
Abstract
The chemotherapeutic agents doxorubicin (dox) or 5-fluorouracil (5FU) are used to treat cancer cells as they cause irreparable DNA damage, inducing these aberrant cells to undergo cell death. The mediator of this process is presumed to be in part the tumor suppressor p53 which regulates genes involved in DNA repair and cell death. When MCF-7 breast cancer cells are treated with these drugs, we observed that the level of p53 and the p53 negative regulator, Mdm2, increased, as seen by others. But contrary to some reports, we observed minimal phosphorylation of p53 at serine 15 in MCF-7 cells after drug treatment. Interestingly, we determined that there was differential regulation of the kinases ATM and Chk2 with the drug treatments, likely the cause for the lack of phosphorylation of p53. We found a dramatic drop in p53 DNA binding affinity for p21 and other gene response elements (RE) after drug treatment. To determine if the p53 that accumulated in the drug treated cells was functionally active, we monitored changes in the protein products of two p53-regulated genes following drug treatment with and without the addition of a p53-specific siRNA. In response to 5FU, both p21 and Mdm2 proteins increased and that increase was alleviated if a p53-specific siRNA was added. This effect was not seen with the addition of dox. Thus, the phosphorylation at serine 15 is not necessary for the functional activation of this transcription factor. We propose a new model for the regulation of p53, Mdm2, and MdmX after drug treatment.
Introduction
p53 is a tumor suppressor protein that accumulates in the nucleus upon the induction of DNA damage to the cell and binds to REs of its gene targets.Citation1 Wild-type p53 controls a number of genes that signal for regulation of important cellular pathways and processes, including classical p53 targets that control the cell cycle and apoptosis, or more recently discovered genes that control pathways involved in protein catabolism, metabolism and ion transport control.Citation1,Citation2 There are many forms of cellular stress known to induce p53 accumulation upon DNA damage, such as gamma irradiation (IR), oxidative stress, UV light, and many commonly used chemotherapeutic agents. These chemotherapeutic agents include the topoisomerase II (an enzyme which unwinds DNA) inhibitors etoposide and doxorubicin (dox), as well as 5-fluorouracil (5FU). Etoposide inhibits the enzyme topoisomerase II that is responsible for unwinding the DNA helix during transcription and DNA replication.Citation3 Dox intercalates into DNA and inhibits the progression of topoisomerase II which halts the process of DNA replication.Citation4 5FU is a thymidylate synthase inhibitor that prevents the production of dTMP, causing rapidly dividing cells to cease replication and die.Citation5 Treatment of cells with these chemotherapeutic agents results in the accumulation of p53, which plays a critical role in maintaining genomic integrity in the cell by activating genes that are responsible for DNA repair, cell cycle arrest, and apoptosis.Citation6-Citation8
The relationship between p53 and its most important negative regulator, Mdm2, has been cataloged extensively over the years, yet continues to evolve as new research emerges that challenges the accepted model for regulation. The mdm2 gene is a direct binding target of p53, and the Mdm2 protein is responsible for ubiquitinating p53, directing the latter protein for proteasomal degradation.Citation9 In addition to the E3 ubiquitin ligase activity of Mdm2, this protein also binds tightly to the transactivation domain (TAD) of p53 and blocks transactivation of genes by p53.Citation9 Upon cellular stress, others have shown that the association between p53 and Mdm2 is abrogated and the p53 protein accumulates.Citation10 The Mdm2 protein is a 491 amino acid polypeptide that is observed as a 90 kDa protein and has demonstrated over 40 spliced variants.Citation11,Citation12 The patterning and numbers of alternatively spliced Mdm2 mRNA transcripts has also been shown to correlate with overall p53 status and survival.Citation13 From a functional standpoint, certain regions of Mdm2 are responsible for discreet activities of this oncoprotein, specifically the N-terminus binding to the p53 TAD,Citation14 as well as the central region and the carboxy-terminus conferring its E3 ubiquitin ligase activity.Citation12,Citation15,Citation16 Blocking the interaction between p53 and Mdm2 has been explored as a possible anti-cancer therapy, as it may allow the tumor suppressive function of p53 to be augmented. The hydrophobic pocket of Mdm2 where p53 TAD peptides bind is the target of Mdm2 antagonists.Citation17 Vassilev and colleagues demonstrate how certain compounds (nutlins) could disrupt the Mdm2-p53 interaction.Citation18 These compounds and their derivatives are currently in clinical trials for treatment of a variety of cancers.
The accumulation of the p53 protein after induction of cellular stress may be negatively regulated in conjunction with or by proteins other than Mdm2. The Mdm2 protein homolog, MdmX, has been reported to be involved in the Mdm2-mediated regulation of p53.Citation19 Chen and colleaguesCitation20 report that Mdm2 polyubiquitinates MdmX and thus results in the degradation of the MdmX protein after DNA damage. They showed that the ataxia telangiectasia mutated (ATM)-mediated phosphorylation of MdmX at serine 403 and subsequent Chk2-mediated phosphorylation of MdmX at serine 342 and serine 367 alter the binding specificity of Mdm2 for MdmX. This group also demonstrated that mutant MdmX lacking these sites for phosphorylation was resistant to polyubiquitination by Mdm2 and therefore was not degraded, which conferred stability of the MdmX protein.Citation20 The inability of the Mdm2 in this case to interact with MdmX is proposed to shift the balance of Mdm2 toward binding p53. These results suggest that the ubiquitin ligase activity of Mdm2 may be redirected to MdmX upon DNA damage since the interaction between p53 and Mdm2 has been blocked.Citation20 If the interaction between Mdm2 and MdmX increases, it may allow p53 to accumulate even in the presence of Mdm2. The interaction between p53 and Mdm2 has been shown to be affected by the phosphorylation status of p53.
Three main studies provide evidence that the phosphorylation of numerous serine and threonine residues in the N-terminal TAD of the p53 protein is an ordered process generally beginning with phosphorylation of serine 15.Citation9,Citation21,Citation22 Based on the results of these studies, it is evident that both serine 15 and serine 20 phosphorylation of p53 may be involved in cooperatively blocking interaction with Mdm2. As noted above, loss of Mdm2 binding enhances the transcriptional activity of p53 by allowing the N-terminal TAD of p53 to interact with the transcriptional machinery in the cell. Phosphorylation of both serine 15 and serine 20 thus promotes the function of the p53 protein as a transcription factor. There are published reports that show how additional proteins, such as ERK2 and Mdm2-ribosomal proteins, may also be involved in posttranslational modification (PTM) or activation of p53.Citation23,Citation24 Serine 15 and serine 20 are first phosphorylated by the kinase Chk2 which in turn is activated by the kinase ATM.Citation25,Citation26 Chehab and colleagues demonstrate in human fibroblasts and U2OS osteosarcoma cells treated with UV that Chk2 phosphorylates p53 at serine 20 in an ATM-independent manner, resulting in dissociation of the p53-Mdm2 complex and accumulation of the p53 protein.Citation27 Decreased levels of ATM or Chk2 or decreases in their activation may be a potential mechanism promoting cancer cell survival in the presence of a functional wild-type p53 protein.
To better understand the regulation of p53 after drug treatment with 5FU or dox, we studied the change in the p53 protein level and PTMs in MCF-7 breast cancer cells that express wild-type p53.Citation26 As the PTM did not follow the accepted model for p53 regulation after DNA damage, we evaluated the ability of p53 to bind DNA after DNA damage and affect transcription. We found that the protein is still able to bind DNA and regulate gene expression following treatment with 5FU. Our observations suggest an alternative model for the regulation of p53 after DNA damage with 5FU and dox in these cancer cells.
