss) induced basal-like as well as claudin-low mammary tumors in transgenic mice.Citation29-Citation32 Thus, we not only established a mouse model for TNBC (including both basal and claudin-low subtypes) through deletion of Lfng, but also identified Met amplification as a prominent cooperative event during TNBC pathogenesis, raising the opportunity for combination therapy targeting both Notch and Met.
In an effort to determine the respective roles for Notch and Met activation in TNBC cells, we established and characterized two distinct TNBC cell lines from the Lfng conditional knockout mouse model, and tested the biological effects of selective Met inhibitor SU11274 and γ-secretase inhibitor MK-0752, as single agents and in combination, in both lines. We observed profound effects on tumor cell phenotypes elicited by pharmacological inhibition of Met and Notch, and revealed crosstalk between these pathways, which may provide rationale for the design of combination therapy to treat TNBC.
Results
Triple negative breast cancer cell lines established from the Lfngcko mouse model
We previously generated the Lfngflox/flox;MMTV-Cre (Lfngcko) mouse model, which develops two major subtypes of TNBC: basal-like and claudin-low.Citation28 In this study, we established two TNBC cell lines, B5725 and C0321, from a basal-like and a claudin-low mammary tumor, respectively, developed in the Lfngcko mice. The two cell lines show different expression/activation of Notch receptors (). Western blot for Notch1 revealed only one band of ~100 kDa, consistent in size with the murine Notch1 intracellular domain (N1IC). B5725 cells showed a much higher level of N1IC compared with C0321. Western blot for Notch2 showed a major band of ~100 kDa (presumable N2IC). In contrast, C0321 cells contained more N2IC compared with B5725 cells. Full-length protein (~250 kDa) and a putative intracellular domain (~100 kDa) of Notch3 receptor were detected by western blot of both cell lines. The overall level of Notch3 was similar in both lines; however C0321 cells had a higher N3IC/full-length ratio compared with B5725 cells, suggesting higher Notch3 activation in C0321 cells. Lastly, Notch4 was detected as a single band of ~90 kDa in size, and its level is slightly higher in C0321 cells. Notably, the differential expression of Notch receptors in the two cell lines is concordant with our previous microarray gene expression analysis in two types of mammary tumors from Lfngflox/flox;MMTV-Cre mice. That is, basal-like tumors express more Notch1 receptor while the claudin-low tumors express more Notch2 and Notch4.Citation28 The Met/Caveolin gene amplicon was co-selected in the vast majority of Lfng deletion-induced mammary tumors,Citation28 we therefore assessed Met gene copy number in the two cell lines by quantitative PCR. Indeed, both lines harbor an increased copy number for Met, more than 12-fold in B5725 and 4-fold in C0321, as compared with splenocytes from syngeneic FVB mice ().
Figure 1. Characterization of two TNBC cell lines B5725 and C0321 established from the Lfngflox/flox;MMTV-Cre mouse model. (A) Western blot analysis for Notch receptors in B5725 and C0321 cells. β-actin is included as loading control. (B) Copy numbers of the Met gene in B5725 and C0321 cells determined by quantitative PCR, and normalized to that of spleen cells in syngeneic FVB mouse. Shown are mean values ± standard errors derived from triplicate PCR for each sample. *P < 0.05, ***P < 0.0005. (C) Phase-contrast images of B5725 and C0321 cells in 2D culture. White arrow points to the filopodium-like protrusion. Scale bars: 50 μm.
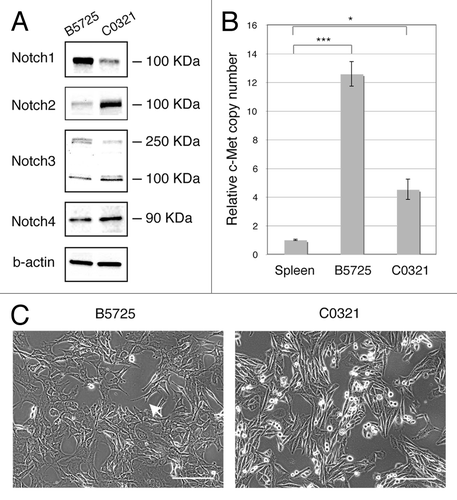
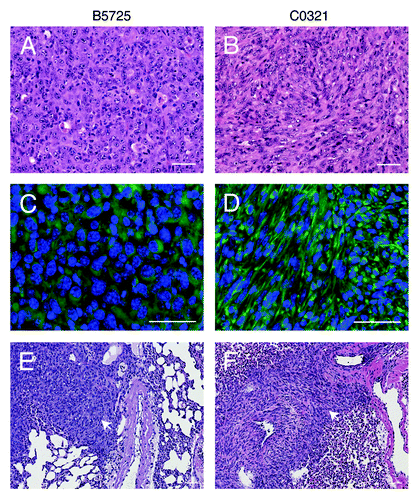
The two cell lines show distinct cell morphology in 2D culture. B5725 cells exhibit abundant filopodium-like protrusions, which have been shown to play critical roles in both tumor formation and metastatic outgrowth.Citation33 Like other claudin-low tumor cells, C0321 cells have a typical spindle-like shape (). As expected, both lines are highly tumorigenic in vivo. Inoculation of as low as 1 × 104 cells from either cell line into the mammary fat pad in syngeneic FVB females resulted in 100% tumor take (n = 10), with tumors reaching 800–1000 mm3 within 3 wk of injection. Tumors formed after injections of each line showed a characteristic and different histology. B5725-derived tumors were composed of cells with larger nucleus compared with the C0321-originated tumors, which show characteristics of spindloid tumor cells (). Anti-vimentin immunostaining showed robust expression in C0321 tumors and lower intensity staining in B5725 tumors (). Visceral metastases are characteristic of human TNBC compared with other subtypes of breast cancer.Citation34 Interestingly, we observed frequent metastasis to distal sites including lung and liver after inoculation of either cell line in the mammary fat pad (, data not shown). The incidence of lung metastasis is one out of six within 6 wk post inoculation. Thus, both cell lines possess high tumorigenicity as well as strong metastatic potential.
