
Abstract
As the knowledge on cancer genetic alterations progresses, it fosters the need for more personalized therapeutic intervention in modern cancer management. Recently, mutations in KRAS, BRAF, and PIK3CA genes have emerged as important mechanisms of resistance to EGFR-targeted therapy in metastatic colorectal cancer (mCRC).
Here we report the first case of a mCRC patient whose disease had progressed on standard lines of treatment and for which we devised a personalized therapeutic approach consisting of vemurafenib (ZelborafTM) and panitumumab (VectibixTM), based on the following molecular profile: BRAFV600E-mutant, amplified EGFR (double positive) and WT KRAS, WT PIK3CA, not-amplified HER2 (triple negative).
This new combination therapy was well tolerated and resulted in a strong control of the disease. In particular, the vemurafenib-panitumumab combination appears to limit the typical toxicity of single agents, since no cutaneous toxic effects typically associated with vemurafenib were observed.
Here we report the first clinical evidence that the combination of an anti-EGFR (panitumumab) and an inhibitor of BRAFV600E (vemurafenib) is well tolerated and results in a strong disease control in an extensively pretreated mCRC patient.
Introduction
Novel biological agents, including anti-epidermal growth factor receptor (EGFR), anti-VEGF targeting therapies, and small-molecule multikinase inhibitors, have recently changed the standard of care of metastatic colorectal cancer (mCRC) patients.Citation1,Citation2 While the use of positive predictive biomarkers for biological therapy would be desirable in order to identify specific subsets of responsive patients, to date the only validated negative predictive biomarker for mCRC is the mutational status of all members of the RAS gene family, which identifies mCRC patients not eligible to monoclonal antibody (moAb) anti-EGFR therapies.Citation3,Citation4 Emphasizing the limitations of negative predictive biomarkers, unfortunately only a subgroup of WT RAS mCRC patients respond to anti-EGFR drugs, being the molecular mechanism/s underlying resistance to anti-EGFR treatment not fully understood.Citation5 Activating mutations in other members of the RAS-BRAF-MEK and PI3K-AKT pathways, both acting downstream of the EGFR signaling cascade, are being investigated as further potential predictive biomarkers.Citation6-Citation8
Apparently, no specific target treatment seems to be available for WT RAS and anti-EGFR resistant mCRC patients. Indeed, the inhibition of the BRAFV600E oncoprotein by the small-molecule drug vemurafenib, which is highly effective in melanoma,Citation9 showed a very limited response in the mCRC setting.Citation7,Citation8 Coherently, only a prognostic significance has been attributed to BRAF mutations in CRC, so far.Citation7 Interestingly however, preclinical studies have indicated that EGFR reactivation contributes to insensitivity of BRAF-mutant CRC to vemurafenib. Thus, the association of BRAF and EGFR inhibitors might effectively target BRAFV600E mutant colon cancers.Citation10,Citation11 We report here the first case of a patient with BRAF-mutant, amplified EGFR (double positive) and KRAS/PIK3CA WT, not-amplified HER2 (triple negative) mCRC whose disease had progressed on standard lines of treatment, but successfully responded to a new combination therapy consisting of vemurafenib (ZelborafTM) and panitumumab (VectibixTM).
Case Report
A 55-y-old man was admitted to our oncology department in July 2007 for a poorly-differentiated adenocarcinoma of the transverse colon. Preoperative carcinoembryonic antigen (CEA) and CA19.9 serum levels were 1.2 ng/mL and 63 U/mL, respectively.
The tumor was completely removed by a right hemicolectomy with lymph node dissection. The patient was staged as IIIB and adjuvant standard treatment with FOLFOX4 (6 mo) was performed. Eleven months later, the patient developed peritoneal carcinomatosis and was treated with FOLFIRI-bevacizumab (9 cycles), discontinued for pulmonary embolism, followed by cytoreductive surgery plus hyperthermic intraperitoneal chemotherapy.
