
Abstract
The inhibitory effect of trastuzumab, a humanized monoclonal antibody directed against the extracellular domain of ErbB2, is associated with its ability to induce ErbB2-Y1248 phosphorylation, and the status of phosphorylated ErbB2-Y1248 (ErbB2-pY1248) may correlate with the sensitivity of breast cancers to trastuzumab. The mechanisms of which remain unclear. Here, we show that binding of trastuzumab to ErbB2 activates ErbB2 kinase activity and enhances ErbB2-Y1248 phosphorylation in trastuzumab-sensitive breast cancer cells. This in turn increases the interaction between ErbB2 and non-receptor Csk-homologous kinase (CHK), leading to growth inhibition of breast cancer cells. Overexpression of CHK mimics trastuzumab treatment to mediate ErbB2-Y1248 phosphorylation, Akt downregulation, and growth inhibition of trastuzumab-sensitive breast cancer cells. CHK overexpression combined with trastuzumab exerts an additive effect on cell growth inhibition. We further demonstrate that positive ErbB2-pY1248 staining in ErbB2-positive breast cancer biopsies correlates with the increased trastuzumab response in trastuzumab neoadjuvant settings. Collectively, this study highlights an important role for ErbB2-pY1248 in mediating trastuzumab-induced growth inhibition and trastuzumab-induced interactions between CHK and ErbB2-pY1248 is identified as a novel mechanism of action that mediates the growth inhibition of breast cancer cells. The novel mechanistic insights into trastuzumab action revealed by this study may impact the design of next generation of therapeutic monoclonal antibodies targeting receptor tyrosine kinases, as well as open new avenues to identify novel targets for the treatment of ErbB2-positive cancers.
Introduction
Trastuzumab is a humanized monoclonal antibody directed against the extracellular domain (subdomain IV) of ErbB2 (also known as human epidermal growth factor receptor 2 [HER2]) and is approved for the treatment of ErbB2/HER2-positive breast and gastric cancers.Citation1,Citation2 It is believed that binding of trastuzumab to ErbB2 inhibits receptor-coupled signaling by (1) prevention of the cleavage of ErbB2 extracellular domain by the metalloproteinase ADAM10 and inhibition of the active p95ErbB2 fragment,Citation3,Citation4 (2) inhibition of either ErbB2 homodimerization or heterodimerization with other ErbB family members,Citation1,Citation5 and (3) induction of ErbB2 endocytosis followed by receptor degradation.Citation6-Citation11 Taken together, binding of trastuzumab to ErbB2 leads to the inhibition of pro-survival and proliferative pathways, such as the phosphatidylinositol 3-kinase (PI3K)/Akt pathwayCitation12,Citation13 and the mitogen-activated protein kinase (MAPK) pathway,Citation14 resulting in growth inhibition of cancer cells.
Unlike other members of ErbB family receptors, a ligand that specifically binds to ErbB2 has not been identified. However, the extracellular domain of ErbB2 can adopt a fixed conformation that resembles a ligand-activated state that permits it to form homo- or heterodimers in the absence of a ligand.Citation15 In trastuzumab-sensitive breast cancer cells such as SKBR3 and BT474, ErbB2 is overexpressed, which leads to the formation of either homo- or heterodimers with other ErbB family members in a ligand-independent manner and the upregulation of AKT activity.Citation5
The agonistic effect of trastuzumab, resulting in ErbB2 phosphorylation that is correlated with inhibition of cell proliferation, was reported previously.Citation16 Diemeier et al. also reported that the inhibitory effect of trastuzumab on trastuzumab-sensitive cells (BT474 and SKBR3 cells) was associated with its ability to induce ErbB2 tyrosine phosphorylation at Y1248.Citation17 A survey of ErbB2-overexpressing breast cancer cell lines showed that trastuzumab sensitivity was frequently associated with the expression of ErbB2 phosphorylated at Y1248.Citation18 Recently, Gijsen et al. found that trastuzumab-induced ErbB2 phosphorylation was due to release of the ligands for ErbB family receptors, which results in ErbB2 heterodimerization and phosphorylation.Citation19
Hudelist et al. showed that the presence of phosphorylation at ErbB1 (also known as human epidermal growth factor receptor 1, HER1 or EGFR)-Y845 and ErbB2-Y1248 was an independent predictor of better progression-free survival following trastuzumab treatment.Citation20 The increased trastuzumab sensitivity in ErbB2 phosphorylation-positive breast cancer is believed to be due to increased tumor cell dependency on activated ErbB family receptors.Citation20 We recently reported that binding of trastuzumab to ErbB2 induced ErbB2 tyrosine phosphorylation in trastuzumab-sensitive cells.Citation11 However, the molecular mechanisms of trastuzumab-induced ErbB2 phosphorylation still remain elusive. Furthermore, it has not been reported whether binding of trastuzumab to ErbB2 modulates ErbB2 kinase activity, which may further impact ErbB2 phosphorylation and downstream of ErbB2-coupled signaling.