Figure 2. Authotopic isograft of B5725 and C0321 cell lines in FVB mice. (A and B) Representative H&E staining of mammary tumors developed from injection of B5725 or C0321 cell line (1 × 105 cells/mouse) in the mammary fat pad of FVB mice. (C and D) Anti-vimentin immunofluorescence staining of the mammary tumor sections. (E and F) Representative H&E staining of metastatic lesions (white arrows) in the lungs after injection of B5725 or C0321 cell line into the mammary fat pad. Scale bars: 50 μm.
Effects of Met inhibitor SU11274 on Met expression and activation, and γ-secretase inhibitor MK-0752 on Notch activation, in TNBC cells
To test for the impact of Met inhibition in our TNBC model cells, we first determined the effects of selective Met inhibitor SU11274 on Met expression and activation. Met has been shown to negatively regulate its own expression through upregulation of Delta ligand and Hes1-mediated transcriptional repression.Citation35 Given that both B5725 and C0321 cells harbor amplification of the Met locus, inhibition of Met signaling may cause de-repression of transcription from multiple copies of the Met gene. Indeed, treatment of B5725 and C0321 cells with SU11274 at a concentration of 6 μM resulted in an increase in total Met protein accumulation in 24 h (). Next, we examined the effect of SU11274 on Met phosphorylation. Western blot analysis with anti-Phospho-Met antibodies showed two bands in B5725 cells, presumably the dually (1234/1235) and triply (1230/1234/1235) phosphorylated forms of Met protein. Interestingly, the upper band was almost absent, while the lower band was largely unchanged, in SU11274-treated B5725 cells. Treatment of C0321 cells with SU11274 caused a dramatic decrease in levels of phospho-Met. The remaining phospho-Met in treated cells appeared to be dually phosphorylated while the control cells showed triply phosphorylated form (). Thus, treatment with SU11274 caused a decrease in phospho-Met but an increase in total Met protein levels in both cell lines.
Figure 3. Effects of Met inhibitor SU11274 on Met expression and activation, and γ-secretase inhibitor MK-0752 on Notch receptor activation. (A) Western blot analysis of Met and phospho-Met in B5725 and C0321 cells incubated with SU11274 at a concentration of 6 μM (in DMSO) for 24 h, or incubated with same amount of DMSO as control. (B) Western blot for Notch receptors in B5725 and C0321 cells treated with 6 μM of MK-0752 for 24 h. Control was incubation with DMSO.
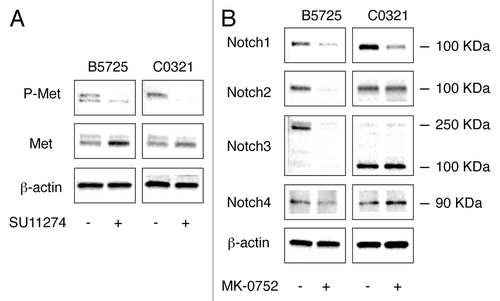
We also tested the effects of γ-secretase inhibitor MK-0752 on Notch receptor activation by measuring the levels of putative Notch intracellular domains (activated Notch receptors) using western blot analysis. In B5725 cells, incubation with MK-0752 at 6 μM concentration for 24 h resulted in dramatically decreased level of N2IC and slightly reduced levels of N1IC and N4IC. Interestingly, MK-0752 treated B5725 cells showed a drastically reduced level of full-length Notch3 protein with very low level of N3IC remaining (). For C0321 cells, incubation with 6 μM MK-0752 for 24 h caused a decrease only in the level of N1IC (). Taken together, γ-secretase inhibitor MK-0752 had an inhibitory effect on multiple Notch receptors in B5725 cells but only inhibited Notch1 in C0321. The differential effects of MK-0752 may be intrinsic to the cell lines.