Results
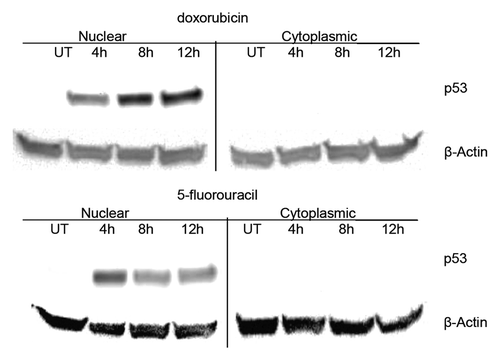
The level of p53 in the cell is maintained at low levels and increases in response to cellular stress and DNA damage.Citation6-Citation8 In order to measure the amount of p53 after drug exposure, the nuclear fractions of treated and untreated MCF-7 cells were analyzed using ELISA (enzyme- linked immunosorbent assay) (data not shown) and western blots. The level of nuclear p53 harvested from untreated cell extracts was determined to be low. However, upon treatment of MCF-7 cell cultures with the DNA damaging agents dox, 5FU or UV, the level of p53 increased significantly (). The cells were incubated with dox or 5FU for 4, 8, or 12 h and in both cases, the level of p53 increased greater than 20-fold after 12 h (based on ELISA results). The p53 protein was found to be almost exclusively localized to the nucleus even after drug treatment ().
Figure 1. Localization of p53 in untreated and drug treated MCF-7 breast cancer cells. MCF-7 cells were treated with doxorubicin or 5-fluorouracil for 4–12 h or untreated (UT), then nuclear and cytoplasmic extracts were produced as described in the Materials and Methods section. Protein from each extract (25 μg) was loaded onto a 10% polyacrylamide gel, transferred to nitrocellulose and then probed for either p53 or β-actin. Western blot analysis shows the distribution of p53 in nuclear vs. cytoplasmic fractions for MCF-7 untreated, doxorubicin, and 5-fluorouracil treated cells for various times. β-actin was used as a loading control. The p53 protein increases dramatically after drug treatment and is nearly exclusively located in the nucleus of MCF-7 breast cancer cells in all conditions.
The p53 protein functions as a tumor suppressor by binding to a wide array of gene targets that control various aspects of cell cycle regulation and apoptosis.Citation6,Citation7 Thus, we analyzed the affinity of p53 for binding to target DNA response elements (REs) using nuclear extracts from untreated and drug treated (dox or 5FU) MCF-7 cells initially using the EMSA (electrophoretic mobility shift assay) normalizing either for p53 or for total protein. We observed differences in the DNA binding detected. When we used the same level of total protein from the nuclear extracts in the DNA binding reaction with the same gene sequences, we found the p53 binds to REs of its gene targets p21, puma, mdm2, bax and surviving, with no significant increase (average P = 0.15) in the shifted band after drug treatment (). However, when the level of p53 is kept constant from each of the extracts, the shifted band for these gene REs showed a significant decrease (average P = 0.006) upon treatment with either dox or 5FU (). We confirmed that p53 is responsible for the shift in the band using siRNA treated MCF-7 cells with a reduction in the shifted band corresponding to the drop in p53 protein (data not shown). The drop in affinity of p53 for the gene target REs over the drug time course was corroborated using the streptavidin magnetic bead assay.
Figure 2. Electrophoretic mobility shift assay (EMSA) using p53 from MCF-7 nuclear extracts and p53 target gene response elements (REs). MCF-7 cells were treated with doxorubicin (dox) or 5-fluorouracil (5FU) for 4–12 h or untreated (UT), then nuclear extracts were produced as described in the Materials and Methods section. DNA binding was performed by EMSA as described by Chandrachud and GalCitation57 and Ray and colleaguesCitation51 using the fluorescently labeled p21 sequence. (A and C) Aliquots of MCF-7 untreated and drug-treated nuclear extracts representing 20 μg total protein were reacted with 5 pmoles of 6-FAM p21 gene RE (A) or the p53 REs from the puma, mdm2, bax, and survivin genes (C). A shifted band was observed for each reaction with a commensurate change in the unbound DNA. The standard deviation of the mean relative to untreated control for each sample was calculated at a 95% confidence level for 3 replicates for the bound DNA sequence. The average change in band density of the shifted band (protein + DNA) was found to be insignificant in each case (avg. P = 0.15). (B and D) Aliquots of MCF-7 untreated (UT) and drug-treated nuclear extracts representing 40 pg of p53 were reacted with 5 pmoles of 6-FAM p21 gene RE (B) or the p53 REs from the puma, mdm2, bax, and survivin genes (D). A shifted band was observed for the reaction with the untreated sample with a commensurate decrease in the unbound DNA compared with the reaction without extract. However, there is minimal detection of a shifted band after drug treatment (either dox or 5FU), as well as an increase in the unbound DNA signal. The standard deviation of the mean relative to untreated control for each sample was calculated at a 95% confidence level for 3 replicates for each bound DNA sequence. The average change in band density relative to the shifted band (protein + DNA) was found to be significant in each case (avg. P = 0.006). DNA only (p21) and extract only (Ext) were used as controls (A and B). Unexpectedly, the increase in level of p53 is associated with a decrease in DNA binding.
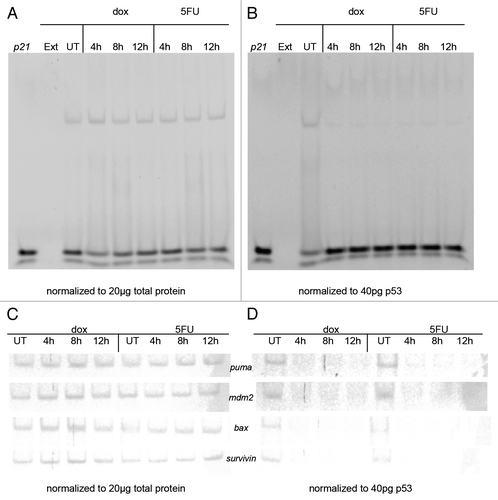
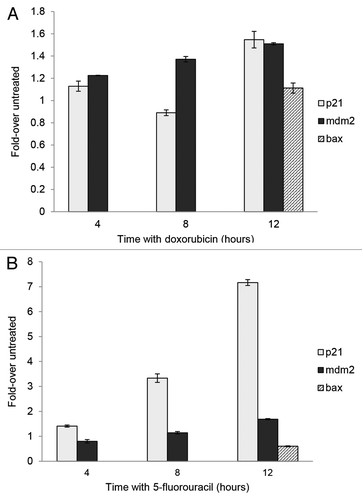
To determine if the association of p53 with chromatin changed over time with the drugs, we performed chromatin immunoprecipitation (ChIP) using chromatin from MCF-7 cells treated for various times with either 5FU or dox. We observed that p53 associates with the p21 RE and that association increases significantly (P < 0.0001) after treatment with 5FU (). The p53 protein association with the RE of mdm2 changed minimally over time with 5FU while we observed a slight change in p53 association to the bax RE after 12 h of drug treatment with dox or 5FU. PCR amplification performed with p21 NS (non-specific) primer and immunoprecipitation (IP) with a normal IgG antibody show negligible detection of bands (data not shown). Interestingly, although we see a dramatic increase in the level of p53 after treatment (), the amount of p53 binding to DNA () and associating with chromatin () is not commensurate with the increase in level of the protein. The reason for this discrepancy is not understood.
Figure 3. Chromatin immunoprecipitation (ChIP) of p53 from MCF-7 cells following treatment with 5-fluorouracil and doxorubicin. MCF-7 cells were treated with doxorubicin or 5-fluorouracil for 4–12 h or untreated, then chromatin was produced as described in the Materials and Methods section. ChIP was performed using anti-p53 antibodies then PCR done on the isolated chromatin using the p21, mdm2, and bax gene RE sequences as described. The raw density values detected in gels (data not shown) were obtained and plotted as compared with that detected with an untreated sample. The standard deviation of the mean relative to input of each sample, shown as the error bars, was calculated at a 95% confidence level for 3 replicates. (A) After a 12 h treatment with doxorubicin, there is a significant increase in the association of p53 with p21 (P = 0.0006) and mdm2 (P = 0.0003). (B) There is a significant increase in the association of p53 with p21 (P < 0.0001) and mdm2 (P = 0.0003) after a 12 h treatment with 5-fluorouracil. The bax gene was not analyzed at intermediate times, only with untreated and 12 h treated samples.
