
Abstract
Limited treatment options are available for chronic myelogenous leukemia (CML) patients who develop imatinib mesylate (IM) resistance. Here we proposed a novel combination regimen, a co-administration of S116836, a novel small molecule multi-targeted tyrosine kinase inhibitor that was synthesized by rational design, and histone deacetylases inhibitor (HDACi) suberoylanilide hydroxamic acid (SAHA), to overcome IM resistance in CML. S116836 at low concentrations used in the present study mildly downregulates auto-tyrosine phosphorylation of Bcr-Abl. SAHA, an FDA-approved HDACi drug, at 1 μM has modest anti-tumor activity in treating CML. However, we found a synergistic interaction between SAHA and S116836 in Bcr-Abl-positive CML cells that were sensitive or resistant to IM. Exposure of KBM5 and KBM5-T315I cells to minimal or non-toxic concentrations of SAHA and S116836 synergistically reduced cell viability and induced cell death. Co-treatment with SAHA and S116838 repressed the expressions of anti-apoptosis proteins, such as Mcl-1 and XIAP, but promoted Bim expression and mitochondrial damage. Of importance, treatment with both drugs significantly reduced cell viability of primary human CML cells, as compared with either agent alone. Taken together, our findings suggest that SAHA exerts synergistically with S116836 at a non-toxic concentration to promote apoptosis in the CML, including those resistant to imatinib or dasatinib.
Introduction
Chronic myelogenous leukemia (CML) is a type of hematopoietic stem cell disorder arising from chromosomal aberration between chromosomes 9 and 22.Citation1 This chromosomal abnormality, known as the Philadelphia chromosome, leads to myeloproliferation.Citation2 The balanced and reciprocal translocation of the chromosomes results in the creation of Bcr-Abl gene, which encodes a Bcr-Abl protein with enhanced tyrosine kinase activity.Citation3 Bcr-Abl is able to activate a wide range of signaling pathways. For example, Bcr-Abl increases the activation and/or expression of a series of anti-apoptotic proteins such as STATs,Citation4 Akt,Citation5 PI3K,Citation6 Mcl-1,Citation7 and Bcl-XL.Citation8 Imatinib is a well-established small molecule tyrosine kinase inhibitor that specifically targets the ATP-binding site of Bcr-Abl to prevent the autophosphorylation of Bcr-Abl itself.Citation9 Despite the specific and remarkable effect of imatinib, an increasing number of CML patients resistant to imatinib are emerging in clinic.Citation10 A frequent cause of the imatinib-resistance is point mutations in the Bcr-Abl relevant domains. There are more than 100 reported mutationsCitation11-Citation13 of which most can be conquered by the second-generation tyrosine kinase inhibitors (e.g., nilotinib, dasatinib), with the exception of the gate-keeper mutation T315I.Citation14-Citation16 Ponatinib, a third-generation of tyrosine kinase inhibitor, has shown activity against refractory CML patients including those harboring T315I Bcr-Abl. However, the long-term benefit of ponatinib has to be balanced against the risk of deleterious side effects in these patients.
S116836 was originally designed and synthesized against T315I Bcr-Abl (). The kinase assay showed that it blocked multiple tyrosine kinases including both wild-type as well as T315I Bcr-Abl. In addition, S116836 showed potent inhibitory effect on the SRC family kinases SRC, LYN, HCK, LCK, and BLK, and receptor tyrosine kinase such as FLT3, TIE2, KIT, PDGFRβ (Pan J et al., unpublished data). The lesson taken from imatinib and the second-generation of small molecule inhibitors (nilotinib and dasatinib) is that resistance will most likely arise after attenuating the function of one target. Disrupting a single pathway will likely be insufficient to eliminate the abnormal myeloid cells. The lack of efficacious and safe therapeutic regimen for the patients with cross-resistance to imatinib and dasatinib calls for a novel therapeutic strategy. The administration of multiple drugs simultaneously, as compared with single agent, exhibits greater anti-tumor activities and overcomes the drug resistance problems. It has been reported that combinations of histone deacetylase inhibitor (HDACi) with a series of small molecule inhibitors show synergistic effects in inducing apoptosis of various cancer cell lines.Citation17-Citation19
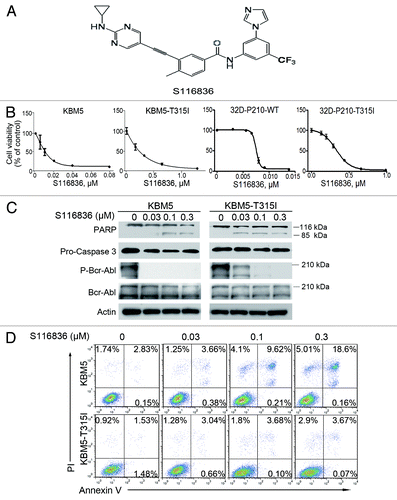
Figure 1. Treatment with S116836 inhibits growth and induces apoptosis of chronic myelogenous leukemia (CML) cells. (A) Chemical structure of compound S116836. (B) Cells were treated with S116836 for 72 h, cells viability was evaluated by using MTS assay. (C) KBM5 or KBM-T315I cells were exposed to increasing concentrations of S116836 for 24 h; western blotting analysis was performed with the indicated antibodies. (D) The CML cells were exposed to escalating concentrations of S116836 for 24 h, the percentages of apoptotic cells were detected by flow cytometry after dual labeling with FITC-Annexin V and propidium iodide (PI).
Histone acetylation plays a pivotal role in the regulation of gene expression. Histone acetyltrasferases transfer the acetyl moieties to the lysine residues of histones to form a relaxed chromatin state. In contrast, histone deacetylases (HDACs) remove the acetyl modification from lysine residues of histone, leading to a condensed chromatin state.Citation20 The balance between acetylation and deacetylation of the chromatin serves as a key epigenetic mechanism for transcription factor-dependent gene expression, which are consequently crucial for numerous fundamental cellular processes including cell cycle,Citation21 apoptosis,Citation22 DNA repair,Citation23 and differentiation.Citation24 However, the abnormal recruitments of HDAC to the promoter of the anti-tumor genes are closely associated with the onset and progression of tumor.Citation25-Citation29 HDACi have been shown to display anti-tumor ability by triggering apoptosis,Citation30 inducing differentiation,Citation31 suppressing cell proliferation,Citation32 and arresting cell cycle.Citation33,Citation34 An additional advantage of HDACi is their low toxicity to the normal tissues.Citation35
Suberoylanilide hydroxamic acid (SAHA), also known as vorinostat, is the first HDACi approved by US FDA for the treatment of cutaneous manifestations in patients with T-cell lymphoma.Citation36 SAHA represses HDACs by directly binding to the catalytic domain of the HDACs, ultimately causing overexpression of pro-apoptosis proteins (e.g., BAX, BAK, and Bim),Citation37,Citation38 and reduced expression of the anti-apoptosis proteins such as XIAP,Citation39 Bcl-2,Citation40 Bcl-XL,Citation41 and Mcl-1.Citation42 In addition, SAHA is able to raise the level of reactive oxygen species and promote the acetylation of non-histone proteins (e.g., p53 and heat shock protein 90, Hsp90).Citation43-Citation45 When HDAC6 is inhibited by SAHA treatment, the chaperone function of Hsp90 was shown to be disrupted through acetylation itself,Citation46 which finally results in the depletion of Bcr-Abl, Akt, and c-Raf. It is of interest that SAHA can work synergistically with various drugs, including the tyrosine kinase inhibitor (imatinib, dasatinib),Citation47 Hsp90 antagonist (7-AAG),Citation48 MEK1/2 inhibitor (PD184352),Citation49 and aurora kinase inhibitor (MK-0457),Citation50 to induce apoptosis of imatinib-resistant leukemia cells.