After a 12 mo disease-free interval, an increment of CA19.9 and a CT scan revealed a peritoneal progression. At this time the patient was characterized for wild-type KRAS mutational status and high EGFR expression by immunohistochemistry and underwent several lines of treatment, such as irinotecan–cetuximab, a second peritoneal cytoreductive surgery, capecitabine–bevacizumab, or sorafenib–panitumumab (off-label use). Every disease progression was exclusively peritoneal and marked by a significant increase in CA19.9 and CEA. An additional line of treatment with regorafenib demonstrated a good control of the disease for 9 mo in an expanded access program. Subsequently, the patient showed a significant rise in serum markers (CA19.9 and CEA) and a multivisceral disease progression (peritoneum, liver, and lung) accompanied by important clinical troubles including diffuse abdominal pain, weight loss, and episodes of sub-ileus.
In order to find additional treatment opportunities dictated by tumor biology, the molecular profile of the tumor was evaluated on a liver metastasis biopsy performed at the time of the latest progression and on previously collected tumor material (primary lesion and peritoneal implants). All samples concordantly revealed the following status: non-amplified HER2, KRAS WT, PIK3CA WT, amplified EGFR, and mutant BRAFV600E. Personalized medicine challenges the ethical issues related to off-label therapies and pharmacogenomics.Citation12,Citation13 The lack of other established therapeutic alternatives together with the above mentioned molecular characterization and preclinical data, met the ethical constraints of a tailored therapy, and prompted us to propose a vemurafenib-panitumumab combined treatment. The protocol was approved by the institutional review board (Policlinico Umberto I, Sapienza University of Rome) on June 2013, and the patient provided written informed consent.
Patient restaging before vemurafenib–panitumumab treatment showed hepatic, pulmonary and peritoneal disease measurable at spiral CT scan (). CEA and CA19.9 serum levels were 558 ng/mL and 4800 U/mL, respectively (). Q-PCR analysis of plasma DNA indicated the presence of circulating tumor DNA (ctDNA) via the identification of the BRAFV600E mutation ().
Figure 1. CT scans of the patient before and after panitumumab-vemurafenib treatment for metastatic CRC. Tumor masses (arrow) can be seen in the liver of the patient before initiation of panitumumab-vemurafenib treatment (A). The masses (arrow) became hypodense, homogenous and significantly reduced in size on CT obtained 3 and 6 mo after treatment (B and C), indicating good response to combination treatment.
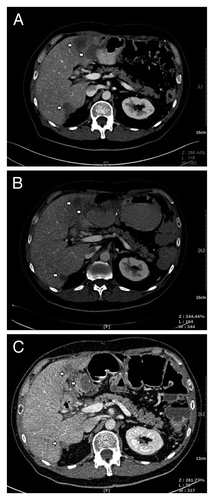
Figure 2. Trend of CEA and CA 19–9 during vemurafenib and panitumumab combination therapy.
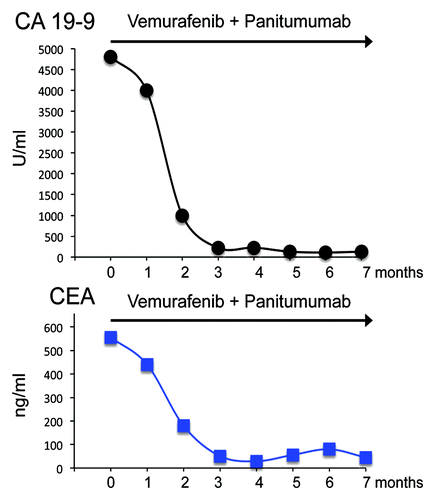
Figure 3. Detection of the BRAFV600E mutation in patient's CRC tissue and plasma. (A) Electropherogram showing the heterozygous BRAFV600E mutation in DNA isolated from patient's CRC tissue. (B) Allele-specific Q-PCR detection of the BRAFV600E mutation in plasma free DNA reveals the presence of circulating tumor DNA before treatment (T0) but not 12 wk after treatment initiation (T1). Data are reported as averages of the threshold cycles (Ct) obtained in two different Q-PCR for the BRAFV600E amplicon and the reference gene amplicon.