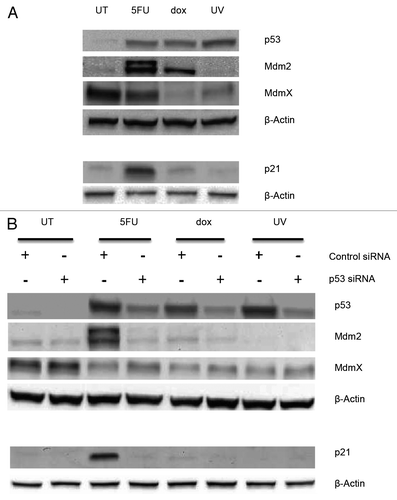
In order to confirm functionality of p53, we measured the level of two proteins whose genes are targets of p53 regulation, namely p21 and Mdm2, following treatment of cells with drugs. We found that the level of both p21 and Mdm2 increase after 5FU treatment, while only the Mdm2 protein was elevated following treatment with dox (). To test the role of the p53 protein in these changes, we treated the cells with siRNA for the TP53 gene and followed that with treatment of the cells with the drugs. In the presence of TP53 siRNA, the levels of both of these proteins are reduced following drug treatment compared with their level with control siRNA (). Thus, it appears that p53 is affecting the level of both of these proteins consistent with its role in affecting the transcription of the p21 and mdm2 genes.Citation28 This would suggest the p53 protein is functional as a transcription factor following drug treatment. MdmX is an Mdm2 homolog known to interact with both p53 and Mdm2 affecting the accumulation of p53.Citation20,Citation29,Citation30 Thus, we were curious whether the level of this protein changed after drug treatment. The MdmX protein is detectable in whole cell extracts from untreated cells and cells treated with 5FU and UV, but minimally from cells treated with dox (). When evaluating the level of the MdmX protein whose gene is not a transcriptional target of p53, the level of this protein does not change with TP53 siRNA treatment and treatment with dox or UV (). Interestingly, the level of the MdmX protein increases following 5FU and TP53 siRNA treatment. We hypothesize that this may be due to the decrease in the level of the Mdm2 protein which is known to induce ubiquitination and degradation of MdmX.Citation30
Figure 4. Whole cell extracts from MCF-7 cells treated with drugs and p53 siRNA. (A) MCF-7 cells were treated with 5-fluorouracil (5FU) or doxorubicin (dox) for 12 h or with UV light (UV) then left for 24 h or left untreated (UT). (B) MCF-7 cells were pretreated with 5 nM p53-specific or non-targeting siRNA for 24 h, followed by treatment with drugs or UV as above. Whole cell extracts were prepared using RIPA buffer and 30 μg total protein separated on either 10% polyacrylamide Tris-glycine SDS gels for blots probed for p53, Mdm2, and MdmX or 16.5% polyacrylamide Tris-tricine SDS gels for blots probed for p21 as described in the Materials and Methods section. The proteins were transferred to nitrocellulose and then probed for the indicated proteins as described in the Materials and Methods section. The results indicate that the p53 protein is required for the accumulation of both Mdm2 and p21 following drug treatment but not for changes in the MdmX protein.
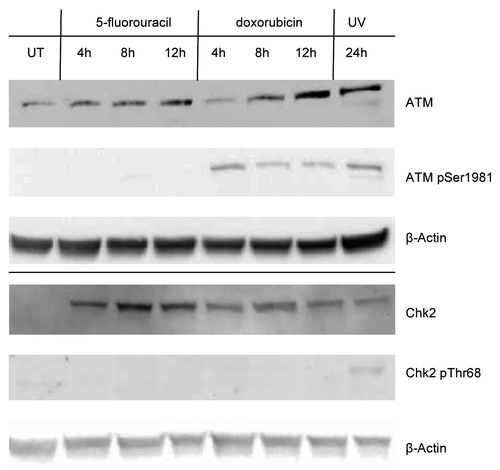
Posttranslational modification (PTM) of p53 is known to affect both the level and function of this transcription factor due to interaction with its negative regulator Mdm2.Citation10 We monitored the level of phosphorylation of p53 at serines 15, 20, 46, and 392, as well as the level of acetylation at lysines 120 and 373 using western blots loading the same amount of p53 from untreated as well as drug or UV treated MCF-7 cells. We found that p53 does not acquire these PTMs at any of these residues upon treatment with either dox or 5FU after 12 h or up to 24 h (data not shown). However, after treatment of MCF-7 cells with UV, we observed robust phosphorylation of serine 15 as well as at serine 392, and acetylation at lysines 120 and 373 on the p53 from nuclear extracts. We only detected phosphorylation at serine 15 once we loaded approximately 6000 pg of p53 from whole cell extracts treated with dox (). This level of serine 15 phosphorylated p53 in the extracts from dox treated cells equated to approximately 7-fold less compared with that found in UV treated cell extracts for the same amount of p53 protein. We did not detect any modifications on the p53 in extracts from the 5FU treated MCF-7 cells. The lack of induction of phosphorylation of p53 at serine 15 led us to investigate the presence of the enzymes ATM and Chk2, both presumed to be required for phosphorylation of p53 at this site.Citation25,Citation26,Citation31 We probed for the presence of total ATM and Chk2, as well as for the phosphorylated (i.e., active) forms of these enzymes in nuclear extracts of drug-treated MCF-7 cells. We detected the presence of both proteins in all extracts, except in the untreated MCF-7 extracts where we did not detect Chk2 (). We were able to visualize the phosphorylated form of ATM after treatment with dox, but not with 5FU. We did not detect the presence of the phosphorylated form of the protein kinase Chk2 after either drug-treatment in nuclear extracts. Upon treatment of MCF-7 cells with UV, we detected the presence of the phosphorylated forms of both protein kinases (). Thus, the lack of p53 phosphorylated at serine 15 in drug treated extracts of MCF-7 cells may be explained as a lack of activation of the kinases, ATM and Chk2, the enzymes believed to make this modification on p53. This indicates that differential serine 15 phosphorylation of p53 is likely caused by alterations of the ATM/Chk2 pathway and results in changes to multiple PTM of the N-terminus of p53 as well as its interaction with Mdm2, although further study into this latter point is required.
Figure 5. Detection of serine 15 phosphorylation in p53 from MCF-7 treated cells MCF-7 whole cell extracts from cells treated with doxorubicin (dox) or 5-fluorouracil (5FU) for 12 or 24 h, or treated with UV followed by incubation for 12 or 24 h, were isolated and used. Extracts containing ~120 μg total protein were separated using a polyacrylamide gel, transferred to a nitrocellulose membrane, and the membrane probed with Phosphodetect™ anti-p53 pSer15. We detect phosphorylation at serine 15 in both UV and dox treated extracts after 12 h, but do not detect the modification in 5FU treated extracts after 24 h. We estimate >6000 pg of p53 was loaded from the dox and 5FU treated extracts after 24 h of treatment, while the amount of p53 in UV treated extracts is estimated to be ~900 pg p53 based on ELISA. The p53 detection is not shown from this gel as it would overwhelm the detection.
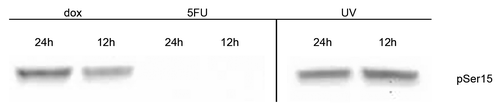
Figure 6. Detection of enzymes potentially responsible for phosphorylation of p53 at serine 15. Nuclear extracts containing 25 μg of total protein from MCF-7 cells treated with doxorubicin or 5-fluorouracil for 4–12 h, 24 h after UV treatment, or untreated (UT) were separated using a 10% polyacrylamide gel, the proteins transferred to nitrocellulose, and the membranes probed with ATM or Chk2, with anti-phospho-ATM pSer1981 or anti-phospho-Chk2 pThr68 antibodies. The blots were subsequently probed for β-actin. ATM and Chk2 proteins are detected in drug- and UV-treated extracts, but the presence of phosphorylated ATM (pSer1981) was only detected in doxorubicin- and UV-treated extracts. Phosphorylated Chk2 (pThr68) was detected only in UV-treated extracts. The ATM kinase, but not Chk2, was detected in untreated cell (UT) extracts.