We hypothesized that SAHA had synergistic effect with S116836 on T315I Bcr-Abl CML cells. This study was to test the hypothesis. We discovered that combination of SAHA and S116836 has synergistic anti-leukemic activity in both wild-type and T315I Bcr-Abl CML cells. Our findings supported that combination of SAHA and S116836 hold promise to overcome imatinib-resistance in CML cells.
Results
S116836 inhibits cell growth and induces apoptosis in imatinib-sensitive and imatinib-resistant CML cells
S116836 is a novel compound to inhibit tyrosine kinase activity (). To determine its effect on CML cells, we first investigated the cell viability of KBM5 and KBM5-T315I under the treatment of S116836. Cells were cultured with escalating concentrations of S116836 for 72 h. MTS assay were performed to detect cell viability. S116836 inhibited growth in KBM5 and KBM5-T315I in a dose-dependent fashion with IC50 values of 15.73 and 407.96 nM, respectively (). Similarly, S116836 dose-dependently inhibited growth in 32D-P210-WT and 32D-P210-T315I with IC50 values of 7.1 and 362 nM, respectively ().
S116836 at lower concentrations (∼0.3 μM) appreciably blocked the phosphorylation of Bcr-Abl, but induced limited apoptosis in CML cells, particularly in imatinib-resistant KBM5-T315I, which was supported by the limited PARP cleavage and Annexin V-positive cells ().
Combination of SAHA and S116836 induces increased acetylated p53 and blockade of Bcr-Abl in CML cells
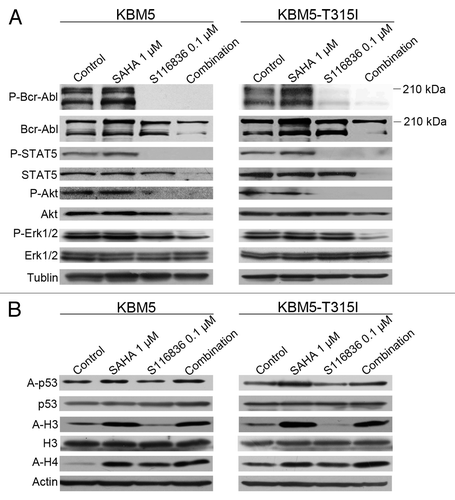
Insurgence of resistance or poor efficacy is a common problem of tyrosine kinase inhibitors in CML. Because many cases of resistance to tyrosine kinase inhibitors can be acquired through the activation of other tyrosine kinases,Citation51 growing attention has been focused on developing combinational approaches to interrupt different pathways. Because HDACi was reported to facilitate the ability of small molecule tyrosine kinase inhibitors,Citation47 we therefore examined the combinational effect of SAHA and S116836. KBM5 or KBM5-T315I cells were exposed to sublethal concentrations of SAHA and S116836 for 24 h, western blotting analysis showed that an increase in acetylation in histone H3 and H4, and representative molecule p53 in SAHA-treated or SAHA plus S116836-treated CML cells (). The phosphorylation of Bcr-Abl and its downstream molecules (e.g., STAT5 and Akt) was completely blocked in S116836-treated or SAHA plus S116836-treated CML cells (). No alteration of the levels of Bcr-Abl was noted. Of interest, further decreases in the phosphorylated Erk1/2 levels were observed in SAHA plus S116836-treated CML cells when compared with each drug alone (), suggesting that SAHA may be involved in the inactivation of Erk1/2. Additionally, the levels of STAT5 and Akt were dramatically reduced in the combination-treated CML cells in comparison with the single drug treated CML cells (). The acetylation in histone H3 and H4 and acetylated p53 were increased in SAHA-treated or SAHA plus S116836-treated CML cells (). Thus, it appears that the combination blocked the Bcr-Abl signaling and pro-survival signaling molecules.
Figure 2. Co-treatment with SAHA and S116836 exerts enhanced activity in blocking downstream signaling of Bcr-Abl. (A) KBM5 or KBM5-T315I cells were cultured with histone deacetylases inhibitor suberoylanilide hydroxamic acid (SAHA) (1 μM), alone or in combination with S116836 (0.1 μM) for 24 h, after which cell lysates were subjected to western blotting analysis. Co-treatment reduces the expression and the phosphorylation of Bcr-Abl, STAT5, Akt, and Erk 1/2. (B) Western blotting analysis was done to monitor the levels of acetylated (acetyl)-histone 3, acetyl-H4 and acetyl-p53/p53 in the cell lysates from KBM5 or KBM5-T315I cells after the cells were treated with SAHA, plus S116836 or not for 24 h.
SAHA and S116836 are synergistic to inhibit cell proliferation of imatinib-resistant CML cells
We next examined the combinational effect of SAHA, a broad spectrum of HDAC inhibitor, and S116836. We discovered that co-treatment of S116836 and SAHA synergistically reduced the cell growth of KBM5 and KBM-T315I cells when compared with SAHA or S116836 alone (). The synergistic suppression of SAHA and S116836 on the proliferation capacity of KBM5 and KBM-T315I cells was further confirmed by colony formation assay. Although S116836 itself was able to lower the cell proliferative activity, SAHA greatly augmented its capacity in reducing anchorage-independent growth ().
Figure 3. SAHA augments the activity of S116836 in inhibiting cell viability, colony formation, and inducing cell cycle disruptions. (A) After treatment with various concentrations of S116836 in the absence or presence of suberoylanilide hydroxamic acid (SAHA) for 72 h, viability of KBM5 or KBM-T315I cells was evaluated using the MTS assay. (B) KBM5 or KBM-T315I cells were exposed with SAHA, S116836 or the combination for 24 h, then the drugs were washed, the cells were seeded in soft agar for colony growth. The number of colony-forming in the absence of drug treatments (control) was defined as 100%. Then the yields of colony from the drug-treated cells were normalized relative to control. The experiments were independently repeated twice. A statistically significant difference was observed. Results are means with SEM; **P < 0.01; ***P < 0.0001, one-way ANOVA, post hoc comparisons, Tukey test. (C) KBM5 or KBM-T315I cells were subjected to treatments of SAHA (1 μM) and S116836 (100 nM) alone or in combination for 24 h, after which flow cytometric analysis of the DNA content was performed to assess cell cycle distribution.