ErbB2 is capable of mediating transformation through distinct effector pathways, and the transformation potential of ErbB2 can be mediated by both positive and negative regulatory tyrosine phosphorylation sites in fibroblasts.Citation21,Citation22 Cell culture data previously showed that tyrosine phosphorylation at ErbB2-Y1248 plays a role in the negative regulation of ErbB2-coupled signaling.Citation23,Citation24 Csk-homologous kinase (CHK), a non-receptor tyrosine kinase, also known as megakaryocyte-associated tyrosine kinase (MATK), binds to phosphorylated ErbB2-Y1248 (ErbB2-pY1248) and negatively regulates the activity of ErbB2.Citation24 It was reported that in addition to phosphorylating and negatively regulating Src kinase activity, CHK also inhibits other Src family tyrosine kinases via binding to their active conformations.Citation25 Binding of CHK to pY1248 of ErbB2 is believed to be important for the suppression of heregulin-activated Src kinase activity, and stable expression of wild-type CHK inhibits the growth of MCF-7 breast cancer cells in soft agar.Citation24,Citation26,Citation27 Among downstream signaling molecules that bind to the C-terminus of ErbB2, CHK is the only effector that is associated with growth inhibition and suppression of oncogenic signaling mediated by ErbB2.Citation24,Citation26,Citation27 This raises the question of whether the function of CHK is associated with trastuzumab-induced phosphorylation of ErbB2-Y1248 and trastuzumab-mediated inhibition of breast cancer cell growth. In the present study, we report that binding of trastuzumab to ErbB2 activates the kinase activity of ErbB2 and increases interaction between ErbB2 and CHK. Overexpression of CHK mimics trastuzumab treatment to mediate ErbB2-Y1248 phosphorylation, Akt downregulation, and growth inhibition of trastuzumab-sensitive breast cancer cells. Our data demonstrate that positive ErbB2-pY1248 staining correlates with trastuzumab sensitivity in trastuzumab clinical neoadjuvant settings and in ErbB2 positive breast cancer cell lines.
Results
Trastuzumab-induced ErbB2-Y1248 phosphorylation correlates with its ability to inhibit Akt activity and downregulate ErbB3 in trastuzumab-sensitive SKBR3 and BT474 cells
To investigate the site-specific phosphorylation of ErbB family receptors upon binding of trastuzumab to ErbB2, we analyzed the relative levels of 17 potential phosphorylation sites among the ErbB family receptors. Trastuzumab-sensitive breast cancer cells were serum-starved overnight and then were treated either with trastuzumab (4 μg/mL) or EGF (100 ng/mL), or left untreated for the indicated times. showed that both trastuzumab and EGF induced phosphorylation of ErbB2-Y1248 (a pair of dots in the blue rectangle) and ErbB1-Y845 (a pair of dots in the red rectangle) compared with the basal levels of the phosphorylation in SKBR3 cells (0 min). EGF also stimulated phosphorylation of ErbB2-Y1112 (a pair of dots in the purple rectangle). Longer exposures revealed several other phosphorylated sites such as S1070, Y1173, and Y1086, in ErbB1 following EGF stimulation, but not trastuzumab treatment (data not shown). The upregulation of phosphorylation at ErbB1-Y845, Y1173, Y1086, and ErbB2-Y1112 following EGF treatment is consistent with previous reports.Citation28-Citation30