Inhibition of Met and Notch in Lfngcko TNBC cells causes suppression of cell proliferation as well as a reduction in tumorsphere and colony formation in vitro
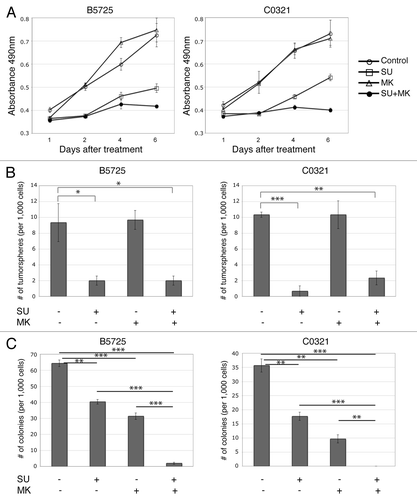
Lower expression of Lfng and amplification/overexpression of Met are common in the basal-like and claudin-low breast cancer subtypes,Citation28-Citation31 suggesting that combination therapy against Met and Notch could be effective for TNBCs with hyper-activation of both pathways. Therefore we tested the efficacy of Met inhibitor SU11274 in combination with MK-0752, a γ-secretase inhibitor currently in clinical trials for advanced or metastatic breast cancer, in suppressing B5725 and C0321 tumor cell growth. Indeed, treatment with SU11274 at a concentration of 6 μM caused dramatically reduced cell growth in both cell lines. While treatment with MK-0752 (6 μM) alone had no effect, combinatory treatment with SU11274 and MK-0752 resulted in almost complete arrest of cell growth in both cell lines (). Next, we tested for the effect of SU11274 and MK-0752 on tumorsphere formation in both lines. Indeed, a significantly reduced numbers of tumorspheres grew when both lines were treated with SU11274. However, treatment with MK-0752 showed no effect, either alone or in combination with SU11274 (). We then examined the effects of Met and γ-secretase inhibitors on colony-forming capability using soft agar assay. Both SU11274 and MK-0752 showed an inhibitory effect on colony formation in both lines. Interestingly, the γ-secretase inhibitor MK-0752 had a stronger effect than the Met inhibitor SU11274. In addition, combination treatment with both inhibitors completely abolished colony-forming capability in C0321 and significantly impaired it in B5725 cells ().
Figure 4. Inhibition of Met and Notch in TNBC cells suppressed cell growth as well as tumorsphere and colony formation. (A) Cell growth curves of B5725 and C0321 cell lines under treatment of SU11274 (6 μM), MK-0752 (6 μM), or SU11274 (6 μM) plus MK-0752 (6 μM). Control is the incubation with DMSO. At each time point, relative numbers of viable cells were determined by MTS assay, and presented as mean values ± standard errors of the absorbance at 490 nm. (B) Quantification of the numbers of tumorspheres formed per 1000 cells in 2 wk. Cells were incubated with SU11274, MK-0752, or DMSO as control, at the same concentration as above. Data are presented as mean values ± standard errors from three experiments. (C) Quantification of the numbers of colonies formed in soft agar per 1000 cells, with the treatment of SU11274, MK-0752, or DMSO control at the same concentration as above. Data are presented as mean values ± standard errors from three experiments. *P < 0.05, **P < 0.005, ***P < 0.0005
Inhibition of Met reversed epithelial-to-mesenchymal transition in TNBC cells
Many TNBC cases, and the claudin-low subtype in particular, show features of epithelial-to-mesenchymal transition (EMT). Met is a potent inducer of EMT, therefore we tested for effects of Met inhibitor on the EMT phenotype in both cell lines. Indeed, western blot analysis showed a dramatic decrease in Twist protein accumulation in both lines after SU11274 treatment (). In addition, C0321 cells showed decreased level of vimentin and B5725 cells showed increased level of E-cadherin after SU11274 treatment. Vimentin level was not changed significantly in B5725 cells, and E-cadherin was undetectable by western blot in C0321 cells (). Next we examined the effect of SU11274 treatment on cell migration in a “wound healing” assay. In both cell lines, after 48 h, control cells had migrated to an extent that scratched areas were nearly “healed”, while SU11274-treated cells barely migrated, leaving the scratches “unhealed”. Interestingly, the inhibitory effect on cell migration was more evident in C0321, a cell line sharing features with claudin-low subtype tumors (). Next we performed immunocytochemistry to compare the expression of cytokeratin 5 (K5) and cytokeratin 8 (K8), markers for the basal and luminal epithelial cells, respectively. Without treatment, both cell lines express very low or undetectable level of cytokeratins. Interestingly, treatment with SU11274 caused a dramatic increase in cytokeratin expression. Indeed, almost all C0321 cells showed coexpression of K5 and K8 upon Met inhibition, and the vast majority of B5725 cells turned on K5 expression (some with weak K8 expression) after SU11274 treatment ().
Figure 5. Inhibition of Met in TNBC cells caused downregulation of Twist, alteration in E-cadherin and vimentin expression, and reduced cell migration. (A) Western blot analysis of E-cadherin, vimentin, and Twist in B5725 and C0321 cells incubated with 6 μM SU11274 for 24, 48, and 72 h. The control cells were incubated with DMSO. (B) Representative images of the wound healing assay. Cells were incubated with SU11274 (6 μM) or DMSO as control.