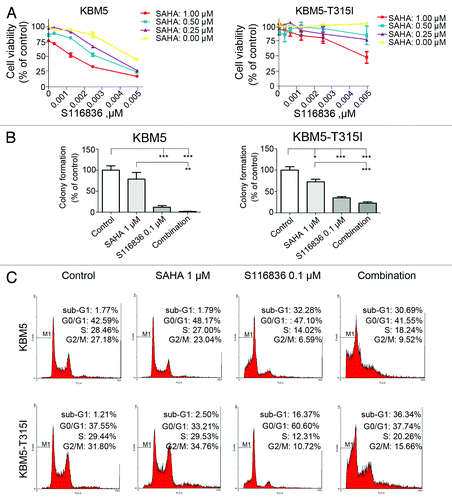
We profiled the cell-cycle distribution of CML cells treated with SAHA and S116836 (). SAHA alone had a limited effect on the cell cycle of KBM5 and KBM5-T315I cells. S116836 for 24 h induced a pronounced decline in the S phase proportion and an accumulation of the G1 phase proportion in imatinib-resistant KBM5-T315I cells. However, addition of SAHA to S116836 culture attenuated the change of G1 and S phase proportion in KBM5-T315I cells. SAHA induced significant increase in apoptosis in both KBM5 and KBM-T315I cells. These data suggest that SAHA may interrupt the cell cycle profile in imatinib-sensitive and imatinib-resistant CML cells.
SAHA enhances S116836-induced lethality in imatinib-sensitive and imatinib-resistant CML cells
We next explored the apoptosis in CML cells exposed to SAHA and S116836. KBM5 and KBM5-T315I cells were treated with the indicated concentrations of S116836 and SAHA alone or in combination for 24 h. The apoptotic cells were stained with Annexin V/PI, and detected by the flow cytometry. As shown in , SAHA (0 μM–1 μM) exhibited no toxicity to CML cells, and S116836 (0 μM–0.1 μM) induced minimal lethality either. However, when KBM5 cells or KBM5-T315I cells were exposed to combinational treatment, a substantial amount of apoptotic cells were observed (). It’s worth noting that the proapoptotic effect was much more significant in KBM5-T315I cells than that in KBM5 cells, indicating the combinational treatment may work in a Bcr-Abl-independent manner.
Figure 4. Co-treatment of SAHA and S116836 leads to significant apoptosis in CML cells. (A and B) KBM5 or KBM-T315I cells were treated with various concentrations of S116836 in combination with different concentrations of SAHA for 24 h, after which they were stained with FITC-Annexin V/propidium iodide. The percentages of the apoptosis cells were determined by the flow cytometry (A). Quantitative analysis of dead cells in 3 independent experiments was shown (B). Columns represent triple respective experiments and the bars denote means with SEM. (C) KBM5 and KBM-T315I cells were treated with various doses of SAHA (0.31 μM~1 μM), S116836 (0.031 μM~0.1 μM), or combination of the 2 drugs. Twenty-four hours later, apoptosis was measured by flow cytometry. Combination index (CI) was analyzed by using the CalcuSyn Software. CI < 1 represents synergism. (D) The CML cells were co-treated with SAHA (1 μM) and S116836 (0.1 μM) for 24 h. The cells were divided into two portions, the first portion was prepared the whole cell lysate with RIPA buffer for western blotting of the cleavage of PARP and the levels of caspase-8, pro-caspase-3, and active caspase-3 (upper). The second portion of the cells was prepared the mitochondria-free cytosolic fractions to monitor the levels of cytochrome c by western blotting (lower). (E) The expression of apoptosis-related proteins in whole cell lysates was detected by western blotting analysis. Actin was used as a loading control.
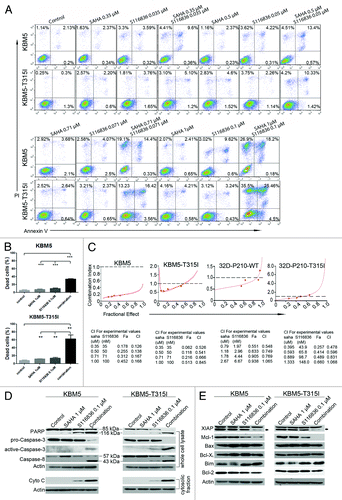
To draw a rigorous conclusion of synergistic effect, we used the widely-accepted CalcuSyn software to assess. The combination index (CI) < 1, = 1, and > 1, represent synergistic, additive, and antagonistic effects, respectively. Median dose effect analysis of apoptosis over various concentrations of S116836 and SAHA showed that the CI values of KBM5 cells were 0.126, 0.138, 0.167, and 0.168. The CI values of KBM5-T315I cells were 0.526, 0.541, 0.666, and 0.845 (), indicating a synergism between SAHA and S116836 (note: CI < 1.0 indicates synergism).
Additional attempts were made to define the pathway of apoptosis. We employed separate or combined treatments of SAHA (1 μM) and S116836 (0.1 μM) to KBM5 or KBM5-T315I cells, and monitored the cytosolic fractions of cytochrome c by western blotting analysis. Although the individual effects of either SAHA or S116836 were minimal, the combination induced a striking increase in cytosolic cytochrome c (). In accordance with the cytochrome c release, a considerable increases in cleavage of PARP procaspase-9, and pro-caspase-3 were observed. A substantial elevation in the levels of activated caspase-3 was also detected by western blotting analysis of the total cell lysate (). Furthermore, the combination of the two drugs also elicited an enhanced cleavage of pro-caspase-8, revealing a synergistic effect to extrinsic apoptotic pathway.
The combinatory effect of SAHA and S116836 were then examined in relation to changes on various proteins involved in apoptosis. As shown in , individual or combinatory treatment did not alter the expressions of Bax and Bcl-XL in both cell lines. In striking contrast, the levels of Mcl-1 and XIAP were dramatically reduced after co-treatment with SAHA and S116836 when compared with alone. In addition, the combined treatments led to a considerable upregulation of the pro-apoptosis protein Bim in both cell lines (). S116836 alone or combined with SAHA induced a slight or modest decrease in Bcl-2 expression in KBM5 but not KBM5-T315I cells. Taken together, the combination of SAHA and S116836 disturbs the expression of Bcl-2 family members.