Figure 1. Trastuzumab induces phosphorylation of ErbB2-Y1248 and ErbB1-Y845, inhibits Akt phosphorylation and downregulates ErbB3 in trastuzumab-sensitive SKBR3 and BT474 cells. (A) SKBR3 cells (2 × 106) were plated in 10 cm dishes, serum-starved overnight and treated either with trastuzumab (4 μg/mL) or EGF (100 ng/mL) for the indicated times or left untreated. After harvesting, the whole cell lysates (WCL) were incubated with RayBio human EGFR phosphorylation antibody array 1 according to the instructions provided by the manufacturer. The pair of dots in red rectangles indicates phosphorylated ErbB1-Y845 (ErbB1-pY845); blue rectangles indicate ErbB2-pY1248; purple rectangles indicate ErbB2-pY1112. The map of EGFR phosphorylation antibody array 1 is available at http://www.raybiotech.com/. (B) Changes in phosphorylation, as detected by RayBio human EGFR phosphorylation antibody array 1, following treatment of BT474 cells with trastuzumab or EGF for the indicated times. The experimental procedures were essentially the same as described under (A). Red rectangles indicate phosphorylation signal for ErbB1-Y845; blue rectangles indicate phosphorylation signal for ErbB2-pY1248. Black rectangles indicate phosphorylation signal for ErbB2-pT686 and pS1113 (from left to right). The experiments with RayBio human EGFR phosphorylation antibody array 1 were repeated at least twice for both cell lines. (C) SKBR3 cells were serum-starved and incubated either with trastuzumab or EGF for the indicated times. The cells were harvested and the levels of ErbB2-pY1248, ErbB1-pY845, P-Akt-T308, and P-Akt-S473 or P-ERK1/2 and total levels of the indicated proteins in WCL were determined by western blot analysis. Actin western blot analysis of WCL was done to control for equal loading (note: from here on, actin western blots were used to control for equal loading in all figures). Bottom two panels, levels of ErbB1-pY845 and total ErbB1 were determined in ErbB1 immunoprecipitate by western blot analysis. (D) Western blot analysis of BT474 cells treated either with trastuzumab or EGF. The experimental procedures were essentially the same as (C). (E) SKBR3 and BT474 cells were serum-starved overnight and treated with trastuzumab for 1 h or left untreated. The levels of phosphorylated ErbB3-Y1289 (ErbB3-pY1289) and total ErbB3 were detected by western blot analysis using antibodies directed against ErbB3-pY1289 or ErbB3. (F) The cells were grown in the media supplemented with 10% FBS and then treated with trastuzumab (10 μg/mL) for the indicated times. WCL harvested from indicated cells were subjected to western blot analysis. The levels of ErbB3 in WCL were detected using an antibody directed against ErbB3.
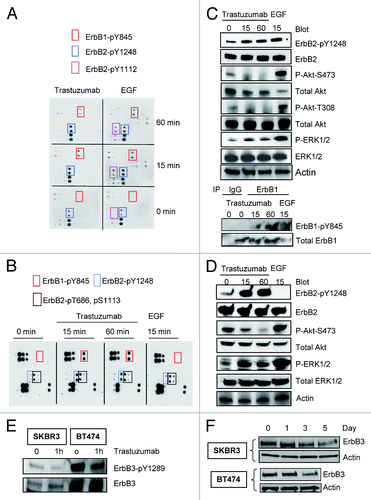
Analysis of phosphorylation in BT474 cells also revealed strong trastuzumab-mediated phosphorylation at ErbB1-Y845 and ErbB2-Y1248 (), similar to that in SKBR3 cells. However, in contrast to SKBR3 cells, treatment of BT474 cells with EGF did not result in the upregulation of phosphorylation at ErbB1-Y845 and ErbB2-Y1248 (). This may be due to very low levels of ErbB1 in BT474 cells. Although phosphorylation of ErbB2-T686 and S1113 (black rectangles) can be detected in BT474 cells, the phosphorylation of these two sites was not increased following treatment with either trastuzumab or EGF, suggesting that these two sites may have relatively higher basal levels of phosphorylation in BT474 cells as compared with that in SKBR3 cells. The different phosphorylation profiles found in SKBR3 and BT474 cells following treatment with either trastuzumab or EGF suggested the heterogeneity in response in trastuzumab-sensitive breast cancer cells. However, these two trastuzumab-sensitive cell lines also shared some common characteristics such as trastuzumab-mediated increase in tyrosine phosphorylation at ErbB2-Y1248 and ErbB1-Y845. Given that trastuzumab inhibits ErbB2-positive breast cancer growth, we propose that trastuzumab-induced phosphorylation at ErbB2-Y1248 may initiate a negative feedback mechanism that inhibits ErbB2-mediated growth signaling in breast cancer cells.