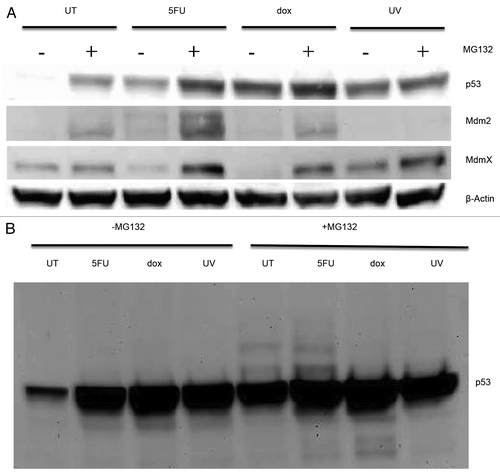
As discussed above, Mdm2 is an ubiquitin ligase modifying the p53 and MdmX proteins so that they are degraded by the proteasome. To obtain evidence for that pathway in our cells, we treated the cells with MG132, a known proteasome inhibitor,Citation30 in the presence of the drugs and analyzed the levels of the p53, Mdm2, and MdmX proteins in whole cell extracts. Blocking the proteasome had a dramatic effect on the accumulation of p53 from the untreated cells increasing the level approximately 15-fold compared with cells without MG132 (). There was a slight, but reproducible increase in the level of p53 in cells treated with drugs or exposed to UV that were also given the proteasome inhibitor. The level of the Mdm2 and MdmX proteins also increased in cells treated with MG132 and the drugs (). To provide evidence for the ubiquitination of p53, we obtained long exposures of the blots exposed to antibodies to p53 (). The p53 blot showed laddering of bands above the normal sized protein from cells that were untreated or treated with 5FU and exposed to MG132, but not in UV or dox treated cells. This laddering could represent ubiquitination of p53 in the untreated and 5FU treated cells consistent with this protein being associated with Mdm2 under these conditions. We also have preliminary evidence for the presence of ubiquitin in immunoprecipitated p53 (unpublished data).
Figure 7. Whole cell extracts from MCF-7 cells treated with MG132 Cells were first treated with 5-fluorouracil (5FU) or doxorubicin (dox) for 6 h or with UV light (UV) then left for 18 h or left untreated (UT), and then treated with 25 μM MG132 or an equivalent volume of the solvent for another 6 h. Whole cell extracts were then produced using RIPA buffer as described in the Materials and Methods section. An aliquot of 30 μg total protein from each extract was loaded onto a 10% polyacrylamide Tris-glycine SDS gel, transferred to nitrocellulose and then probed for p53, Mdm2, MdmX, and β-Actin as described in the Materials and Methods section. (A) Short exposure of the blots to measure the levels of each protein. (B) Longer exposure of the blot for p53 allowed observation of laddering pattern potentially due to ubiquitin modification of this protein in some of the treatments. Treatment of cells with MG132 increases the levels of the p53, Mdm2, and MdmX proteins indicating that each are subject to proteasome degradation although the degradation of p53 in untreated cells appears to be more than in the other treatments consistent with significant levels of laddering of p53 isoforms in the MG132 treated samples.
Discussion
Our research was directed at understanding the function and regulation of the p53 tumor suppressor in MCF-7 breast cancer cells after treatment with chemotherapeutic drugs doxorubicin (dox) or 5-fluorouracil (5FU). The battery of experiments performed here allowed us to assess the level and localization of p53 in MCF-7 breast cancer cells, the regulation of p53 through phosphorylation of serine 15 via the ATM/Chk2 kinase cascade and implications on the involvement of Mdm2 in p53 accumulation and degradation. Early studies of p53 showed that there is a significant accumulation in the level of the protein after induction of DNA damage;Citation20,Citation32-Citation35 our data are consistent with this literature. It is possible that an increase in TP53 mRNA is partially responsible for the increase in the p53 protein at longer time points, but studies by Yuan et al.Citation36 and Mendonca et al.Citation37 show that there is little to no change in TP53 mRNA in cancer cells treated for less than 24 h with cisplatin. Therefore, we did not pursue evaluation of TP53 mRNA message levels. We found significant differences in the effect of 5FU and dox on the DNA binding and the PTM of p53 by ATM and Chk2 that suggest potential alterations in the standard models for p53 regulation in MCF-7 breast cancer cells. Additionally, we believe that p53 is functionally active as a DNA binding protein and transcription factor, as demonstrated by showing the effect of p53 siRNA with UV and drug treatments on p53 target proteins. DNA binding in general appears to be independent of whether or not the protein is posttranslationally modified at serine 15.
We found minimal detection of serine 15 phosphorylated p53 after 5FU or dox treatment that was consistent with a lack of full activation of the ATM/Chk2 pathway ( and ). Other groups reported detection of posttranslationally modified p53 following these same drugs.Citation7,Citation8,Citation38 While some of the difference in the detection of serine 15 modification could be due to differences in cell type, detection antibody, or extraction protocol, we feel that a significant explanation could be the way we normalize our western blots to the same levels of total p53. In most cases, the other groups normalize to total protein which results in the loading of significantly different levels of p53 on the blots since the levels of this protein increase at least 20-fold after the treatment (). For this reason, we postulate that the increase in the detection of phosphorylated p53 noted in the other studies may be the result of a total increase in p53.Citation10,Citation21,Citation39,Citation40 In comparison, we found robust phosphorylation of p53 4 h after treatment with UV light (). Also, for our studies, we used p53 from nuclear extracts and not whole cell lysates as used by Ju and colleagues.Citation7 It is possible that a small amount of the p53 in the cytoplasm (or in a complex only extracted using whole cell lysis) could be modified. In fact, when we did a western blot of a whole cell lysate, we were able to detect the phosphorylated p53 in the dox-treated cell extracts ().
It appears that the differential regulation of the ATM/Chk2 pathway corresponds directly to the state of phosphorylation of serine 15 on p53 we observed, namely that we detect robust phosphorylation of serine 15 on p53 after UV and that’s the only condition where both ATM and Chk2 are activated. This indicates that phosphorylation of the Chk2 kinase (in the case of dox treatment) or both the ATM kinase and the Chk2 kinase (in the case of 5FU treatment) does not occur and thus, these enzymes are not becoming activated. These data indicate that the ATM/Chk2 pathway is differently regulated upon treatment of MCF-7 cells, and this differential regulation is likely affecting phosphorylation events in the N-terminal transactivation domain (TAD) of p53. To our knowledge, there are no other studies showing the lack of activation of the ATM/Chk2/p53 pathway in MCF-7 cells as a potential modality for p53 regulation. It has been reported that the forkhead box protein (FOXO3), a transcription factor and tumor suppressor, is necessary for phosphorylation of serine 1981 on ATM (in the absence of serine 1981 auto-phosphorylation) upon DNA damage induced by IR in MCF-7 cells.Citation41 It is possible that the DNA damage signal is not being transmitted to either FOXO3 or ATM and thus, not to Chk2 upon drug treatment.
We have evidence that the accumulation of the p53 occurs despite the lack of serine 15 phosphorylation. As noted above, following treatment of cells with 5FU, we observed a significant accumulation of the p53 protein (), yet this protein does not contain any detectable phosphorylation at serine 15 (). The inhibition of the proteasome with MG132 produced a slight laddering of p53 forms in the 5FU treated cells that would be consistent with the addition of the ubiquitin peptide on p53 (). These results suggest that p53 is being degraded in this system and that may be caused by Mdm2, one of the best characterized E3 ubiquitin ligase enzymes associated with p53. Using immunoprecipitation, we have obtained preliminary evidence in our lab to support the association of Mdm2 and p53 after 5FU treatment. This is consistent with published literature indicating that serine 15 phosphorylation status of p53 affects interaction with Mdm2.Citation10,Citation21,Citation42 Interestingly, p53 accumulates following 5FU treatment of cells despite the lack of serine 15 phosphorylation and the potential for continued association with Mdm2. The mechanism for this is not known.