SAHA/S116836 regimens synergistically inhibits the growth of primary CML cells
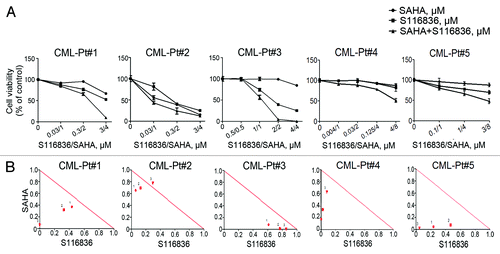
The findings of synergistic effects of SAHA and S116836 suggest a potential combination of two therapeutic agents against CML. In an attempt to translate our observations into a clinical model system, we investigated the effects of co-treatment with both agents on the growth of primary cells from patients with CML. The primary cells isolated from the peripheral blood or bone marrow from 5 patients with CML () were treated with drugs with indicated concentrations for 72 h, the cell viability monitored by MTS assay revealed an inhibition of S116836 on the growth of CML cells (), and addition of SAHA enhanced this inhibition. The CI values of these 5 patients was calculated by using the CalcuSyn software further supported a synergistic effect of the combination of SAHA and S116836 on the growth of primary CML cells ().
Table 1. Characteristics of patients with chronic myelogenous leukemia
Figure 5. The combination of SAHA and S116836 synergistically inhibits the cell viability in primary CML cells. (A) Peripheral blood mononuclear cells from 5 CML patients were treated with various concentrations of SAHA in the presence or absence of S116836 for 72 h, after which the cell viability of each agent alone and of combination was analyzed by the MTS assay. (B) The combination index (CI) about the viability of the cells treated with SAHA and S116836 was calculated with CalcuSyn Software.
Discussion
Despite the success of imatinib in Bcr-Abl-positive CML,Citation52 the development of resistance to IM and dasatinib, especially for those in accelerated and blast phases, remains a major challenge for treatments.Citation13,Citation15,Citation53 A promising approach to tackle the challenge is combining targeted drugs to interrupt multiple signaling pathways simultaneously. In this study, we demonstrate that combination of the multi-targeted tyrosine kinase inhibitor S116836 and HDACi SAHA causes significant mitochondrial damage and apoptosis in IM-sensitive or -resistant CML cells. Our data show that using either agent alone causes none or very minimum apoptosis; however, combining these two drugs together induces an enhanced lethality.
Our results were consistent with the findings of Fiskus et al., who reported that dasatinib and SAHA have synergism to kill imatinib-resistant cells.Citation47 However, S116836 is a novel compound that is active against T315I Bcr-Abl. Based on the median-effect method of Chou and Talalay, we compared the synergistic effect of TKI and SAHA in a pair of human chronic myelogenous leukemia (CML) patient-derived imatinib-sensitive KBM5 cells expressing the 210 kDa native Bcr-Abl and imatinib-resistant KBM5-T315I cells harboring a threonine-to-isoleucine substitution at position 315 of Abl. In addition, the synergism was observed with a pair of 32D myeloid cells stably expressing either 210 kDa wild-type Bcr-Abl or T315I Bcr-Abl was the synergistic effect of TKI and SAHA.
The dual targeting strategy has been witnessed widely in many different types of cancers. A recent research in lung cancer model showed that interrupting a single pathway failed to induce cell death, and instead suppressing multiple pathways was essential to induce cell death as a consequence of inhibiting overlapping or redundant pathways.Citation54 In the case of acute myeloid leukemia (AML), Gilliland and Griffin have laid out a two-hit concept of leukemogenesis which serves as a theoretical basis for combining targeted agents for AML therapy. This theory posits that leukemogenesis acts through collaboration between two types of proteins: Type I, their dysfunction leads to disarranged cell differentiation; Type II, their dysfunction promotes cell survival.Citation55 Thus, in the present study, one potential mechanism underlying this S116836/SAHA-mediated apoptosis may be simultaneous suppressions on the dysregulated differentiation pathway and cell survival pathway. It is also possible that disrupting these pathways lowers the threshold for SAHA-mediated cell apoptosis. The precise mechanisms need to be further studied.
In our study, one key protein with a role in the synergistic process is Mcl-1. Inoue et al. has reported that HDACi mediated Mcl-1 upregulation and bring out the limited efficacy of apoptosis.Citation56 However, either co-administration with a Mcl-1 inhibitor or downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in CML cells and CLL (chronic lymphocytic leukemia) cells.Citation56 SAHA will may upregulate Mcl-1 in CML cells, although our data show SAHA has no apparent effect on CML cells. S116836 has a modest effect in inhibiting Mcl-1. Thus, the synergistic effect of SAHA and S116836 may also work through the repression of Mcl-1 by S116836 and offset the side effect cause by SAHA.
While further studies are warranted to determine the mechanism by which S116836 regulate Mcl-1, several different pathways have been documented to modulate the expression of Mcl-1, including PI3K/Akt, STAT5, and MEK/ERK. PI3K/Akt regulates the Mcl-1 through a transcription factor complex containing CREB.Citation57 In CLL, sustained activation of Akt led to the increased expression of Mcl-1, Bcl-XL, and XIAP, greatly increasing leukemic cell viability.Citation58 Our results showed that S116836 attenuated the phosphorylation of Akt, and addition of SAHA reduced not only the activity but also the total expression of Akt. The expression level of total Akt is consistent with that of Mcl-1. Therefore, a potential mechanism may be that, when treated with SAHA and S116836 together, downregulation of Akt contributes to the reduction of Mcl-1, and further leads to the synergistic effect of apoptosis. In addition to Akt, recent evidence suggests STAT5 may also play an crucial role in regulating the expression of Mcl-1.Citation59 In Bcr-Abl+ cells, abnormal activation of STAT5 elevated the expression of Mcl-1, while silencing of STAT5 resulted in a decrease in the expression level of Mcl-1.Citation7,Citation60 In our results, expression and activation of STAT5 is similar to Akt and Mcl-1, suggesting that STAT may also play a role in the regulation of Mcl-1. We also observed that combined treatment with SAHA and S118936 led to a reduction of activated Erk1/2, which is related to downregulation of Mcl-1. It has been reported that actived Erk1/2 stabilizes Mcl-1 through phosporylating its PEST region.Citation61 Sorafenib directly inhibited Erk1/2 to lower the expression of Mcl-1.Citation62
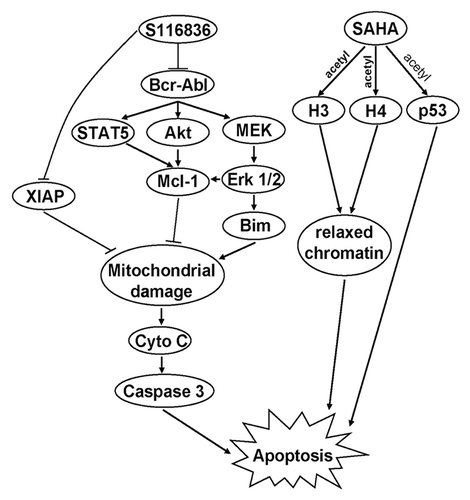
In addition, XIAP and Bim also showed synergism as a result of SAHA/S116836 combined treatments. We speculate that all of these proteins contribute to the induction of mitochondrial damage, which is reflected on the increasing release of cytochrome c and the PARP cleavage. In summary, our finding uncovered a new strategy of dual treatments that combining the S116836 and SAHA significantly induces apoptosis in CML cells. This synergistic effect may involve multiple mechanisms shown in .