To confirm data obtained from the antibody array, we analyzed the phosphorylation status of ErbB2-Y1248 and ErbB1-Y845 following either trastuzumab or EGF treatment in SKBR3 and BT474 cells. EGF treatment resulted in an increase in phosphorylation of ErbB2-Y1248 in SKBR3 cells (), but not in BT474 cell (), consistent with the data shown in . However, EGF induced strong upregulation of Akt-S473 and/or T308 and ERK1/2 phosphorylation in both cell lines (). In both cell lines, trastuzumab induced strong phosphorylation of ErbB2-Y1248, consistent with data shown in . In contrast to EGF, trastuzumab-mediated increase in ErbB2-Y1248 phosphorylation was associated with a strong suppression in Akt phosphorylation in both SKBR3 and BT474 cells ( and ). Taken together, these data suggest that trastuzumab-induced ErbB2-Y1248 phosphorylation may be associated with a mechanism, yet unidentified, that mediates inhibition of Akt signaling. Surprisingly, trastuzumab enhanced phosphorylation of ERK1/2 in both cell lines ( and ). This may be the consequence of ErbB1-Y845 phosphorylation induced by trastuzumab since it has been reported that ErbB1-Y845 phosphorylation is associated with ERK activity.Citation31 Increased ErbB1-Y845 phosphorylation was confirmed in SKBR3 cells following trastuzumab treatment (), consistent with data shown in .
We then addressed the effect of trastuzumab on ErbB3 phosphorylation and degradation. As shown in , trastuzumab treatment resulted in the inhibition of ErbB3-Y1289 phosphorylation in both SKBR3 and BT474 cells, consistent with previous reports.Citation5,Citation19 We then addressed whether trastuzumab was able to induce ErbB3 degradation in trastuzumab-sensitive cells. As shown in , we found that the levels of ErbB3 in both SKBR3 and BT474 cells were reduced as a function of time of trastuzumab treatment, indicating that trastuzumab can also induce ErbB3 degradation in trastuzumab-sensitive cells ().
Trastuzumab activates ErbB2 kinase activity and is able to induce ErbB2-Y1248 phosphorylation in the presence of the tyrosine kinase inhibitor lapatinib
Data shown in proved that although treatment with either trastuzumab or EGF resulted in tyrosine phosphorylation of ErbB family receptors, the phosphorylation profiles induced by trastuzumab or EGF are different. This may lead to different outcomes of cellular response. To further explore the relationship between trastuzumab-induced growth inhibition and ErbB2 phosphorylation, we asked the question of whether trastuzumab treatment might lead to the enhancement of ErbB1/ErbB2 heterodimer formation in ErbB2 overexpressing cells. Serum-starved SKBR3 cells were treated with either trastuzumab (4 μg/mL) for 1 h or EGF for 15 min. As shown in , in SKBR3 cells, while ErbB1/ErbB2 heterodimers can be detected at the steady-state, treatment with either trastuzumab or EGF did not increase heterodimer formation between ErbB1 and ErbB2. However, both trastuzumab and EGF enhanced the phosphorylation of ErbB1-Y845. We then asked whether similar results would be obtained in BT474 cells. Since the levels of ErbB1 expression are very low in BT474 cells (data not shown), detection of ErbB2/ErbB1 heterodimers was performed by receptor crosslinking with the cross linking reagent DTSSP following trastuzumab treatment (4 μg/mL) of serum-starved BT474 cells. The experiments were performed as previously described.Citation5 Data obtained from BT474 cells were consistent with the results obtained from SKBR3 cells ().