Mdm2 has been reported to interact with the N-terminus of p53, so we suspected the accumulation of p53 may be also be affected by the presence of MdmX, a homolog of Mdm2 that functions as a stable heterodimer with Mdm2.Citation43-Citation45 MdmX stabilizes the Mdm2 protein by blocking Mdm2 auto-ubiquitination when the two proteins are bound.Citation46,Citation47 Interestingly, MdmX is also ubiquitinated by Mdm2Citation30 and has been proposed to inhibit Mdm2-mediated p53 degradation by competing with p53 for Mdm2 binding.Citation48,Citation49 Consistent with that, we found that the level of MdmX goes down after drug treatment and this drop is dependent on the proteasome since addition of MG132, a proteasome inhibitor, increases its level (). Although MdmX is considered a negative regulator of p53,Citation50 this protein may also protect p53 from Mdm2-mediated degradation, as shown by Jackson and Berberich.Citation48 Interestingly, a report from Chen and colleagues showed that DNA damage induced by IR in MCF-7 breast cancer cells and U2OS osteosarcoma cells results in ATM-dependent phosphorylation of MdmX.Citation20 Subsequent phosphorylation of MdmX by Chk2 promotes the interaction of Mdm2 with MdmX, releasing Mdm2 from p53. Mdm2 could then ubiquitinate MdmX and promote its degradation. Both Mdm2 and MdmX can functionally inactivate p53.Citation50 We found ATM and Chk2 to be in various states of activation depending on the treatment inducing DNA damage in MCF-7 cells (). So, we postulate that the ATM/Chk2 pathway may partly be responsible for the regulation of not only p53 phosphorylation but also modification of MdmX and Mdm2, which then impacts the accumulation of p53. Chen and colleaguesCitation20 propose that the phosphorylation of serine 403 on MdmX by ATM promotes the interaction of MdmX with Mdm2 and redirects the ubiquitin activity of Mdm2 from p53 to MdmX. The idea that the E3 ubiquitin ligase activity of Mdm2 can be redirected to a different substrate (MdmX) away from p53 is a plausible explanation for our observation that p53 continues to accumulate without serine 15 phosphorylation and we feel this is an avenue worth further exploration.
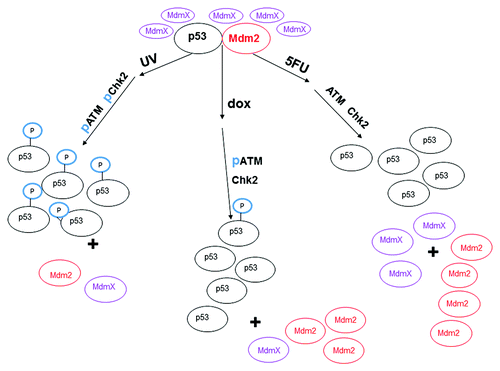
In , we show a proposed model for the regulation of p53 by the ATM/Chk2 pathway. We illustrate that upon treatment of MCF-7 cells with 3 different DNA damaging agents, there is differential phosphorylation of ATM and Chk2. Cells treated with UV show phosphorylation of ATM and Chk2, with subsequent phosphorylation on serine 15 of p53. Treatment of cells with 5FU shows that there is no phosphorylation of ATM or Chk2, which results in unmodified p53. Lastly, treatment of cells with dox results in phosphorylation of ATM but not Chk2, which results in minimal phosphorylation of p53. We postulate that the activated ATM can phosphorylate MdmX (as shown by Chen and colleaguesCitation20) and that modified MdmX protein redirects Mdm2 away from p53 (as shown by Jackson and BerberichCitation48). Our data support this hypothesis in that the levels of MdmX increase when the production of Mdm2 is abrogated in the presence of siRNA for p53, particularly in the presence of drug (). Separation of Mdm2 from p53 allows the latter protein to accumulate even in the presence of Mdm2 in the nucleus.
Figure 8. Model of changes in ATM and Chk2 and the effect on the interaction between p53, MdmX, and Mdm2 after treatment of MCF-7 cells with various DNA damaging agents. After treatment of MCF-7 cells with UV, ATM and Chk2 are activated by phosphorylation (represented as “p”) and phosphorylate p53 at serine 15 (represented as “P” sphere on p53). The phosphorylated p53 no longer interacts with Mdm2 which allows accumulation of the p53 protein. ATM is phosphorylated upon treatment of the cells with doxorubicin (dox) for 12 h, but Chk2 is not phosphorylated after this treatment. The p53 protein harvested from dox treated MCF-7 cells is minimally phosphorylated at serine 15 and does not interact with Mdm2. Neither ATM, nor Chk2 are activated in MCF-7 cells treated with 5-fluorouracil (5FU), and the p53 protein harvested from 5FU treated MCF-7 cells is not phosphorylated at serine. The difference in protein levels is approximately represented by the different number of ovals.
Conclusions
Our observations beg the question as to whether or not the PTM play a paramount role in p53 protein stability, since we observe a significant accumulation of the p53 protein after treatment of MCF-7 cells with 5FU, yet there is no evidence of phosphorylation of the serine 15. While we discuss above the fact that the accumulation of p53 is not associated with modification of serine 15, we have also provided evidence that this modification is independent of the function of the protein as a transcription factor. We observed DNA binding of the p53 protein from 5FU-treated cells using ChIP (). We also found that addition of p53-specific siRNA blocks the accumulation of 2 proteins whose genes are regulated by p53 following 5FU (). As we show this p53 is not phosphorylated at serine 15, these two pieces of evidence suggest that the protein lacking this modification is still functional, consistent with studies by Ray et al.Citation51 and Huovinen et al.Citation52 However, there is conflicting evidence regarding the role of serine 15 phosphorylation on the functionality of p53 as shown by other groups, including Ashcroft et al.Citation53 and Zerbini et al.Citation54 Thus, there is still considerable controversy as to the role of the phosphorylation of serine 15 on the function of p53 as a transcription factor. Some work has been published where this serine residue has been substituted for either a non-phosphorylated alanine residue or one that mimics continuous phosphorylation (aspartic acid). But, as these modified proteins are then overexpressed in the cells, it is not clear the applicability of the information obtained on the role of the wild-type p53 in its native context. Future experiments with isolated proteins with different phosphorylation states at serine 15 might clarify the situation.
It is still curious that the level of p53 increases without the serine 15 phosphorylation and despite preliminary evidence for the association with Mdm2. Perhaps the gross amount of p53 that accumulates over time with drug simply overwhelms its negative regulators, such as Mdm2, and that the PTM serves a different purpose. Kaeser and colleagues transfected HCT116 colon cancer cells with a panel of p53 mutant proteins harboring alanine substitutions at serine 6, 9, 15, 20, 33, and 37 and found that the p53 was stabilized and accumulated after treatment with dox or 5FU.Citation55 Their findings are contrary to previously published literature, but are consistent with our work. This allows us to conclude phosphorylation of serine 15 is clearly not required for the accumulation of p53 as had previously been reported.Citation55 We were also able to determine that there appears to be differential activation of the ATM/Chk2 pathway in MCF-7 cells treated with dox or 5FU. We feel that the lack of ATM/Chk2 activation is a potential root cause for why p53 is not phosphorylated at serine 15. This work may have important implications for understanding the responsiveness of the MCF-7 breast cancer cells to the chemotherapeutic drugs dox and 5FU. In total, our studies have helped better elucidate the regulation of p53 in MCF-7 breast cancer cells via the ATM/Chk2 pathway. In the future, we hope to determine the specific mechanism for p53 accumulation independent of phosphorylation at serine 15.