Figure 6. The proposed model of SAHA and S116836 to mediate apoptosis in CML cells. Co-treatment with SAHA and S116836 inhibits the activity of Bcr-Abl, Akt, Erk 1/2, and STAT5, which result in reduction of Mcl-1 and XIAP, and upregulation of Bim. These effects may contribute to mitochondrial damage and subsequently induce the apoptosis of imatinib-sensitive or resistant cells.
It is worthy to note that enhanced lethality of SAHA/S116836 was not only seen in IM-sensitive cells, but also in the resistant ones. Several mechanisms often associated with IM resistance consist of Bcr-Abl overexpression, reduced uptake of IM, and acquisition of point mutation (e.g., T315I) in the Bcr-Abl kinase domain known to be essential for IM binding.Citation63 As showed in our results, S116836 is a potent compound in preventing the activation of Bcr-Abl no matter Bcr-Abl bears a T315I mutation or not. However, the effect of S116836 in inducing apoptosis of KBM5 or KBM5-T315I cells is still mild, indicating that, apart from the inhibition of Bcr-Abl, other mechanisms may have a role in the synergistic effect. Donato et al. found undetectable expression of BCR-ABL protein in imabinib-resistant cells from CML patients, suggesting that IM resistance may stem from a Bcr-Abl-independent manner.Citation64 Some other aberrantly expressed proteins beside Bcr-Abl may also cause this resistance in IM-resistant CML. Although Src-related Lyn kinase and Erk1/2 has been demonstrated that associated with the Bcr-Abl-independent resistance,Citation65,Citation66 it is still unclear whether any other proteins participate in this resistance. In this context, a multi-targets therapy is an ideal approach.
Although the precise mechanisms responsible for this phenomenon are elusive, HDACi has been well accepted to make leukemia cells more susceptible to apoptosis, and there are several successful examples of combining tyrosine kinase inhibitors with HDACi to induce apoptosis of CML cells resistant to IM—for instance, SAHA and IM, SAHA and dasatinib, LBH589 and AMN107.Citation47,Citation67,Citation68 Here we present the combination of SAHA and S116836 as a new therapy for treating IM-resistant CML.
Materials and Methods
Chemicals and antibodies
S116836 (structure shown in ) was dissolved in DMSO and 20 mM stock solution was stored at −20 °C. Antibodies against c-Abl (C-19), p53 (DO-7), Mcl-1 (S-19), Bcl-XL, histone H3, caspase-3, and Bax were from Santa Cruz Biotechnology. Antibodies against poly (ADP)-ribose polymerase (PARP; clone 4C10-5), cytochrome c, XIAP were from BD Biosciences. Antibodies against phospho-c-Abl (Y245), phospho-Erk 1/2 (T202/Y204), Erk 1/2, Akt, acetyl-histone H3 (K9), acetyl-histone H4 (K16), acetyl-p53 (K382), and caspase-8 were from Cell Signaling Technology. Antibodies against phospho-STAT5, STAT5, and Bcl-2 were from EMD Millipore Upstate.
Cell culture
KBM5 cells bearing 210 kDa wild-type Bcr-Abl were sensitive to IM. KBM5-T315I cells bearing T315I mutation in Bcr-Abl have the resistance to IM. Both of these cell lines were cultured in IMDM (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum (FCS), as described previously.Citation69 In addition, KBM5-T315I cells were also grown with 1.0 μM IM. imatinib was removed before experiments followed by a wash-out period of 2–3 d. The 32D myeloid cells stably expressing either 210 kDa wild-type Bcr-Abl (32D-P210-WT) or T315I Bcr-Abl (32D-P210-T315I) were maintained in RPMI 1640 with 10% FCS. Peripheral blood cells were obtained from 5 CML patients in the First Affiliated Hospital and Guangdong Provincial People’s Hospital (). The research is consistent with the institutional guidelines and the Declaration of Helsinki principles. The isolation of mononuclear cells has been described in previous work.Citation70,Citation71 Cells were suspended in RPMI 1640 supplemented with 10% FCS.
Cell viability assay
Cell viability was determined by MTS assay (CellTiter 96 Aqueous One Solution reagent; Promega).Citation70,Citation71 Cells were plated in quadruplicate onto the 96-well plates, and treated with the escalated concentrations of drugs for 72 h before performing the MTS assay. The optical density (O.D.) values for untreated group were set as 100% viability; the values of treated groups were normalized against the control group. The drug concentration that induced 50% inhibition of cell growth (IC50) was calculated by regression fitting of a dose-response curve.
Flow cytometry analysis of cell cycle
Cells were exposed to various concentrations of drugs, and then the cells were collected and fixed in 66% (V/V) cold ethanol at −20 °C. After more than 16 h, cell were washed twice with cold PBS and suspended with propidium iodide and RNase A (0.5 mg/mL) (in the dark) for 1 h. Data of cell cycle distribution was collected by using FACSCalibur flow cytometer and CellQuestPro software.
Clonogenicity assay
Cells treated with the indicated concentrations of SAHA and S116836 for 24 h, were then collected and washed twice with PBS, then, cultured in drug-free Iscove’s medium containing 0.3% agar and 20% FCS. After the ∼10 d incubation, the number of colonies was counted.
Assessment of apoptosis
Apoptosis was evaluated by using of Annexin V/propidium iodide (PI) binding assay according to the instruction of the manufacturer (Sigma-Aldrich). Samples were analyzed by using of FACSCalibur flow cytometer and CellQuestPro software as previously described.Citation72,Citation73
Western blotting analysis
Western analyses were performed as previously described.Citation74
Statistical analysis
Experiments were performed at least three times. GraphPad Prism 5.0 software (GraphPad Software) was used for statistical analysis. Combination Index was calculated by CalcuSyn software 2.0 according to the software’s instruction.
| Abbreviations: | ||
| CML | = | chronic myelogenous leukemia |
| IM | = | imatinib mesylate |
| HDACs | = | histone deacetylases |
| HDACi | = | HDAC inhibitor |
| SAHA | = | suberoylanilide hydroxamic acid |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported by grants from the National Basic Research Program of China (973, 2009CB825506 to J. Pan), National Natural Science Funds (81025021, U1301226, 81373434, and 91213304 to J. Pan), the Research Foundation of Education Bureau of Guangdong Province, China (cxzd1103) to J. Pan, the Research Foundation of Guangzhou Bureau of Science and Technology, the National Hi-Tech Research and Development Program of China (863, 2008AA02Z420 to J. Pan), and the Fundamental Research Funds for the Central Universities (to J. Pan).