Figure 2. Trastuzumab activates ErbB2 tyrosine kinase and induces ErbB2-Y1248 phosphorylation in the presence of lapatinib. (A) Increased ErbB1-Y845 phosphorylation was detected following trastuzumab treatment of SKBR3 cells. Serum-starved SKBR3 cells were treated for 1 h. After harvesting the WCL, the immunoprecipitation reaction with antibody recognizing ErbB2 (29D8) was performed to detect the heterodimer between ErbB1 and ErbB2. (B) The experiments were performed similar to those described in (A) except that prior to harvesting the WCL, trastuzumab-treated BT474 cells were crosslinked with DTSSP reagent according to the modified protocol provided in the literature.Citation5 (C) Trastuzumab induces the activation of ErbB2 kinase activity in BT474 and SKBR3 cells. After serum-starving overnight, SKBR3 and BT474 cells were either treated with trastuzumab for 1 h or EGF for 15 min, or left untreated as indicated. WCL were harvested and subjected to immunoprecipitation using either human control IgG or trastuzumab. ErbB2 tyrosine kinase activity in each immunoprecipitate was determined using a universal tyrosine kinase assay kit (Takara Bio Inc.) according to manufacturer’s instructions. Data are expressed as mean ± SEM. Statistical significance was determined by the Student t test. *P < 0.05. (D) SKBR3 cells were plated and grown in the serum-containing media and then serum-starved overnight. Cells were either pre-treated with lapatinib (200 nM) for 4 h or not pretreated and then either treated with trastuzumab (4 μg/mL) or left untreated. Analysis of phosphorylation levels of ErbB family phosphorylation sites was done by RayBio human EGFR phosphorylation antibody array 1 according to the manufacturer’s instructions. (E) Analysis of phosphorylation level in ErbB family receptors following treatment of BT474 cells with trastuzumab, lapatinib, or trastuzumab plus lapatinib. The experimental procedures were essentially the same as those described in (D).
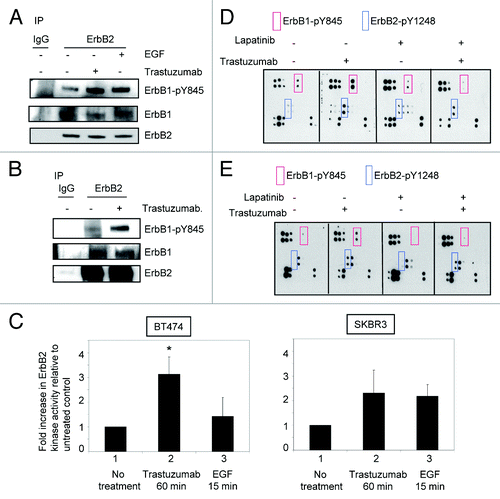
These data raised a question of whether binding of trastuzumab to ErbB2 enhanced its kinase activity, which may lead to the enhanced phosphorylation of ErbB2-Y1248. As shown in , a significant increase (~3-fold increase) in ErbB2 kinase activity was observed in BT474 cells treated with trastuzumab for 1h compared with the untreated control cells (P = 0.019), while ErbB2 kinase activity was only slightly increased in BT474 cells treated with EGF compared with the untreated control (P = 0.26). This may be due to the low levels of ErbB1 in BT474 cells. Data shown in may also explain why EGF did not stimulate phosphorylation of ErbB2-Y1248 in BT474 cells (). Approximately 2.5-fold increase in ErbB2 kinase activity was also observed in SKBR3 cells treated with trastuzumab for 1h compared with the untreated control cells (P = 0.053). However, in SKBR3 cells, the levels of EGF-induced ErbB2 kinase activity were similar to that induced by trastuzumab (P = 0.14). These data suggest that phosphorylation of ErbB2-Y1248 induced by trastuzumab may be the consequence of the upregulated ErbB2 kinase activity upon trastuzumab treatment.