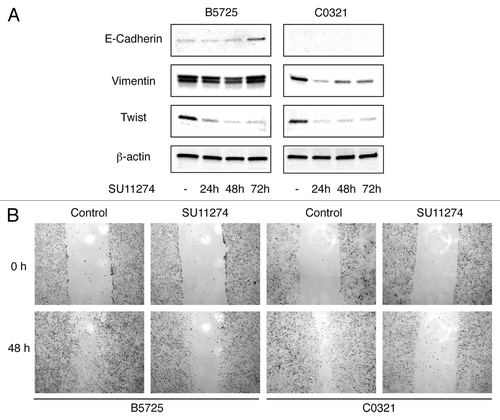
Figure 6. Increased cytokeratin expression in TNBC cells treated with Met inhibitor. Shown are representative images of the immunofluorescence staining for cytokeratin 5 (K5) and cytokeratin 8 (K8) in B5725 and C0321 cells after 48h treatment of SU11274 (6 μM) or DMSO as control. Dapi staining is for the nucleus. Merged images for anti-K5 (green) and anti-K8 (red) staining are shown at the right. Scale bar: 20 μm.
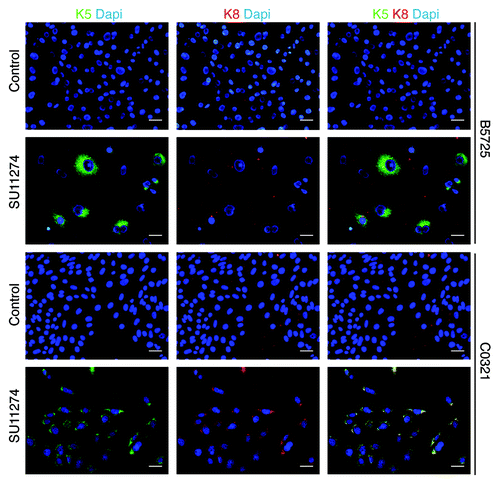
Taken together, these results indicate that inhibition of Met by SU11274 reversed the EMT phenotype in B5725 and induced MET (mesenchymal-to-epithelial transition) in C0321 cells, shown by reduced expression of mesenchymal cell markers and upregulation of epithelial cell markers, as well as reduced cell migration. These results are in agreement with a recent report that treatment of claudin-low breast cancer cells with Met inhibitors resulted in reversal of epithelial-to-mesenchymal transition.Citation31
Met modulates Notch signaling through differential regulation of Delta and Jagged/Serrate ligands
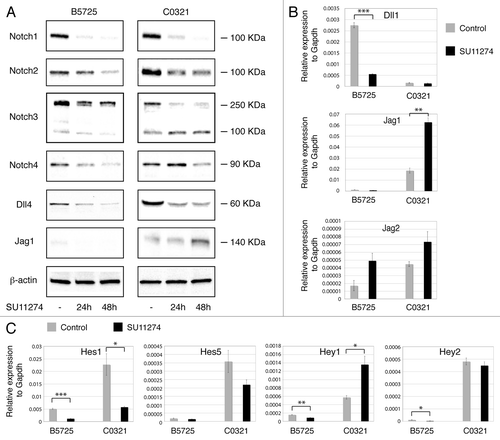
Combined inhibition of Met and Notch synergistically affected TNBC cell proliferation, suggesting possible crosstalk between both signaling pathways. Therefore we tested for effects of Met inhibition on Notch signaling in B5725 and C0321 cells. Interestingly, treatment with SU11274 dramatically altered Notch activation in both cell lines. Western blot analysis in SU11274-treated B5725 cells showed significantly decreased levels of putative Notch1, Notch2, and Notch4 intracellular domains while the level of Notch3 intracellular fragment was slightly reduced. SU11274-treated C0321 cells showed decreased levels of Notch1, Notch2, and Notch4 intracellular domains, but increased accumulation of Notch3 intracellular peptide (). We next tested for changes in Notch ligand expression in these cells. Treatment with SU11274 caused a decrease in Dll4 protein levels in both lines, but an increase in Jag1 protein accumulation in C0321 cells only (). We also determined Dll1, Jag1, and Jag2 mRNA levels by quantitative RT-PCR. Met inhibition caused downregulation of Dll1 in B5725 cells and upregulation of Jag1 in C0321 cells. In addition, Jag2 mRNA was elevated in both lines after 48 h of SU11274 treatment (). Thus, Met signaling differentially regulates expression of Delta and Jagged/Serrate ligands in TNBC cells, leading to differential modulation of Notch signaling. Indeed, SU11274-treated B5725 cells showed decreased levels of Hes1, Hey1, and Hey2 Notch target genes, while C0321 cells treated with SU11274 expressed less Hes1 mRNA but more Hey1 message (). Elevated Hey1 expression in C0321 is most likely due to upregulation of Jag1 ligand and enhanced Jag1/Notch3 signaling. These results are in agreement with a previous study showing that Met activation leads to transcriptional induction of Dll1 and Dll4 in MDA-MB-435-β4 human mammary carcinoma cells.Citation35 Notably, our data show that Met activation can repress Jag/Serrate ligand expression in TNBC cells, especially those with claudin-low characteristics (C0321 cells in this case). Therefore, inhibition of Met signaling may lead to enhanced Jagged-mediated Notch activation in TNBC, likely making it necessary to target both Met and Notch simultaneously for a better outcome.