Materials and Methods
Cell culture and treatment
The MCF-7 breast cancer cell line was obtained from American Type Culture Collection (HTB-22). The cells were grown and maintained in Dulbecco’s modified Eagle’s medium (American Type Culture Collection, 30-2002) with appropriate supplements of 10% fetal bovine serum (Thermo Fisher Scientific, SH30088.03), penicillin/streptomycin (Lonza, 17-602E), and insulin (Sigma, I3536) at 37 °C in the presence of 5% CO2.
p53 siRNA treatment of MCF-7 cells
MCF-7 breast cancer cell cultures were treated with either the ON-TARGETplus SMARTpool human TP53 siRNA (Thermo Fisher Scientific Dharmacon, L-003329-00) or non-targeting pool siRNA (Thermo Fisher Scientific Dharmacon, D-001810-10-20) which were received as lyophilized powders and reconstituted in 1× siRNA buffer as described in the product manual. Cells were maintained as described above, except fresh antibiotic-free complete media was used for cell culture when the siRNAs were added. The siRNA was prepared as described in the product manual using the DharmaFECT reagent (Thermo Fisher Scientific, T-2001-02) and was added to the growth media in the cell flasks for a final siRNA concentration of 5 nM. The cells were allowed to grow in a 37 °C, 5% CO2 incubator for 24 h (includes drug or UV treatment) prior to extraction.
Cell extract preparation
The cells were grown to 80–90% confluency in T75 or T150 flasks and cytoplasmic and nuclear proteins were harvested following a protocol by Jagleská and colleaguesCitation56 from “untreated” cells, cells treated for 4, 8, or 12 h with 1.5 μg/mL doxorubicin (Sigma, D1515) or 500 μM 5-fluorouracil (Sigma, F6627), or cells exposed to 50 J/m2 UV light for 15 s followed by a 24 h incubation prior to extraction. For total cell extracts, 100 000 MCF-7 cells were seeded into each well of a 6-well plate, than treated 24 h later for 4, 8, 12, or 24 h with 1.5 μg/mL doxorubicin, 500 μM 5-fluorouracil, or 50 J/m2 UV light for 15 s followed by a 24 h incubation prior to extraction. The cells were extracted using IP Lysis/Wash buffer provided with the Pierce Crosslink IP Kit (Thermo Fisher Scientific, 21647) or RIPA buffer (Thermo Scientific, 89900). The extraction buffer (100 μL) was added to a single well and allowed to incubate for 5 min. The cells were scraped and transferred to the next well. This was repeated in a third well, finishing with a total of 100 μL of extract from 3 wells of each plate for each condition. The samples were then spun at 13 000 × g for 10 min and the resulting supernate was used for analysis. Total protein concentration for both the nuclear and cytoplasmic fractions was determined using the Pierce Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo Fisher Scientific, 23225) using bovine serum albumin as a standard. The p53 concentration for both fractions was determined using the Pantropic p53 enzyme-linked immunosorbent assay (ELISA) kit from Roche Diagnostics (11828789001).
MG132 treatment of MCF-7 cells
MCF-7 cells to be treated with the proteasome inhibitor MG132 (Selleckchem, S2619) were first treated with 5-fluorouracil or doxorubicin for 6 h or with 50 J/m2 UV light for 15 s followed by 18 h or left untreated as described above. At this time, MG132 was added to each culture flask to a final concentration of 25 μM. An equivalent volume of the solvent, dimethyl sulfoxide (Sigma, D8418), was added to those flasks not treated with MG132. Cells were left to incubate for the remaining 6 h and were then extracted with RIPA buffer as described above.
Western blots
Following separation of proteins on 10% or 16.5% (w/v) polyacrylamide gels (BioRad, 456-1033 or 456-3063) in the presence of sodium dodecyl sulfate (SDS) (Laemmli buffer), the gel was transferred to nitrocellulose membranes using the iBlot system (Invitrogen, IB1001, IB301001), then the membrane blocked in the buffer to be used for the chosen primary antibody. Specific proteins were visualized using the appropriate antibodies, as follows: anti-p53 (Ab-12) DO-7 (Calbiochem-EMD, OP140); PhosphoDetect™anti-p53 (pSer15) (Cell Signaling, 9284); anti-Mdm2 (SMP14)(Santa Cruz, sc-965); anti-Mdm2 mouse supernate from anti-Mdm2 4B2, 4B11, 2A9 (J. Bargonetti Lab, Hunter College); anti-ATM (Y170) (Abcam, ab32420); Anti-ATM (phosphoS1981) (EP1890Y) (Abcam, ab81292); Chk2 (Cell Signaling, 2662); Anti-Phospho-Chk2 (T68)(R&D Systems, AF1626); Anti-β-Actin Clone AC-15 (Sigma Aldrich, A5441). The primary antibody was incubated with the nitrocellulose membrane in either phosphate buffered saline plus 1% Tween (PBST) or Tris buffered saline plus 1% Tween (TBST) with 5% bovine serum albumin overnight at 4 °C on a rocker. For all western blots performed, the membranes were washed 3× with 1× PBST or 1× TBST and then incubated with either horse radish peroxidase (HRP)-linked anti-rabbit IgG (1:10 000) (Cell Signaling, 7074S) or HRP-linked anti-mouse IgG (1:10 000) (Cell Signaling, 7076S) secondary antibody in 5% bovine serum albumin in 1× PBST for 1 h and visualized with SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, 34078). Signal intensities were observed by means of densitometry and analyzed using Quantity One software (BioRad).
Electrophoretic mobility shift assay (EMSA)
For visualizing the fluorescent DNA-p53 protein complex in the gel, an aliquot of a nuclear extract sample normalized either for p53 (40 pg or 50 pg, as estimated from ELISA) or total protein (20 μg, as estimated by BCA assay) was allowed to react with 5 pmoles of 5′ 6-carboxyfluorescein conjugated 30 base-pair double stranded p21, bax, puma, pcna, or mdm2 DNA sequence (custom synthesis by Integrated DNA Technologies) using sequences previously described (Chandrachud and Gal).Citation55 Poly(A)x(dT) (1 μg) (Roche Diagnostics, 10108677001) was added to each reaction mixture to minimize DNA binding of non-specific proteins. Low Salt Buffer (20 mM HEPES pH 7.5, 1 mM EDTA, 1 mM DTT, 10 mM (NH4)2SO4, 10 mM KCl, 0.2% [v/v] Tween-20) was also added to each mixture to stabilize the DNA and buffer the binding reaction. The reaction mixture was incubated for 30 min at 20 °C before separation on a 5% TBE polyacrylamide gel (BioRad, 456-5013) at 95 V for 50 min. A fluorescent image of the gel was then taken at 0.5–1 s exposures using a Quantity One Fluorescent UV scanner (BioRad). Signal intensities were analyzed via densitometry using Quantity One software (BioRad).
Streptavidin magnetic bead assay
This assay was performed as described previously (Chandrachud and Gal)Citation57 utilizing 50 pg (as estimated from ELISA) of wild-type p53 from the nuclear extract from the MCF-7 cell line and 20 pmoles of the biotinylated p21 gene regulatory sequence (custom synthesis by Integrated DNA Technologies). Following each binding reaction, the “unbound” and “bound” fractions were obtained using streptavidin magnetic beads and a magnet (Roche Diagnostics, 11641778001), separated by SDS-PAGE (Bio-Rad, 456-1033) along with a pre-bound sample and then transferred by iBlot (Invitrogen) onto a nitrocellulose membrane for detection of p53 using anti-p53(Ab12)(DO-7) (Calbiochem, OP140). The signal intensities were quantitated with the Quantity One software (Bio-Rad).