We next investigated the effects of blocking ErbB1/ErbB2 kinase activity on trastuzumab-mediated ErbB1-Y845 and ErbB2-Y1248 phosphorylation. Serum-starved SKBR3 and BT474 cells were pretreated with 200 nM lapatinib, a dual tyrosine kinase inhibitor of ErbB1 and ErbB2, for 4 h followed by trastuzumab treatment at 4 μg/mL for 1 h. As shown in , lapatinib pretreatment effectively blocked trastuzumab-mediated phosphorylation at ErbB1-Y845 (, red rectangles), suggesting that upregulated ErbB2 kinase activity induced by trastuzumab was responsible for the transphosphorylation of ErbB1-Y845. However, trastuzumab was still capable of inducing phosphorylation of ErbB2-Y1248 in the presence of lapatinib in SKBR3 cells although the extent of phosphorylation of ErbB2-Y1248 was slightly lower than that in the absence of lapatinib (, blue rectangles). Similar results were obtained when BT474 cells were used for this experiment (). Taken together, these data suggested that trastuzumab-mediated ErbB2-Y1248 phosphorylation was, at least partially, independent of ErbB1/ErbB2 kinase activities and that a tyrosine kinase, yet unidentified, plays a role in trastuzumab-mediated ErbB2-Y1248 phosphorylation.
Trastuzumab treatment increases interaction between ErbB2 and CHK
It has been reported that the ErbB2-pY1248 is a docking site for downstream effectors.Citation21,Citation24,Citation32 CHK, a non-receptor tyrosine kinase, has been reported to bind to ErbB2 directly and to act as a negative regulator of breast cancer cell growth.Citation27 Kim et al. demonstrated that the CHK SH2 domain binds directly to phosphorylated ErbB2-Y1248 and that this interaction is critical for the inhibition of heregulin-stimulated Src kinase activity.Citation24 We also confirmed that ErbB2 interacted with CHK in BT474 cells. As shown in , using an antibody directed against ErbB2 (trastuzumab), CHK was co-immunoprecipitated with ErbB2 in BT474 cells ().
Figure 3. Trastuzumab treatment increases the interaction between ErbB2 and CHK in BT474 cells. (A) BT474 cells were electroporated with either empty pCMV6-entry vector or pCMV-entry vector encoding DDK-tagged CHK. After transfection, cells were grown in the serum-containing media to recover for 24 h, and then were serum-starved overnight. Immunoprecipitation was performed as indicated and the immunoprecipitates were run in parallel with WCL from the indicated reactions to detect CHK and ErbB2 expression by western blot analysis. (B) BT474 cells were plated, serum-starved, fixed, and permeabilized. Following permeabilization, Duolink proximity ligation assay was performed according to the manufacturer’s instructions as indicated in the Materials and Methods. Representative images of samples incubated with anti-ErbB2 and anti-CHK antibodies (top row), and negative control samples (no primary antibodies, middle; anti-ErbB2 antibody only, bottom row).
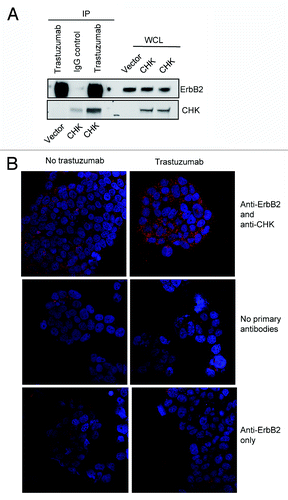
To address if CHK plays a role in trastuzumab-mediated ErbB2-Y1248 phosphorylation and growth inhibition, we investigated whether trastuzumab treatment increased the interaction between CHK and ErbB2 using Duolink proximity ligation assay (PLA). Duolink, based on in situ PLA, is designed to visualize endogenous protein interactions in fixed cells or tissues.Citation33 BT474 cells were seeded and serum-starved overnight. Cells were then treated with trastuzumab for 1 h or left untreated. Two primary antibodies raised in different species were used to detect ErbB2 and CHK protein complex. As shown in (top left panel), there was a basal level of ErbB2/CHK interaction (red dots) detected in BT474 cell. However, the interaction between ErbB2 and CHK was dramatically enhanced in cells treated with trastuzumab (top right panel). These data suggested that trastuzumab treatment increased the formation of a CHK/ErbB2 protein complex. The second and third panels in showed that few signals (red dots) were detected in cells that were labeled with either no primary antibodies or only one primary antibody (anti-ErbB2 only). Using Duolink assay, a trastuzumab-mediated increase in ErbB2/CHK interaction was also detected in SKBR3 cells (Fig. S1).