Figure 7. Met modulates Notch signaling in TNBC cells through differential regulation of Dll and Jagged ligands. (A) Western blot analysis of Notch receptors and Dll4, Jag1 ligands in B5725 and C0321 cells incubated with SU11274 at a concentration of 6 μM (in DMSO) for 24 and 48 h, or incubated with same amount of DMSO as control. β-actin is included as the loading control. (B) Relative expression levels of Dll1, Jag1, and Jag2 in B5725 and C0321 cells treated with 6 μM of SU11274 (or DMSO as control) for 48 h. mRNA levels of each gene were determined by quantitative RT-PCR and normalized to Gapdh in each sample. Shown are mean values ± standard errors from three reactions. (C) Relative mRNA levels of Notch downstream targets Hes1, Hes5, Hey1, and Hey2 in B5725 and C0321 cells treated with 6 μM of SU11274 (or DMSO as control) for 48 h. Shown are mean values ± standard errors from three reactions. *P < 0.05, **P < 0.005, ***P < 0.0005.
Discussion
The two mouse breast cancer cell lines we established and characterized are unique in two aspects. First, B5725 and C0321 both carry Met gene amplification in addition to deletion of Lfng. Since Met amplification/overexpression and downregulation of Lfng are common in basal and claudin-low molecular subtype tumors, these lines have direct relevance to human disease and are uniquely suitable for investigation of Notch and Met function in TNBC cells as well as for efficacy tests on inhibitors for both receptor systems. While the vast majority of human TNBC (basal subtype in particular) express very low level of Lfng, only a small fraction of them harbor Notch-activating translocations.Citation24,Citation28 Therefore Lfng conditional knockout represents a more generalized TNBC model compared with others that overexpress constitutively active forms of Notch (Notch intracellular domain). In addition, the latter models are unsuitable for testing of most Notch-targeting agents (such as GSI) that block cleavage of Notch. Second, B5725 and C0321 cell lines were established from mammary tumors developed in mice of pure FVB background, which enabled us to perform isograft experiment by injecting tumor cells into the mammary fat pad of syngeneic FVB mice. Unlike xenografts using immunodeficient NOD/SCID mice, isografts in immunocompetent mice will be a valuable tool for the study of tumor/host interaction and the effects of pharmacological agents in this context.
We previously identified Met amplification as a prevailing genetic event cooperating with the Lfng deficiency in driving basal-like and claudin-low breast cancer pathogenesis.Citation28 Lfng is known to enhance Dll-mediated Notch activation and inhibit Notch signaling activated by Jagged/Serrate ligands.Citation25-Citation27 Interestingly, here we show that activation of Met upregulates Dll1, Dll4 and downregulates Jag1, Jag2 gene expression in TNBC cells. Hence, Met amplification appears to compensate for the Lfng deletion in the differential modulation of ligand-mediated Notch signaling. This could be one of the reasons for selection of Met gene amplification during the course of Lfng deletion-induced breast cancer pathogenesis.
Met inhibitors and GSIs are both being tested for efficacy in ongoing clinical trials for multiple cancers, including TNBC. Our study with these novel TNBC cell lines demonstrate potent anti-tumor effects of a Met inhibitor in vitro, and warrants further investigation using isografts in syngeneic FVB mice as a preclinical model. Compared with the Met inhibitor, the GSI (MK0752) appeared less effective as a single agent in inhibiting tumor cell growth in 2D culture, but more efficient in blocking colony formation, suggesting an essential role for Notch activation in tumor growth through cell-cell communication. Interestingly, the GSI used here, MK0752, appeared to have different potencies against cleavage of different Notch receptors in different cell lines. The mechanistic reasons for these differences deserve further investigation. Possible explanations may include differences in the intracellular half-lives for each NIC, different isoforms of γ-secretase (or different subcellular pools of γ-secretase) preferentially cleaving different Notch receptors and Notch post-transcriptional modifications. Finally, it is noteworthy that inhibition of Met caused upregulation of Jag1 in C0321 cells, leading to elevated Jag1-mediated Notch3 activation. Thus, combination targeting of Met and Notch may be necessary for patients with Met overexpression/hyperactivation, especially those showing lower Lfng and higher Jag1 expression.
Materials and Methods
Cell culture and treatment
Cell lines B5725 and C0321 were established from independent primary mammary tumors developed in the Lfngflox/flox;MMTV-Cre (Lfngcko) mice.Citation28 Briefly, mouse mammary tumors were dissociated in Epicult-B medium plus collagenase/hyaluronidase (StemCell Technologies), and single cell suspensions were generated according to manufacturer’s protocols. Non-epithelial cells were removed using the EasySep Mouse Epithelial Cell Enrichment Kit (StemCell Technologies). Mammary tumor epithelial cells were then cultured in DMEM containing 10% fetal bovine serum (FBS) with antibiotics, and were maintained in a humidified environment containing 5% CO2. Both cell lines were treated with SU11274 (Selleck, S1080) and MK-0752 (Selleck, S2660) at a final concentration of 6 μM (dissolved in DMSO). Control cells were incubated with an equivalent amount of DMSO alone for the same period of time.