The patient was treated with panitumumab 6 mg/kg IV every 14 d and vemurafenib 960 mg orally twice daily. Soon after 4 wk, a significant clinical benefit with complete regression of the clinical symptoms occurred. Twelve weeks later, the programmed disease restaging with CT scan showed a strong reduction of all metastatic lesions (PR according to RECIST1.1 criteria) (). Consistent with the CT scan, tumor markers also showed a significant decrease: CEA, 51 ng/mL, CA19.9, 221 U/mL (). Most importantly, ctDNA harboring the BRAFV600E mutation disappeared from plasma. To date, the patient is still asymptomatic 28 wk after starting the treatment when the CT scan showed a further reduction of all metastatic lesions (PR according to RECIST1.1 criteria) (). Tumor markers are stably low () and circulating BRAFV600E DNA absent. Treatment regimen is being well tolerated and the patient only presents minor skin-related toxicity (maximum grade 2). In particular no cutaneous toxicity (such as squamous cell carcinomas and keratoacanthomas) typically associated with vemurafenib are observed.
Discussion
Our work represents the first clinical evidence that the combination of an inhibitor of BRAFV600E (vemurafenib) and an EGFR antagonist (panitumumab) is well tolerated and results in a highly effective control of the disease, in a mCRC patient extensively pretreated with several standard and off-label lines of treatment.
BRAF belongs to a gene family also including CRAF (or RAF1) and ARAF, encoding serine–threonine kinases that heterodimerize each other and constitute, together with downstream MEK and ERK proteins, a powerful mitogen-activated protein kinase (MAPK) pathway. RAF kinases are activated by several receptor tyrosine kinases (RTKs) such as EGFR. Notably, while mutations of ARAF and CRAF are rare in human cancer, mutated BRAF frequently occurs in papillary thyroid carcinoma, melanoma, hairy cell leukemia, hepatocellular carcinoma, and 11% of CRC.Citation14 To this regard, BRAFV600E is an ideal druggable target in BRAF mutated cancers (i.e., CRC) since it is active as a monomer in an upstream EGFR/RAS-independent way, thus providing an explanation to the observed resistance to inhibitors of EGFR/RAS pathway (i.e., anti-EGFR antibodies) (, left panel). The resistance to EGFR-targeted therapies eventually developing along progression of lung, colorectal, pancreatic, or head and neck cancers, is challenging because of the various mechanisms underlying anti-EGFR insensitivity (summarized in , right panel). Understanding these various resistance pathways would provide an opportunity to develop new inhibitors to prevent or overcome therapeutic resistance.Citation15 In this context, it can be predicted that BRAFV600E-containing CRCs would be inhibitable by BRAFV600E antagonists (, right panel). Nevertheless, BRAFV600E CRCs fail to respond to mutated BRAF inhibitors as single agents.Citation8 Likewise a short-term sensitivity to anti-BRAFV600E monotherapy has been reported in melanoma.Citation16
Figure 4. Model of BRAFV600E-dependent cell growth and mechanisms of resistance to BRAF inhibition and restoring of drug sensitivity through combination therapies. BRAF mutation confers constitutive pathway activation (red arrows) independent of upstream RTKs (EGFR, IGF-1R)/RAS signaling (left panel), providing an explanation to the observed insensitivity to anti-EGFR antibody treatment (panitumumab, cetuximab) in BRAF mutant CRC. Such a process is blunted by BRAFV600E (BRAFmut) inhibitors (blue symbols and drugs) (right panel). Additional mechanisms of EGFR primary or secondary resistance described in lung, head-neck cancer and CRC (i.e., EGFR mutation; oncogenic shift or activation of a bypass pathway such as KRAS, BRAF, PIK3CA, secondary EGFR mutation or a parallel/alternative pathway [PTEN, IGF1] is also indicated). Right panel also illustrates how bypass and resistance to BRAF-inhibitors may occur through the rescue of the BRAFV600E-mediated attenuation of ERK negative feedback induced by BRAF inhibitors and subsequent reactivation of ligand-dependent signaling from EGFR or IGF-1R or via wild type RAS/RAF or PIK3CA/AKT or PIK3CA or EGFR or MEK1 or further BRAF gain-of-function mutations (PIK3CAmut or asterisks) in several tumor types. Drugs under development in preclinical models or clinical trials to overcome BRAF inhibitor resistance are labeled in blue. References are quoted in the text.