CHK contributes to trastuzumab-induced ErbB2-Y1248 phosphorylation and ErbB2-degradation
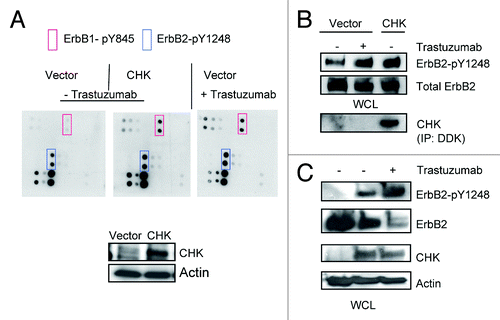
Data presented in suggested that CHK might play a role in trastuzumab-induced ErbB2-Y1248 phosphorylation. As shown in , in the absence of trastuzumab, overexpression of CHK in BT474 cells resulted in a tyrosine phosphorylation profile that was similar to that induced by trastuzumab in that tyrosine phosphorylation of ErbB2-Y1248 and ErbB1-Y845 was enhanced (, blue and red rectangles). We next investigated the functional connections between trastuzumab and CHK. As expected, trastuzumab induced an increase in ErbB2-Y1248 phosphorylation (). Consistent with data shown in , overexpression of CHK also stimulated ErbB2-Y1248 phosphorylation (). showed that the phosphorylation of ErbB2-Y1248 induced by CHK overexpression was further increased by trastuzumab treatment. Importantly this enhanced phosphorylation of ErbB-Y1248 was accompanied by a reduction in the total levels of ErbB2 in CHK-expressing BT474 cells that were treated with trastuzumab for 1 h (). Taken together, data presented on and implicate that trastuzumab-mediated recruitment of CHK to ErbB2 receptor is associated with increases in ErbB2-Y1248 phosphorylation and ErbB2 degradation. These data may also explain why lapatinib did not completely block trastuzumab-induced phosphorylation of ErbB2-Y1248 (). It should be noted that we still do not know whether CHK directly phosphorylates ErbB2 at Y1248.
Figure 4. Overexpression of CHK induces ErbB2-Y1248 phosphorylation and mediates ErbB2 degradation following treatment with trastuzumab. (A) BT474 cells were electroporated with either empty pCMV6-entry vector or pCMV-entry vector encoding DDK-tagged CHK. Cells were then grown in serum-containing media to recover for 24 h, followed with serum starvation for 24 h. Vector control cells were either treated with trastuzumab (4 μg/mL) for 1 h or left untreated. The RayBiotech antibody array assay was used to detect the phosphorylation levels among different EGFR family members. Red rectangles, ErbB1-pY845; blue rectangles, ErbB2-pY1248. The expression of DDK-tagged CHK in WCL was detected using an antibody directed against DDK. (B) The experimental procedures were similar to those described in (A) except that WCL obtained from BT474 cells were subjected to western blot analysis to detect ErbB2-pY1248 and total ErbB2. The overexpressed DDK-tagged CHK was immunoprecipitated and detected using antibody directed against DDK. (C) BT474 cell were transiently transfected with either empty pCMV6- entry vector or pCMV6-entry vector encoding DDK-tagged CHK. The cells were grown in the serum-containing media for 24 h and were then serum-starved for 24 h. The cells were either treated with trastuzumab (4 μg/mL) for 1 h or left untreated. The levels of ErbB2-pY1248 and total ErbB2 in WCL were detected using western blot analysis. DDK-tagged CHK expression was detected using an antibody directed against CHK.