MTS cell viability assay
Cell growth in the presence of SU11274 and/or MK-0752 was measured by MTS assay (CellTiter 96 one solution, Promega). Cells were seeded in a 96-well plate at 1 × 103 cells/well in DMEM containing 10% FBS, and subsequently incubated with drugs. Assays were performed by adding 20 μL of the MTS reagent to each culture well, incubating for 2 h, and then absorbance recorded at 490 nm with a multi-well plate reader (Synergy4, BioTek). Assays were repeated at least three times.
Western blot analysis
Cells were lysed in RIPA buffer (Boston Bioproducts) with protease inhibitor cocktail (Roche). Protein concentrations were determined using the BCA protein assay kit (Thermo). Samples containing an equal amount of total protein were resolved by SDS-PAGE and electrotransferred to a nitrocellulose membrane (Biorad). Specific proteins were detected with antibodies against Notch1 (1:1000; Cell Signaling, 3608), Notch2 (1:2000; DSHB, University of Iowa, C651.6DbHN), Notch3 (1:1000; Proteintech, 55114-1-AP), Notch4 (1:1000; Millipore, 09-089), Dll4 (1:1000; Abcam, ab7280), Jagged-1 (1:500; Santa Cruz, sc-6011), Twist (1:500; Santa Cruz, sc-81417), vimetin (1:1000; Cell Signaling, 5741), E-cadherin (1:1000; Cell Signaling, 3195), Met (1:1000; Cell Signaling, 3127), Phospho-Met (Tyr1234/1235) (1:1000; Cell Signaling, 3077) or β-actin (1:4000; Santa Cruz, sc-81178).
Quantitative RT-PCR
Total RNA was prepared from cells using the RNeasy Mini Kit (Qiagen) and reverse-transcribed using an iScript cDNA Synthesis Kit (Bio-Rad). Quantitative RT-PCR was performed on a BioRad CFX96 Real-Time System using the RT2 SYBR Green qPCR Master Mixes (Qiagen). Primer sequences for Dll1, Jag1, Jag2, Hes1, Hes5, Hey1, and Hey2 were as previously reported.Citation36 The relative abundance of mRNA for each gene, with respect to GAPDH, was determined using the equation 2−ΔCT, where ΔCT = CTTested Gene − CTGAPDH. Data were derived from triple reactions for each sample.
Determination of Met gene copy number
Genomic DNA was prepared from B5725 and C0321 cell lines, as well as from an FVB mouse spleen, using DNeasy Blood and Tissue Kit (Qiagen). Relative copy numbers for Met in B5725 and C0321 cells (as compared with the copy number in spleen) were determined by quantitative PCR. Copy number for Edem1, a gene on chromosome 6 that is genomically stable was also determined as a control. The sequence of PCR primers used for Met and Edem1 were previously described.Citation37 Q-PCR for each sample was performed in triplicates and relative Met copy numbers were derived from standardizing input DNA to control signal for Edem1.
Tumorsphere culture
Cells were harvested from 2D culture using trypsin-EDTA, plated at 1 × 103 cells/well in a 24-well ultra-low attachment plate (Corning, 3473), and then expanded in serum-free DMEM/F12 with MEGM SingleQuot Kit Suppl. and Growth Factors (Lonza, CC-4136) for 2–3 wk. Spheres were quantified in a cytometer (Celigo) by counting the number of embryoid bodies per culture well.
Soft agar colony formation assay
Cells were trypsinized, counted, and then mixed in DMEM medium containing 0.35% agarose. Twenty-five hundred cells for each well were plated on top of 0.5% agarose layer in a 12-well plate. Plated cells were allowed to form colonies for about 2 wk. The colonies formed were then visualized by staining in 0.005% crystal violet for 4 h.
Cell migration assay
Collective cell migration was measured in a wound healing assay. Cells were seeded in a 6-well plate at a concentration of 5 × 105 cells per well. After 12 h, a portion of the monolayer was scratched with a 1000 μL pipette tip, and examined for resealing of the “wounded” monolayer 48 h later.
Tumor cell transplantation
Briefly, 1 × 104 to 1 × 106 B5725 or C0321 cells were resuspended in 10 μL DMEM medium and mixed at 1:1 ratio with 20 μL Matrigel (BD Bioscience) on ice. A total of 40 μL cell sample each were then injected into the fourth inguinal mammary fat pad of syngeneic FVB mice at 4 wk of age. Animals were anesthetized with isoflurane for the injection. All animal care and procedures were performed according to NIH guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Histology and immunofluorescence staining
Formalin-fixed paraffin-embedded mouse tissues were processed for histology and immunofluorescence staining by standard procedures. For immunofluorescence, cells were cultured on glass coverslips, and then fixed with 4% paraformaldehyde. Primary antibodies used for immunostaining were: vimentin (1:100; Cell Signaling, 5741), K5 (1:1600; Covance, PRB-160P) and K8 (1:800; Covance, MMS-162P). Representative images were acquired with a Nikon Eclipse 80i microscope.