![Figure 4. Model of BRAFV600E-dependent cell growth and mechanisms of resistance to BRAF inhibition and restoring of drug sensitivity through combination therapies. BRAF mutation confers constitutive pathway activation (red arrows) independent of upstream RTKs (EGFR, IGF-1R)/RAS signaling (left panel), providing an explanation to the observed insensitivity to anti-EGFR antibody treatment (panitumumab, cetuximab) in BRAF mutant CRC. Such a process is blunted by BRAFV600E (BRAFmut) inhibitors (blue symbols and drugs) (right panel). Additional mechanisms of EGFR primary or secondary resistance described in lung, head-neck cancer and CRC (i.e., EGFR mutation; oncogenic shift or activation of a bypass pathway such as KRAS, BRAF, PIK3CA, secondary EGFR mutation or a parallel/alternative pathway [PTEN, IGF1] is also indicated). Right panel also illustrates how bypass and resistance to BRAF-inhibitors may occur through the rescue of the BRAFV600E-mediated attenuation of ERK negative feedback induced by BRAF inhibitors and subsequent reactivation of ligand-dependent signaling from EGFR or IGF-1R or via wild type RAS/RAF or PIK3CA/AKT or PIK3CA or EGFR or MEK1 or further BRAF gain-of-function mutations (PIK3CAmut or asterisks) in several tumor types. Drugs under development in preclinical models or clinical trials to overcome BRAF inhibitor resistance are labeled in blue. References are quoted in the text.](/cms/asset/a3535e64-cb32-49f5-bd2c-8b9c7f96df43/kcbt_a_10928878_f0004.gif)
Several mechanisms have been reported to underlie the resistance to BRAF inhibitors, as summarized in .Citation17,Citation18 While inhibitors of mutant BRAF monomers suppress the ERK signaling, they relieve ERK-dependent negative feedback and reactivate ligand-dependent EGFR/RAS signaling upon wild-type RAF dimers in melanoma cells.Citation19 Likewise, recent preclinical evidence of the feedback reactivation of EGFR in CRC by vemurafenib-mediated blockade of BRAF has been reportedCitation10,Citation11 (). In turn, this triggers sustained RAS- and CRAF-mediated MAPK signaling and cell proliferation. Blocking EGFR activity with a monoclonal antibody to EGFR has been shown to restore the sensitivity to vemurafenib in preclinical models,Citation10,Citation11 providing a strong rationale for the combination of vemurafenib and anti-EGFR antibodies in BRAF-mutated CRC.
Resistance to BRAF inhibitors has also been reported through further activation of RAS/MAPK pathway (e.g., mutations of NRAS or MEK1, loss of NF1, alternatively spliced variants of BRAF, amplification of CRAF or BRAF) as well as activation of additional parallel signaling pathways such as the IGF1R/PI3K/AKT (e.g., resulting from PTEN loss) or PDGFRβ pathwaysCitation17,Citation20-Citation24 (). Although this drug resistance mechanism and its overcoming by combined treatments is potentially operating in several other tumors harboring BRAF mutations, no clinical efficacy has been reported either.
In addition to the clinical efficacy of the combined therapy with vemurafenib and panitumumab described here, Al-Marrawi et al.Citation25 recently reported that treatment of a single patient with a combination of sorafenib and cetuximab led to a mixed radiographic response with some areas showing dramatic improvement and other areas showing stable disease over a 7-mo period. These findings are consistent with the modest disease control we also observed in our case treated with panitunumab and sorafenib. However, sorafenib, a multi-kinase inhibitor with limited activity on BRAFV600E, cannot be considered an optimal drug for the specific purpose. Indeed, weak inhibition of BRAF by sorafenib would transactivate RAF dimers maintaining a still consistent downstream MEK activity.Citation18
Our case report raises several considerations that need to be exploited in subsequent studies performed on large series. The first one is related to the observation of BRAF mutation together with EGFR amplification, suggesting that vemurafenib might be an effective drug in BRAF mutant mCRC patients if used in association with EGFR inhibitors. This would blunt escaping signaling pathways (i.e., PI3K-AKT) downstream of the amplified EGFR responsible for the maintenance of the disease progression, suggesting that the dramatic response observed in our case might have been further influenced by EGFR amplification. Thus, a careful selection of the patients eligible to this treatment might be performed on the basis of two distinct positive predictive markers.