Chromatin immunoprecipitation (ChIP)
For chromatin immunoprecipitation, the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling, 9004) was used. Following the treatment of MCF-7 cells for 4, 8, or 12 h with dox (1.5 μg/mL) or 5FU (500 μM) or 24 h after a 15 s UV (50 J/m2) exposure, the chromatin was cross-linked with 1% (v/v) formaldehyde for 10 min at room temperature. Subsequent nuclei preparation and Nuclease S7 digestion of chromatin was performed as per the kit protocol. An aliquot representing the 2% input sample of cross-linked chromatin was removed prior to the addition of antibodies. The cross-linked chromatin was diluted 10-fold in the kit 1× ChIP buffer and incubated overnight at 4 °C with rotation with 1 μg of Normal Rabbit IgG (Cell Signaling, 2729) or 1 μg of anti-p53 DO-1 (Calbiochem-EMD, OP43). The conjugation of immunoprecipitated (IPed) chromatin to ChIP-Grade protein G agarose beads, the elution of captured chromatin from the antibody/protein G beads, the reversal of cross-links for all IPed and 2% input samples by proteinase K (20 mg/mL) and DNA purification were performed as per the kit protocol to yield a final volume of 50 μL in DNA Elution Reagent C. The primers used for analysis of ChIP DNA by standard PCR (custom synthesized by Integrated DNA Technologies) represented the following p53 gene recognition sequences (5′ to 3′): p21, −2293 base pairs (position relative to the transcription start site), GCTGTGGCTC TGATTGGCTT T, forward; p21, −2192 base pairs, ACAGGCAGCC CAAGGACAAA, reverse; mdm2, −188 base pairs, TGCCTGTCGG GTCACTAGTG TGAA, forward; mdm2, +24 base pairs, TACCCTCCAA TCGCCACTGA A CA, reverse; bax, +484 base pairs, AGAATGTAGG ATACAGGCCC AGCC, forward; bax, +685 base pairs, AGA GGA GAA AGG AAG GGA AGA GGT, reverse; p21NS, −4421 base pairs, GAGTCCTGTT TGCTTCTGGG CA, forward; p21NS, −4178 base pairs, CTGCATTGGG GCTGCCTATG TA, reverse. Standard PCR conditions were: 30 s denaturing at 94 °C, 30 s annealing at gene specific temperatures, and 2 min extension at 72 °C for a gene specific number of cycles. Gene-specific amplifications conditions were as follows: p21, 55 °C annealing temperature, 32 cycles; mdm2, 55 °C annealing temperature, 31 cycles; bax, 60 °C annealing temperature, 33 cycles; p21 NS, 55 °C annealing temperature, 30 cycles. PCR products were resolved on 1.8% (w/v) agarose gels and visualized with ethidium bromide staining. Signal intensities were observed by means of densitometry and analyzed using Quantity One software (BioRad).
| Abbreviations: | ||
| 5FU | = | 5-fluorouracil |
| ATM | = | ataxia telangiectasia mutated |
| BCA | = | bicinchoninic acid |
| ChIP | = | chromatin immunoprecipitation |
| dox | = | doxorubicin |
| dTMP | = | deoxythymidine monophosphate |
| ELISA | = | enzyme linked immunosorbant assay |
| EMSA | = | electrophoretic mobility shift assay |
| FOXO3 | = | forkhead box protein |
| HEPES | = | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| HRP | = | horseradish peroxidase |
| IP | = | immunoprecipitation |
| IR | = | ionizing radiation (gamma radiation) |
| MEF | = | mouse embryo fibroblasts |
| ng | = | nanogram |
| NS | = | non-specific |
| PAGE | = | polyacrylamide gel electrophoresis |
| PBST | = | phosphate buffered saline plus Tween |
| PCR | = | polymerase chain reaction |
| pg | = | picogram |
| PTM | = | posttranslational modification |
| RE | = | response element |
| RIPA | = | radio-immunoprecipitation assay buffer |
| SA | = | streptavidin magnetic bead assay |
| SDS | = | sodium dodecyl sulfate |
| siRNA | = | small interfering ribonucleic acid |
| TAD | = | transactivation domain |
| TBE | = | Tris/borate/EDTA |
| TBST | = | Tris buffered saline plus Tween |
| TRIS | = | Tris(hydroxymethyl)aminomethane |
| μg | = | microgram |
| UT | = | untreated |
| UV | = | ultraviolet light |
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors would like to acknowledge John Mallon for his assistance with protein extractions, DNA binding assays and thoughtful conversations; Diane Messina for assistance with maintenance of tissue culture; Mark Gerger for his help with statistical analyses; Amy Fedele for her assistance with the preparation of figures; and Sally Stem for her assistance with procurement of laboratory reagents. We would also like to acknowledge Sanofi Pasteur, Inc. for the use of laboratory supplies and facilities when needed.
References
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 12; http://dx.doi.org/10.1038/nrm2395; PMID: 18431400
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207 - 19; http://dx.doi.org/10.1016/j.cell.2005.10.043; PMID: 16413492
- Hande KR. Etoposide: four decades of development of a topoisomerase II inhibitor. Eur J Cancer 1998; 34:1514 - 21; http://dx.doi.org/10.1016/S0959-8049(98)00228-7; PMID: 9893622
- Ottewell PD, Woodward JK, Lefley DV, Evans CA, Coleman RE, Holen I. Anticancer mechanisms of doxorubicin and zoledronic acid in breast cancer tumor growth in bone. Mol Cancer Ther 2009; 8:2821 - 32; http://dx.doi.org/10.1158/1535-7163.MCT-09-0462; PMID: 19789217
- Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003; 3:330 - 8; http://dx.doi.org/10.1038/nrc1074; PMID: 12724731
- Collavin L, Lunardi A, Del Sal G. p53-family proteins and their regulators: hubs and spokes in tumor suppression. Cell Death Differ 2010; 17:901 - 11; http://dx.doi.org/10.1038/cdd.2010.35; PMID: 20379196
- Ju J, Schmitz JC, Song B, Kudo K, Chu E. Regulation of p53 expression in response to 5-fluorouracil in human cancer RKO cells. Clin Cancer Res 2007; 13:4245 - 51; http://dx.doi.org/10.1158/1078-0432.CCR-06-2890; PMID: 17634554
- Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther 2011; 10:69 - 79; http://dx.doi.org/10.1158/1535-7163.MCT-10-0581; PMID: 21075910
- Kubbutat MHG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299 - 303; http://dx.doi.org/10.1038/387299a0; PMID: 9153396
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997; 91:325 - 34; http://dx.doi.org/10.1016/S0092-8674(00)80416-X; PMID: 9363941
- Jeyaraj S, O’Brien DM, Chandler DS. MDM2 and MDM4 splicing: an integral part of the cancer spliceome. Front Biosci (Landmark Ed) 2009; 14:2647 - 56; http://dx.doi.org/10.2741/3402; PMID: 19273224
- Okoro DR, Rosso M, Bargonetti J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer 2012; 3:311 - 9; http://dx.doi.org/10.1177/1947601912455323; PMID: 23150764
- Kalnina Z, Zayakin P, Silina K, Linē A. Alterations of pre-mRNA splicing in cancer. Genes Chromosomes Cancer 2005; 42:342 - 57; http://dx.doi.org/10.1002/gcc.20156; PMID: 15648050
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol 1993; 13:4107 - 14; PMID: 7686617
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420:25 - 7; http://dx.doi.org/10.1016/S0014-5793(97)01480-4; PMID: 9450543
- Rodriguez MS, Desterro JMP, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol Cell Biol 2000; 20:8458 - 67; http://dx.doi.org/10.1128/MCB.20.22.8458-8467.2000; PMID: 11046142
- García-Echeverría C, Chène P, Blommers MJ, Furet P. Discovery of potent antagonists of the interaction between human double minute 2 and tumor suppressor p53. J Med Chem 2000; 43:3205 - 8; http://dx.doi.org/10.1021/jm990966p; PMID: 10966738
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 8; http://dx.doi.org/10.1126/science.1092472; PMID: 14704432