Statistics
All data are presented as mean ± standard error. Statistical analysis was performed using the two-tailed Student t test. A P value of 0.05 or less was considered statistically significant.
| Abbreviations: | ||
| TNBC | = | triple negative breast cancer |
| GSI | = | γ-secretase inhibitor |
| ERα | = | estrogen receptor α |
| PR | = | progesterone receptor |
| HER2 | = | human epidermal growth factor receptor 2 |
| EMT | = | epithelial-to-mesenchymal transition |
| Lfng | = | Lunatic Fringe |
| NIC | = | Notch intracellular domain |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by NIH grants R21 CA175136 to K.X. and P01 AG025531 to L.M. The authors would like to thank Dr Sean Egan at the Hospital for Sick Children, Toronto for reagents and advice.
References
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience?. Nat Rev Clin Oncol 2010; 7:683 - 92; http://dx.doi.org/10.1038/nrclinonc.2010.154; PMID: 20877296
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med 2010; 363:1938 - 48; PMID: 21067385
- Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist 2011; 16:Suppl 1 1 - 11; PMID: 21278435
- Podo F, Buydens LM, Degani H, Hilhorst R, Klipp E, Gribbestad IS, Van Huffel S, van Laarhoven HW, Luts J, Monleon D, et al, FEMME Consortium. Triple-negative breast cancer: present challenges and new perspectives. Mol Oncol 2010; 4:209 - 29; PMID: 20537966
- Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist 2011; 16:Suppl 1 61 - 70; http://dx.doi.org/10.1634/theoncologist.2011-S1-61; PMID: 21278442
- Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res 2008; 14:1368 - 76; http://dx.doi.org/10.1158/1078-0432.CCR-07-1658; PMID: 18316557
- Perou CM, Børresen-Dale AL. Systems biology and genomics of breast cancer. Cold Spring Harb Perspect Biol 2011; 3:a003292; http://dx.doi.org/10.1101/cshperspect.a003293; PMID: 21047916
- Creighton CJ, Chang JC, Rosen JM. Epithelial-mesenchymal transition (EMT) in tumor-initiating cells and its clinical implications in breast cancer. J Mammary Gland Biol Neoplasia 2010; 15:253 - 60; http://dx.doi.org/10.1007/s10911-010-9173-1; PMID: 20354771
- Hennessy BT, Gonzalez-Angulo AM, Stemke-Hale K, Gilcrease MZ, Krishnamurthy S, Lee JS, Fridlyand J, Sahin A, Agarwal R, Joy C, et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res 2009; 69:4116 - 24; http://dx.doi.org/10.1158/0008-5472.CAN-08-3441; PMID: 19435916
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008; 133:704 - 15; http://dx.doi.org/10.1016/j.cell.2008.03.027; PMID: 18485877
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res 2010; 12:R68; http://dx.doi.org/10.1186/bcr2635; PMID: 20813035
- Bouras T, Pal B, Vaillant F, Harburg G, Asselin-Labat ML, Oakes SR, Lindeman GJ, Visvader JE. Notch signaling regulates mammary stem cell function and luminal cell-fate commitment. Cell Stem Cell 2008; 3:429 - 41; http://dx.doi.org/10.1016/j.stem.2008.08.001; PMID: 18940734
- Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K, Honjo T, Hennighausen L. The canonical Notch/RBP-J signaling pathway controls the balance of cell lineages in mammary epithelium during pregnancy. Dev Biol 2006; 293:565 - 80; http://dx.doi.org/10.1016/j.ydbio.2006.02.043; PMID: 16581056
- Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, Iscove N, Jones S, McKinney S, Emerman J, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell 2008; 3:109 - 18; http://dx.doi.org/10.1016/j.stem.2008.05.018; PMID: 18593563
- Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets 2010; 11:745 - 51; http://dx.doi.org/10.2174/138945010791170860; PMID: 20041844
- Lee CW, Simin K, Liu Q, Plescia J, Guha M, Khan A, Hsieh CC, Altieri DC. A functional Notch-survivin gene signature in basal breast cancer. Breast Cancer Res 2008; 10:R97; http://dx.doi.org/10.1186/bcr2200; PMID: 19025652
- Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res 2005; 65:8530 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-05-1069; PMID: 16166334
- Reedijk M, Pinnaduwage D, Dickson BC, Mulligan AM, Zhang H, Bull SB, O’Malley FP, Egan SE, Andrulis IL. JAG1 expression is associated with a basal phenotype and recurrence in lymph node-negative breast cancer. Breast Cancer Res Treat 2008; 111:439 - 48; http://dx.doi.org/10.1007/s10549-007-9805-3; PMID: 17990101
- Cohen B, Shimizu M, Izrailit J, Ng NF, Buchman Y, Pan JG, Dering J, Reedijk M. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res Treat 2010; 123:113 - 24; http://dx.doi.org/10.1007/s10549-009-0621-9; PMID: 19915977