The second one is related to drugs adverse effects. Skin is largely the most frequent target of adverse events for both drugs. However, vemurafenib toxicity spectrum, (e.g., squamous cell carcinomas/keratoacanthomas, maculopapular rashes, hyperkeratosis) is significantly distinct from that of panitumumab (acneiform rash, paronychia, xerosis).Citation26 In the case described, no cutaneous toxic effects typically associated with vemurafenib were observed. In particular, squamoproliferative lesions appearing at early times (median time 8 wk) during vemurafenib treatmentCitation27 were completely absent. Therefore the vemurafenib-panitumumab combination appears to limit the typical toxicity of single agents.
Besides overcoming BRAF inhibitor resistance through co-targeting activated parallel or upstream RTK mitogenic or survival pathways (i.e., EGFR, MET, IGF1R), additional therapeutic strategies include the combined use of MEK or ERK or AKT inhibitorsCitation17,Citation28,Citation29 as well as the intermittent administration of BRAF inhibitors described in preclinical models.Citation30 However, to date no clinical evidence substantiates this hypothesis.
Conclusions and Future Directions
Here we report the first clinical evidence that the combination of an anti-EGFR (panitumumab) and an inhibitor of BRAFV600E (vemurafenib) is well tolerated and results in a highly effective disease control in an extensively pretreated mCRC patient. The possibility to select patients eligible to this therapeutic approach on the basis of two distinct positive predictive markers (EGFR gene amplification/overexpression and mutant BRAF) and the apparent reciprocal limitation of the side effects of the two drugs are interesting hypotheses to be tested in prospective studies. Furthermore, unraveling the various mechanisms of resistance to BRAF inhibition is expected to pinpoint novel combination therapies. To this regard, recent advances in CRC patients classification based on gene expression profilesCitation31 may help identifying new strategies for personalized treatment.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by AIRC (Associazione Italiana Ricerca Cancro) IG10610 and IG12116, AIRC 5XMILLE, MIUR FIRB and PRIN projects, Ministry of Health, Pasteur Institute/Cenci Bolognetti Foundation and Italian Institute of Technology (IIT).
References
- Moorcraft SY, Smyth EC, Cunningham D. The role of personalized medicine in metastatic colorectal cancer: an evolving landscape. Therap Adv Gastroenterol 2013; 6:381 - 95; http://dx.doi.org/10.1177/1756283X13491797; PMID: 24003339
- Price TJ, Segelov E, Burge M, Haller DG, Ackland SP, Tebbutt NC, Karapetis CS, Pavlakis N, Sobrero AF, Cunningham D, et al. Current opinion on optimal treatment for colorectal cancer. Expert Rev Anticancer Ther 2013; 13:597 - 611; http://dx.doi.org/10.1586/era.13.37; PMID: 23617351
- Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004; 351:337 - 45; http://dx.doi.org/10.1056/NEJMoa033025; PMID: 15269313
- Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369:1023 - 34; http://dx.doi.org/10.1056/NEJMoa1305275; PMID: 24024839
- Blanke CD, Goldberg RM, Grothey A, Mooney M, Roach N, Saltz LB, Welch JJ, Wood WA, Meropol NJ, NCI GI Steering Committee Colon Cancer Task Force. KRAS and colorectal cancer: ethical and pragmatic issues in effecting real-time change in oncology clinical trials and practice. Oncologist 2011; 16:1061 - 8; http://dx.doi.org/10.1634/theoncologist.2011-0011; PMID: 21737577
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and panitumumab in colorectal cancer. J Clin Oncol 2010; 28:1254 - 61; http://dx.doi.org/10.1200/JCO.2009.24.6116; PMID: 20100961
- Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 2009; 27:5931 - 7; http://dx.doi.org/10.1200/JCO.2009.22.4295; PMID: 19884549
- Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, Nolop KB, Saltz LD. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol [Internet] 2010; suppl 3534. Available from: http://meetinglibrary.asco.org/content/50595-74