References
- Rowley JD. Letter: A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature 1973; 243:290 - 3; http://dx.doi.org/10.1038/243290a0; PMID: 4126434
- McLaughlin J, Chianese E, Witte ON. In vitro transformation of immature hematopoietic cells by the P210 BCR/ABL oncogene product of the Philadelphia chromosome. Proc Natl Acad Sci U S A 1987; 84:6558 - 62; http://dx.doi.org/10.1073/pnas.84.18.6558; PMID: 3498165
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science 1986; 233:212 - 4; http://dx.doi.org/10.1126/science.3460176; PMID: 3460176
- de Groot RP, Raaijmakers JA, Lammers JW, Jove R, Koenderman L. STAT5 activation by BCR-Abl contributes to transformation of K562 leukemia cells. Blood 1999; 94:1108 - 12; PMID: 10419904
- Gesbert F, Sellers WR, Signoretti S, Loda M, Griffin JD. BCR/ABL regulates expression of the cyclin-dependent kinase inhibitor p27Kip1 through the phosphatidylinositol 3-Kinase/AKT pathway. J Biol Chem 2000; 275:39223 - 30; http://dx.doi.org/10.1074/jbc.M007291200; PMID: 11010972
- Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol 2000; 20:1179 - 86; http://dx.doi.org/10.1128/MCB.20.4.1179-1186.2000; PMID: 10648603
- Aichberger KJ, Mayerhofer M, Krauth MT, Skvara H, Florian S, Sonneck K, Akgul C, Derdak S, Pickl WF, Wacheck V, et al. Identification of mcl-1 as a BCR/ABL-dependent target in chronic myeloid leukemia (CML): evidence for cooperative antileukemic effects of imatinib and mcl-1 antisense oligonucleotides. Blood 2005; 105:3303 - 11; http://dx.doi.org/10.1182/blood-2004-02-0749; PMID: 15626746
- Gesbert F, Griffin JD. Bcr/Abl activates transcription of the Bcl-X gene through STAT5. Blood 2000; 96:2269 - 76; PMID: 10979976
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 2000; 289:1938 - 42; http://dx.doi.org/10.1126/science.289.5486.1938; PMID: 10988075
- Gambacorti-Passerini CB, Gunby RH, Piazza R, Galietta A, Rostagno R, Scapozza L. Molecular mechanisms of resistance to imatinib in Philadelphia-chromosome-positive leukaemias. Lancet Oncol 2003; 4:75 - 85; http://dx.doi.org/10.1016/S1470-2045(03)00979-3; PMID: 12573349
- Branford S. Chronic myeloid leukemia: molecular monitoring in clinical practice. Hematology Am Soc Hematol Educ Program 2007; 2007:376 - 83; http://dx.doi.org/10.1182/asheducation-2007.1.376; PMID: 18024654
- Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, Pane F, Müller MC, Ernst T, Rosti G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood 2011; 118:1208 - 15; http://dx.doi.org/10.1182/blood-2010-12-326405; PMID: 21562040
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293:876 - 80; http://dx.doi.org/10.1126/science.1062538; PMID: 11423618
- Cortes J, Rousselot P, Kim DW, Ritchie E, Hamerschlak N, Coutre S, Hochhaus A, Guilhot F, Saglio G, Apperley J, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood 2007; 109:3207 - 13; http://dx.doi.org/10.1182/blood-2006-09-046888; PMID: 17185463
- Redaelli S, Piazza R, Rostagno R, Magistroni V, Perini P, Marega M, Gambacorti-Passerini C, Boschelli F. Activity of bosutinib, dasatinib, and nilotinib against 18 imatinib-resistant BCR/ABL mutants. J Clin Oncol 2009; 27:469 - 71; http://dx.doi.org/10.1200/JCO.2008.19.8853; PMID: 19075254
- Müller MC, Cortes JE, Kim DW, Druker BJ, Erben P, Pasquini R, Branford S, Hughes TP, Radich JP, Ploughman L, et al. Dasatinib treatment of chronic-phase chronic myeloid leukemia: analysis of responses according to preexisting BCR-ABL mutations. Blood 2009; 114:4944 - 53; http://dx.doi.org/10.1182/blood-2009-04-214221; PMID: 19779040
- Nimmanapalli R, Fuino L, Bali P, Gasparetto M, Glozak M, Tao J, Moscinski L, Smith C, Wu J, Jove R, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or -refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res 2003; 63:5126 - 35; PMID: 12941844
- Rosato RR, Almenara JA, Yu C, Grant S. Evidence of a functional role for p21WAF1/CIP1 down-regulation in synergistic antileukemic interactions between the histone deacetylase inhibitor sodium butyrate and flavopiridol. Mol Pharmacol 2004; 65:571 - 81; http://dx.doi.org/10.1124/mol.65.3.571; PMID: 14978235
- Hideshima T, Bradner JE, Wong J, Chauhan D, Richardson P, Schreiber SL, Anderson KC. Small-molecule inhibition of proteasome and aggresome function induces synergistic antitumor activity in multiple myeloma. Proc Natl Acad Sci U S A 2005; 102:8567 - 72; http://dx.doi.org/10.1073/pnas.0503221102; PMID: 15937109
- Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem 2001; 70:81 - 120; http://dx.doi.org/10.1146/annurev.biochem.70.1.81; PMID: 11395403
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009; 325:834 - 40; http://dx.doi.org/10.1126/science.1175371; PMID: 19608861
- van Bavel CC, Dieker J, Muller S, Briand JP, Monestier M, Berden JH, van der Vlag J. Apoptosis-associated acetylation on histone H2B is an epitope for lupus autoantibodies. Mol Immunol 2009; 47:511 - 6; http://dx.doi.org/10.1016/j.molimm.2009.08.009; PMID: 19747733
- Yu S, Teng Y, Waters R, Reed SH. How chromatin is remodelled during DNA repair of UV-induced DNA damage in Saccharomyces cerevisiae. PLoS Genet 2011; 7:e1002124; http://dx.doi.org/10.1371/journal.pgen.1002124; PMID: 21698136
- Popova EY, Krauss SW, Short SA, Lee G, Villalobos J, Etzell J, Koury MJ, Ney PA, Chasis JA, Grigoryev SA. Chromatin condensation in terminally differentiating mouse erythroblasts does not involve special architectural proteins but depends on histone deacetylation. Chromosome Res 2009; 17:47 - 64; http://dx.doi.org/10.1007/s10577-008-9005-y; PMID: 19172406
- Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol 2009; 23:1963 - 72; http://dx.doi.org/10.1210/me.2009-0188; PMID: 19855091
- Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate 2004; 59:177 - 89; http://dx.doi.org/10.1002/pros.20022; PMID: 15042618
- Wilson AJ, Byun DS, Popova N, Murray LB, L’Italien K, Sowa Y, Arango D, Velcich A, Augenlicht LH, Mariadason JM. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J Biol Chem 2006; 281:13548 - 58; http://dx.doi.org/10.1074/jbc.M510023200; PMID: 16533812
- Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S, Iwase H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res 2004; 10:6962 - 8; http://dx.doi.org/10.1158/1078-0432.CCR-04-0455; PMID: 15501975
- Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell 2004; 5:455 - 63; http://dx.doi.org/10.1016/S1535-6108(04)00114-X; PMID: 15144953
- Tang YA, Wen WL, Chang JW, Wei TT, Tan YH, Salunke S, Chen CT, Chen CS, Wang YC. A novel histone deacetylase inhibitor exhibits antitumor activity via apoptosis induction, F-actin disruption and gene acetylation in lung cancer. PLoS One 2010; 5:e12417; http://dx.doi.org/10.1371/journal.pone.0012417; PMID: 20856855
- Liu S, Klisovic RB, Vukosavljevic T, Yu J, Paschka P, Huynh L, Pang J, Neviani P, Liu Z, Blum W, et al. Targeting AML1/ETO-histone deacetylase repressor complex: a novel mechanism for valproic acid-mediated gene expression and cellular differentiation in AML1/ETO-positive acute myeloid leukemia cells. J Pharmacol Exp Ther 2007; 321:953 - 60; http://dx.doi.org/10.1124/jpet.106.118406; PMID: 17389244
- Venkataramani V, Rossner C, Iffland L, Schweyer S, Tamboli IY, Walter J, Wirths O, Bayer TA. Histone deacetylase inhibitor valproic acid inhibits cancer cell proliferation via down-regulation of the alzheimer amyloid precursor protein. J Biol Chem 2010; 285:10678 - 89; http://dx.doi.org/10.1074/jbc.M109.057836; PMID: 20145244
- Simboeck E, Sawicka A, Zupkovitz G, Senese S, Winter S, Dequiedt F, Ogris E, Di Croce L, Chiocca S, Seiser C. A phosphorylation switch regulates the transcriptional activation of cell cycle regulator p21 by histone deacetylase inhibitors. J Biol Chem 2010; 285:41062 - 73; http://dx.doi.org/10.1074/jbc.M110.184481; PMID: 20952396
- Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, Fornace AJ Jr., Pospelova TV, Pospelov VA. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010; 9:3945 - 55; http://dx.doi.org/10.4161/cc.9.19.13160; PMID: 20935470
- Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target?. Cancer Cell 2003; 4:13 - 8; http://dx.doi.org/10.1016/S1535-6108(03)00165-X; PMID: 12892709
- Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007; 12:1247 - 52; http://dx.doi.org/10.1634/theoncologist.12-10-1247; PMID: 17962618
- Rosato RR, Almenara JA, Dai Y, Grant S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol Cancer Ther 2003; 2:1273 - 84; PMID: 14707268
- Zhao Y, Tan J, Zhuang L, Jiang X, Liu ET, Yu Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc Natl Acad Sci U S A 2005; 102:16090 - 5; http://dx.doi.org/10.1073/pnas.0505585102; PMID: 16243973
- Rosato RR, Maggio SC, Almenara JA, Payne SG, Atadja P, Spiegel S, Dent P, Grant S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid sphingomyelinase-dependent generation of ceramide. Mol Pharmacol 2006; 69:216 - 25; PMID: 16189296
- Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors down-regulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cell Biol 2005; 25:1608 - 19; http://dx.doi.org/10.1128/MCB.25.5.1608-1619.2005; PMID: 15713621
- Neuzil J, Swettenham E, Gellert N. Sensitization of mesothelioma to TRAIL apoptosis by inhibition of histone deacetylase: role of Bcl-xL down-regulation. Biochem Biophys Res Commun 2004; 314:186 - 91; http://dx.doi.org/10.1016/j.bbrc.2003.12.074; PMID: 14715264
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541 - 52; http://dx.doi.org/10.1038/sj.onc.1210620; PMID: 17694093
- Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol Cancer Ther 2009; 8:3075 - 87; http://dx.doi.org/10.1158/1535-7163.MCT-09-0254; PMID: 19887547
- Rao R, Lee P, Fiskus W, Yang Y, Joshi R, Wang Y, Buckley K, Balusu R, Chen J, Koul S, et al. Co-treatment with heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin (DMAG) and vorinostat: a highly active combination against human mantle cell lymphoma (MCL) cells. Cancer Biol Ther 2009; 8:1273 - 80; http://dx.doi.org/10.4161/cbt.8.13.8726; PMID: 19440035
- Zhao Y, Lu S, Wu L, Chai G, Wang H, Chen Y, Sun J, Yu Y, Zhou W, Zheng Q, et al. Acetylation of p53 at lysine 373/382 by the histone deacetylase inhibitor depsipeptide induces expression of p21(Waf1/Cip1). Mol Cell Biol 2006; 26:2782 - 90; http://dx.doi.org/10.1128/MCB.26.7.2782-2790.2006; PMID: 16537920
- Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, Mandawat A, Atadja P, Bradner JE, Bhalla K. HDAC6 inhibition enhances 17-AAG--mediated abrogation of hsp90 chaperone function in human leukemia cells. Blood 2008; 112:1886 - 93; http://dx.doi.org/10.1182/blood-2008-03-143644; PMID: 18591380
- Fiskus W, Pranpat M, Balasis M, Bali P, Estrella V, Kumaraswamy S, Rao R, Rocha K, Herger B, Lee F, et al. Cotreatment with vorinostat (suberoylanilide hydroxamic acid) enhances activity of dasatinib (BMS-354825) against imatinib mesylate-sensitive or imatinib mesylate-resistant chronic myelogenous leukemia cells. Clin Cancer Res 2006; 12:5869 - 78; http://dx.doi.org/10.1158/1078-0432.CCR-06-0980; PMID: 17020995
- Rahmani M, Yu C, Dai Y, Reese E, Ahmed W, Dent P, Grant S. Coadministration of the heat shock protein 90 antagonist 17-allylamino- 17-demethoxygeldanamycin with suberoylanilide hydroxamic acid or sodium butyrate synergistically induces apoptosis in human leukemia cells. Cancer Res 2003; 63:8420 - 7; PMID: 14679005
- Yu C, Dasmahapatra G, Dent P, Grant S. Synergistic interactions between MEK1/2 and histone deacetylase inhibitors in BCR/ABL+ human leukemia cells. Leukemia 2005; 19:1579 - 89; http://dx.doi.org/10.1038/sj.leu.2403868; PMID: 16015388
- Dai Y, Chen S, Venditti CA, Pei XY, Nguyen TK, Dent P, Grant S. Vorinostat synergistically potentiates MK-0457 lethality in chronic myelogenous leukemia cells sensitive and resistant to imatinib mesylate. Blood 2008; 112:793 - 804; http://dx.doi.org/10.1182/blood-2007-10-116376; PMID: 18505786
- Milojkovic D, Apperley J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin Cancer Res 2009; 15:7519 - 27; http://dx.doi.org/10.1158/1078-0432.CCR-09-1068; PMID: 20008852