- Biderman L, Poyurovsky MV, Assia Y, Manley JL, Prives C. MdmX is required for p53 interaction with and full induction of the Mdm2 promoter after cellular stress. Mol Cell Biol 2012; 32:1214 - 25; http://dx.doi.org/10.1128/MCB.06150-11; PMID: 22290440
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J 2005; 24:3411 - 22; http://dx.doi.org/10.1038/sj.emboj.7600812; PMID: 16163388
- Saito S, Yamaguchi H, Higashimoto Y, Chao C, Xu Y, Fornace AJ Jr., Appella E, Anderson CW. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J Biol Chem 2003; 278:37536 - 44; http://dx.doi.org/10.1074/jbc.M305135200; PMID: 12860987
- Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 transactivation but does not directly influence interaction with HDM2. EMBO J 1999; 18:7002 - 10; http://dx.doi.org/10.1093/emboj/18.24.7002; PMID: 10601022
- Yeh PY, Chuang SE, Yeh KH, Song YC, Chang LL, Cheng AL. Phosphorylation of p53 on Thr55 by ERK2 is necessary for doxorubicin-induced p53 activation and cell death. Oncogene 2004; 23:3580 - 8; http://dx.doi.org/10.1038/sj.onc.1207426; PMID: 15116093
- Sun XX, Dai MS, Lu H. 5-fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J Biol Chem 2007; 282:8052 - 9; http://dx.doi.org/10.1074/jbc.M610621200; PMID: 17242401
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998; 281:1677 - 9; http://dx.doi.org/10.1126/science.281.5383.1677; PMID: 9733515
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 2000; 14:289 - 300; PMID: 10673501
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 2000; 14:278 - 88; PMID: 10673500
- Wu L, Levine AJ. Differential regulation of the p21/WAF-1 and mdm2 genes after high-dose UV irradiation: p53-dependent and p53-independent regulation of the mdm2 gene. Mol Med 1997; 3:441 - 51; PMID: 9260156
- Okamoto K, Taya Y, Nakagama H. Mdmx enhances p53 ubiquitination by altering the substrate preference of the Mdm2 ubiquitin ligase. FEBS Lett 2009; 583:2710 - 4; http://dx.doi.org/10.1016/j.febslet.2009.07.021; PMID: 19619542
- Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol 2003; 23:5113 - 21; http://dx.doi.org/10.1128/MCB.23.15.5113-5121.2003; PMID: 12860999
- Liang X, Guo Y, Figg WD, Fojo AT, Mueller MD, Yu JJ. The role of wild-type p53 in cisplatin-induced Chk2 phosphorylation and the inhibition of platinum resistance with a Chk2 inhibitor. Chemother Res Pract 2011; 2011:715469; http://dx.doi.org/10.1155/2011/715469; PMID: 22312557
- Kern SE, Kinzler KW, Bruskin A, Jarosz D, Friedman P, Prives C, Vogelstein B. Identification of p53 as a sequence-specific DNA-binding protein. Science 1991; 252:1708 - 11; http://dx.doi.org/10.1126/science.2047879; PMID: 2047879
- Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991; 51:6304 - 11; PMID: 1933891
- Maltzman W, Czyzyk L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol Cell Biol 1984; 4:1689 - 94; PMID: 6092932
- Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M, Vassilev A, Anderson CW, Appella E. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998; 12:2831 - 41; http://dx.doi.org/10.1101/gad.12.18.2831; PMID: 9744860
- Yuan JM, Li XD, Liu ZY, Hou GQ, Kang JH, Huang DY, Du SX. Cisplatin induces apoptosis via upregulating Wrap53 in U-2OS osteosarcoma cells. Asian Pac J Cancer Prev 2011; 12:3465 - 9; PMID: 22471498
- Mendonça LM, da Silva Machado C, Teixeira CC, de Freitas LA, Bianchi MdeL, Antunes LM. Curcumin reduces cisplatin-induced neurotoxicity in NGF-differentiated PC12 cells. Neurotoxicology 2013; 34:205 - 11; http://dx.doi.org/10.1016/j.neuro.2012.09.011; PMID: 23036615
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, Wahl GM, Heimbrook DC, Vassilev LT. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem 2004; 279:53015 - 22; http://dx.doi.org/10.1074/jbc.M410233200; PMID: 15471885
- Tack LC, Wright JH. Altered phosphorylation of free and bound forms of monkey p53 and simian virus 40 large T antigen during lytic infection. J Virol 1992; 66:1312 - 20; PMID: 1310751
- Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella E, Kastan MB. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev 1997; 11:3471 - 81; http://dx.doi.org/10.1101/gad.11.24.3471; PMID: 9407038
- Chung YM, Park SH, Tsai WB, Wang SY, Ikeda MA, Berek JS, Chen DJ, Hu MC. FOXO3 signalling links ATM to the p53 apoptotic pathway following DNA damage. Nat Commun 2012; 3:1000; http://dx.doi.org/10.1038/ncomms2008; PMID: 22893124
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281:1674 - 7; http://dx.doi.org/10.1126/science.281.5383.1674; PMID: 9733514
- Shvarts A, Steegenga WT, Riteco N, van Laar T, Dekker P, Bazuine M, van Ham RCA, van der Houven van Oordt W, Hateboer G, van der Eb AJ, et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 1996; 15:5349 - 57; PMID: 8895579
- Stad R, Little NA, Xirodimas DP, Frenk R, van der Eb AJ, Lane DP, Saville MK, Jochemsen AG. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep 2001; 2:1029 - 34; http://dx.doi.org/10.1093/embo-reports/kve227; PMID: 11606419
- Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett 2012; 586:1390 - 6; http://dx.doi.org/10.1016/j.febslet.2012.02.049; PMID: 22673503
- Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 1999; 274:38189 - 96; http://dx.doi.org/10.1074/jbc.274.53.38189; PMID: 10608892
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett 1999; 447:5 - 9; http://dx.doi.org/10.1016/S0014-5793(99)00254-9; PMID: 10218570
- Jackson MW, Berberich SJ. MdmX protects p53 from Mdm2-mediated degradation. Mol Cell Biol 2000; 20:1001 - 7; http://dx.doi.org/10.1128/MCB.20.3.1001-1007.2000; PMID: 10629057
- Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ, Jochemsen AG. Hdmx stabilizes Mdm2 and p53. J Biol Chem 2000; 275:28039 - 44; PMID: 10827196
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem 2002; 277:19251 - 4; http://dx.doi.org/10.1074/jbc.C200150200; PMID: 11953423
- Ray D, Murphy KR, Gal S. The DNA binding and accumulation of p53 from breast cancer cell lines and the link with serine 15 phosphorylation. Cancer Biol Ther 2012; 13:848 - 57; http://dx.doi.org/10.4161/cbt.20835; PMID: 22785213
- Huovinen M, Loikkanen J, Myllynen P, Vähäkangas KH. Characterization of human breast cancer cell lines for the studies on p53 in chemical carcinogenesis. Toxicol In Vitro 2011; 25:1007 - 17; http://dx.doi.org/10.1016/j.tiv.2011.03.018; PMID: 21457773
- Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol 2000; 20:3224 - 33; http://dx.doi.org/10.1128/MCB.20.9.3224-3233.2000; PMID: 10757806
- Zerbini LF, Wang Y, Correa RG, Cho JY, Libermann TA. Blockage of NF-kappaB induces serine 15 phosphorylation of mutant p53 by JNK kinase in prostate cancer cells. Cell Cycle 2005; 4:1247 - 53; http://dx.doi.org/10.4161/cc.4.9.1966; PMID: 16082226
- Kaeser MD, Pebernard S, Iggo RD. Regulation of p53 stability and function in HCT116 colon cancer cells. J Biol Chem 2004; 279:7598 - 605; http://dx.doi.org/10.1074/jbc.M311732200; PMID: 14665630
- Jagelská E, Brázda V, Pospisilová S, Vojtesek B, Palecek E. New ELISA technique for analysis of p53 protein/DNA binding properties. J Immunol Methods 2002; 267:227 - 35; http://dx.doi.org/10.1016/S0022-1759(02)00182-5; PMID: 12165443
- Chandrachud U, Gal S. Three assays show differences in binding of wild-type and mutant p53 to unique gene sequences. Technol Cancer Res Treat 2009; 8:445 - 53; PMID: 19925028