- Ling H, Sylvestre JR, Jolicoeur P. Notch1-induced mammary tumor development is cyclin D1-dependent and correlates with expansion of pre-malignant multipotent duct-limited progenitors. Oncogene 2010; 29:4543 - 54; http://dx.doi.org/10.1038/onc.2010.186; PMID: 20562911
- Sansone P, Storci G, Giovannini C, Pandolfi S, Pianetti S, Taffurelli M, Santini D, Ceccarelli C, Chieco P, Bonafé M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007; 25:807 - 15; http://dx.doi.org/10.1634/stemcells.2006-0442; PMID: 17158237
- Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res 2010; 70:709 - 18; http://dx.doi.org/10.1158/0008-5472.CAN-09-1681; PMID: 20068161
- Speiser J, Foreman K, Drinka E, Godellas C, Perez C, Salhadar A, Erşahin Ç, Rajan P. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int J Surg Pathol 2012; 20:139 - 45; http://dx.doi.org/10.1177/1066896911427035; PMID: 22084425
- Robinson DR, Kalyana-Sundaram S, Wu YM, Shankar S, Cao X, Ateeq B, Asangani IA, Iyer M, Maher CA, Grasso CS, et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat Med 2011; 17:1646 - 51; http://dx.doi.org/10.1038/nm.2580; PMID: 22101766
- Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol 2003; 4:786 - 97; http://dx.doi.org/10.1038/nrm1228; PMID: 14570055
- Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat Cell Biol 2000; 2:515 - 20; http://dx.doi.org/10.1038/35019553; PMID: 10934472
- Yang LT, Nichols JT, Yao C, Manilay JO, Robey EA, Weinmaster G. Fringe glycosyltransferases differentially modulate Notch1 proteolysis induced by Delta1 and Jagged1. Mol Biol Cell 2005; 16:927 - 42; http://dx.doi.org/10.1091/mbc.E04-07-0614; PMID: 15574878
- Xu K, Usary J, Kousis PC, Prat A, Wang DY, Adams JR, Wang W, Loch AJ, Deng T, Zhao W, et al. Lunatic fringe deficiency cooperates with the Met/Caveolin gene amplicon to induce basal-like breast cancer. Cancer Cell 2012; 21:626 - 41; http://dx.doi.org/10.1016/j.ccr.2012.03.041; PMID: 22624713
- Graveel CR, DeGroot JD, Su Y, Koeman J, Dykema K, Leung S, Snider J, Davies SR, Swiatek PJ, Cottingham S, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci U S A 2009; 106:12909 - 14; PMID: 19567831
- Ponzo MG, Lesurf R, Petkiewicz S, O’Malley FP, Pinnaduwage D, Andrulis IL, Bull SB, Chughtai N, Zuo D, Souleimanova M, et al. Met induces mammary tumors with diverse histologies and is associated with poor outcome and human basal breast cancer. Proc Natl Acad Sci U S A 2009; 106:12903 - 8; http://dx.doi.org/10.1073/pnas.0810402106; PMID: 19617568
- Knight JF, Lesurf R, Zhao H, Pinnaduwage D, Davis RR, Saleh SM, Zuo D, Naujokas MA, Chughtai N, Herschkowitz JI, et al. Met synergizes with p53 loss to induce mammary tumors that possess features of claudin-low breast cancer. Proc Natl Acad Sci U S A 2013; 110:E1301 - 10; http://dx.doi.org/10.1073/pnas.1210353110; PMID: 23509284
- Gastaldi S, Sassi F, Accornero P, Torti D, Galimi F, Migliardi G, Molyneux G, Perera T, Comoglio PM, Boccaccio C, et al. Met signaling regulates growth, repopulating potential and basal cell-fate commitment of mammary luminal progenitors: implications for basal-like breast cancer. Oncogene 2013; 32:1428 - 40; http://dx.doi.org/10.1038/onc.2012.154; PMID: 22562252
- Shibue T, Brooks MW, Weinberg RA. An integrin-linked machinery of cytoskeletal regulation that enables experimental tumor initiation and metastatic colonization. Cancer Cell 2013; 24:481 - 98; http://dx.doi.org/10.1016/j.ccr.2013.08.012; PMID: 24035453
- Rakha EA, Chan S. Metastatic triple-negative breast cancer. Clin Oncol (R Coll Radiol) 2011; 23:587 - 600; http://dx.doi.org/10.1016/j.clon.2011.03.013; PMID: 21524569
- Stella MC, Trusolino L, Pennacchietti S, Comoglio PM. Negative feedback regulation of Met-dependent invasive growth by Notch. Mol Cell Biol 2005; 25:3982 - 96; http://dx.doi.org/10.1128/MCB.25.10.3982-3996.2005; PMID: 15870272
- Zhang S, Loch AJ, Radtke F, Egan SE, Xu K. Jagged1 is the major regulator of Notch-dependent cell fate in proximal airways. Dev Dyn 2013; 242:678 - 86; http://dx.doi.org/10.1002/dvdy.23965; PMID: 23526493
- Smolen GA, Muir B, Mohapatra G, Barmettler A, Kim WJ, Rivera MN, Haserlat SM, Okimoto RA, Kwak E, Dahiya S, et al. Frequent met oncogene amplification in a Brca1/Trp53 mouse model of mammary tumorigenesis. Cancer Res 2006; 66:3452 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-05-4181; PMID: 16585167