- Sawyers CL, Hochhaus A, Feldman E, Goldman JM, Miller CB, Ottmann OG, Schiffer CA, Talpaz M, Guilhot F, Deininger MW, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood 2002; 99:3530 - 9; http://dx.doi.org/10.1182/blood.V99.10.3530; PMID: 11986204
- Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med 2006; 354:2531 - 41; http://dx.doi.org/10.1056/NEJMoa055229; PMID: 16775234
- Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, Peng Z, Herbst RS, Papadimitrakopoulou V, Minna JD, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One 2010; 5:e14124; http://dx.doi.org/10.1371/journal.pone.0014124; PMID: 21124782
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002; 100:1532 - 42; http://dx.doi.org/10.1182/blood-2002-02-0492; PMID: 12176867
- Inoue S, Walewska R, Dyer MJ, Cohen GM. Downregulation of Mcl-1 potentiates HDACi-mediated apoptosis in leukemic cells. Leukemia 2008; 22:819 - 25; http://dx.doi.org/10.1038/leu.2008.1; PMID: 18239621
- Wang JM, Chao JR, Chen W, Kuo ML, Yen JJ, Yang-Yen HF. The antiapoptotic gene mcl-1 is up-regulated by the phosphatidylinositol 3-kinase/Akt signaling pathway through a transcription factor complex containing CREB. Mol Cell Biol 1999; 19:6195 - 206; PMID: 10454566
- Longo PG, Laurenti L, Gobessi S, Sica S, Leone G, Efremov DG. The Akt/Mcl-1 pathway plays a prominent role in mediating antiapoptotic signals downstream of the B-cell receptor in chronic lymphocytic leukemia B cells. Blood 2008; 111:846 - 55; http://dx.doi.org/10.1182/blood-2007-05-089037; PMID: 17928528
- Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, Iino T, Rocnik JL, Kikushige Y, Mori Y, Shima T, Iwasaki H, Takenaka K, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009; 114:5034 - 43; http://dx.doi.org/10.1182/blood-2008-12-196055; PMID: 19808698
- Shuai K, Halpern J, ten Hoeve J, Rao X, Sawyers CL. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996; 13:247 - 54; PMID: 8710363
- Domina AM, Vrana JA, Gregory MA, Hann SR, Craig RW. MCL1 is phosphorylated in the PEST region and stabilized upon ERK activation in viable cells, and at additional sites with cytotoxic okadaic acid or taxol. Oncogene 2004; 23:5301 - 15; http://dx.doi.org/10.1038/sj.onc.1207692; PMID: 15241487
- Ding Q, Huo L, Yang JY, Xia W, Wei Y, Liao Y, Chang CJ, Yang Y, Lai CC, Lee DF, et al. Down-regulation of myeloid cell leukemia-1 through inhibiting Erk/Pin 1 pathway by sorafenib facilitates chemosensitization in breast cancer. Cancer Res 2008; 68:6109 - 17; http://dx.doi.org/10.1158/0008-5472.CAN-08-0579; PMID: 18676833
- Jabbour E, Parikh SA, Kantarjian H, Cortes J. Chronic myeloid leukemia: mechanisms of resistance and treatment. Hematol Oncol Clin North Am 2011; 25:981 - 95, v; http://dx.doi.org/10.1016/j.hoc.2011.09.004; PMID: 22054730
- Donato NJ, Wu JY, Stapley J, Lin H, Arlinghaus R, Aggarwal BB, Shishodia S, Albitar M, Hayes K, Kantarjian H, et al. Imatinib mesylate resistance through BCR-ABL independence in chronic myelogenous leukemia. Cancer Res 2004; 64:672 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-03-1484; PMID: 14744784
- Donato NJ, Wu JY, Stapley J, Gallick G, Lin H, Arlinghaus R, Talpaz M. BCR-ABL independence and LYN kinase overexpression in chronic myelogenous leukemia cells selected for resistance to STI571. Blood 2003; 101:690 - 8; http://dx.doi.org/10.1182/blood.V101.2.690; PMID: 12509383
- Nambu T, Araki N, Nakagawa A, Kuniyasu A, Kawaguchi T, Hamada A, Saito H. Contribution of BCR-ABL-independent activation of ERK1/2 to acquired imatinib resistance in K562 chronic myeloid leukemia cells. Cancer Sci 2010; 101:137 - 42; http://dx.doi.org/10.1111/j.1349-7006.2009.01365.x; PMID: 19843070
- Fiskus W, Pranpat M, Bali P, Balasis M, Kumaraswamy S, Boyapalle S, Rocha K, Wu J, Giles F, Manley PW, et al. Combined effects of novel tyrosine kinase inhibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl-expressing human leukemia cells. Blood 2006; 108:645 - 52; http://dx.doi.org/10.1182/blood-2005-11-4639; PMID: 16537804
- Yu C, Rahmani M, Almenara J, Subler M, Krystal G, Conrad D, Varticovski L, Dent P, Grant S. Histone deacetylase inhibitors promote STI571-mediated apoptosis in STI571-sensitive and -resistant Bcr/Abl+ human myeloid leukemia cells. Cancer Res 2003; 63:2118 - 26; PMID: 12727828
- Zhang H, Trachootham D, Lu W, Carew J, Giles FJ, Keating MJ, Arlinghaus RB, Huang P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008; 22:1191 - 9; http://dx.doi.org/10.1038/leu.2008.74; PMID: 18385754
- Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, Sun X, Wu Y, Zhou J, Pan J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res 2010; 70:2516 - 27; http://dx.doi.org/10.1158/0008-5472.CAN-09-3950; PMID: 20215516
- Wu Y, Chen C, Sun X, Shi X, Jin B, Ding K, Yeung SC, Pan J. Cyclin-dependent kinase 7/9 inhibitor SNS-032 abrogates FIP1-like-1 platelet-derived growth factor receptor α and bcr-abl oncogene addiction in malignant hematologic cells. Clin Cancer Res 2012; 18:1966 - 78; http://dx.doi.org/10.1158/1078-0432.CCR-11-1971; PMID: 22447844
- Pan J, Quintás-Cardama A, Kantarjian HM, Akin C, Manshouri T, Lamb P, Cortes JE, Tefferi A, Giles FJ, Verstovsek S. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. Blood 2007; 109:315 - 22; http://dx.doi.org/10.1182/blood-2006-04-013805; PMID: 16912224
- Pan J, Quintás-Cardama A, Manshouri T, Giles FJ, Lamb P, Tefferi A, Cortes J, Kantarjian H, Verstovsek S. The novel tyrosine kinase inhibitor EXEL-0862 induces apoptosis in human FIP1L1-PDGFR-alpha-expressing cells through caspase-3-mediated cleavage of Mcl-1. Leukemia 2007; 21:1395 - 404; http://dx.doi.org/10.1038/sj.leu.2404714; PMID: 17495975
- Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q, Lu Z, Chen Q, Lai Y, Pan J. Triptolide inhibits Bcr-Abl transcription and induces apoptosis in STI571-resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 2009; 15:1686 - 97; http://dx.doi.org/10.1158/1078-0432.CCR-08-2141; PMID: 19240172