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et al, BRIM-3 Study Group. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507 - 16; http://dx.doi.org/10.1056/NEJMoa1103782; PMID: 21639808
- Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2012; 2:227 - 35; http://dx.doi.org/10.1158/2159-8290.CD-11-0341; PMID: 22448344
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483:100 - 3; http://dx.doi.org/10.1038/nature10868; PMID: 22281684
- Lewis JR, Lipworth WL, Kerridge IH, Day RO. The economic evaluation of personalised oncology medicines: ethical challenges. Med J Aust 2013; 199:471 - 3; http://dx.doi.org/10.5694/mja13.10046; PMID: 24099207
- Nardini C.. Review: the ethics of clinical trials. ecancer 2014; 8:387 - 96
- Huang T, Karsy M, Zhuge J, Zhong M, Liu D. B-Raf and the inhibitors: from bench to bedside. J Hematol Oncol 2013; 6:30; http://dx.doi.org/10.1186/1756-8722-6-30; PMID: 23617957
- Chong CR, Jänne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389 - 400; http://dx.doi.org/10.1038/nm.3388; PMID: 24202392
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, McArthur GA, Hutson TE, Moschos SJ, Flaherty KT, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med 2012; 366:707 - 14; http://dx.doi.org/10.1056/NEJMoa1112302; PMID: 22356324
- Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med 2013; 19:1401 - 9; http://dx.doi.org/10.1038/nm.3392; PMID: 24202393
- Poulikakos PI, Rosen N. Mutant BRAF melanomas–dependence and resistance. Cancer Cell 2011; 19:11 - 5; http://dx.doi.org/10.1016/j.ccr.2011.01.008; PMID: 21251612
- Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012; 22:668 - 82; http://dx.doi.org/10.1016/j.ccr.2012.10.009; PMID: 23153539
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468:973 - 7; http://dx.doi.org/10.1038/nature09626; PMID: 21107323
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010; 464:427 - 30; http://dx.doi.org/10.1038/nature08902; PMID: 20179705
- Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, Kolinsky K, Packman K, Kim MJ, Trunzer K, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res 2012; 72:969 - 78; http://dx.doi.org/10.1158/0008-5472.CAN-11-1875; PMID: 22205714
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010; 18:683 - 95; http://dx.doi.org/10.1016/j.ccr.2010.11.023; PMID: 21156289
- Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ, Schostack K, Simcox ME, Kopetz S, Heimbrook D, et al. Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 2012; 72:779 - 89; http://dx.doi.org/10.1158/0008-5472.CAN-11-2941; PMID: 22180495
- Al-Marrawi MY, Saroya BS, Brennan MC, Yang Z, Dykes TM, El-Deiry WS. Off-label use of cetuximab plus sorafenib and panitumumab plus regorafenib to personalize therapy for a patient with V600E BRAF-mutant metastatic colon cancer. Cancer Biol Ther 2013; 14:703 - 10; http://dx.doi.org/10.4161/cbt.25191; PMID: 23792568
- Belum VR, Fischer A, Choi JN, Lacouture ME. Dermatological adverse events from BRAF inhibitors: a growing problem. Curr Oncol Rep 2013; 15:249 - 59; http://dx.doi.org/10.1007/s11912-013-0308-6; PMID: 23463215
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med 2010; 363:809 - 19; http://dx.doi.org/10.1056/NEJMoa1002011; PMID: 20818844
- Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R Jr., Dayyani F, Gopal YN, Jiang ZQ, Wistuba II, Tang XM, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 2013; 19:657 - 67; http://dx.doi.org/10.1158/1078-0432.CCR-11-1446; PMID: 23251002
- Morris EJ, Jha S, Restaino CR, Dayananth P, Zhu H, Cooper A, Carr D, Deng Y, Jin W, Black S, et al. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov 2013; 3:742 - 50; http://dx.doi.org/10.1158/2159-8290.CD-13-0070; PMID: 23614898
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013; 494:251 - 5; http://dx.doi.org/10.1038/nature11814; PMID: 23302800
- Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LC, Lannon WA, Grotzinger C, Del Rio M, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med 2013; 19:619 - 25; http://dx.doi.org/10.1038/nm.3175; PMID: 23584089