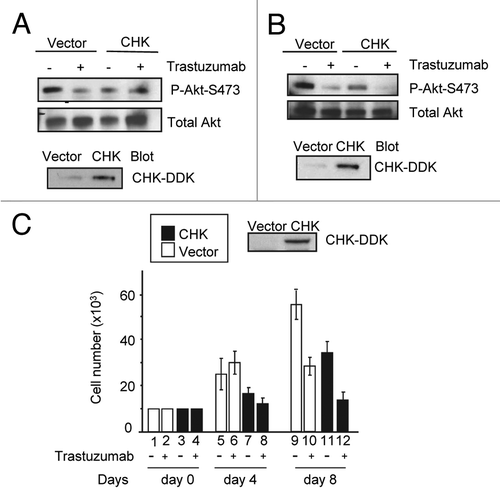
Overexpression of CHK results in downregulation of Akt activity and inhibition of BT474 cell growth
We next examined the effects of CHK overexpression on Akt phosphorylation. As expected, trastuzumab treatment of BT474 cells transiently transfected with control vector resulted in the inhibition of Akt-S473 phosphorylation (). Transient overexpression of CHK also resulted in a decrease in Akt-S473 phosphorylation independent of trastuzumab treatment (). Trastuzumab treatment of CHK expressing cells did not result in further downregulation of Akt-S473 phosphorylation (). Similar results were obtained from SKBR3 cells transfected with either the control vector or the vector encoding DDK-tagged CHK, except that in SKBR3 cells trastuzumab treatment led to a further inhibition in Akt-S473 phosphorylation (). Transient overexpression of DDK-tagged CHK in BT474 and SKBR3 cells was detected using an antibody directed against the DDK tag (, bottom panels). We next asked if overexpression of CHK inhibited the growth of BT474 cells. As shown in , transient expression of CHK in BT474 cells inhibited the growth of BT474 cells. We observed about a 30% reduction in cell number on days 4 and 8, respectively for cells overexpressing CHK as compared with cells expressing empty vector (, cf. column 5 with column 7 for day 4; cf. column 9 with column 11 for day 8). Addition of trastuzumab further inhibited the growth of BT474 cells overexpressing CHK such that about a 55% reduction in cell numbers occurred at both time points in cells overexpressing CHK compared with cells expressing the vector control (, cf. column 6 with column 8 for day 4; column 10 with column 12). Taken together, data shown in suggested that overexpression of CHK mimicked the inhibitory effect of trastuzumab on Akt activity and BT474 cell growth. Additionally, at day 8 the combination of CHK overexpression and trastuzumab treatment had an additive effect on growth inhibition, in that a 49% growth inhibition was observed for cells treated with trastuzumab alone whereas a 75.2% growth inhibition occurred when CHK-expressing cells were treated with trastuzumab on day 8 (, cf. columns 9, 10, and 12).
Figure 5. CHK overexpression reduces Akt phosphorylation and inhibits BT474 cell growth. (A) Western blot analysis of Akt phosphorylation in BT474 cells overexpressing either empty pCMV6 vector or DDK-tagged CHK. BT474 cells were electroporated and left to recover in serum containing media for 24 h. Cells were then serum-starved for 24 h and were either treated with trastuzumab (4 μg/mL) or left untreated. WCL were subjected to western blot analysis using antibodies directed against pAkt-S473 or Akt. Anti-DDK antibody was used to detect overexpression of DDK-tagged CHK in WCL. (B) Western blot analysis of Akt-S473 phosphorylation in SKBR3 cells overexpressing either empty pCMV6 vector or DDK-tagged CHK. The experimental procedures were essentially the same as those described in (A). (C) Analysis of growth of BT474 cells transiently overexpressing empty vector or vector encoding DDK-tagged CHK. After transfection, cells were plated in triplicate at 10 × 103/well in 12 well plates in media containing either trastuzumab (10 μg/mL) or no trastuzumab. Cells were trypsinized, mixed with trypan blue and counted using BioRad TC10 automated cell counter on the indicated days. Data represent the mean ± SEM from one of the two independent experiments each performed in triplicate. DDK-tagged CHK expression in BT474 cells was detected by western blot analysis using an anti-DDK antibody.
Trastuzumab does not induce ErbB2-Y1248 phosphorylation, but is capable of downregulating Akt phosphorylation in trastuzumab-resistant JIMT1 cells
Results shown in the previous sections suggest that the ability of trastuzumab to mediate phosphorylation of ErbB2-Y1248, which correlates with Akt inhibition, is an important part of mechanism of action for trastuzumab-induced growth inhibition in trastuzumab-sensitive cells. We then asked whether trastuzumab was able to induce ErbB2-Y1248 phosphorylation in trastuzumab-resistant cells. Unlike SKBR3 and BT474 cells, little or no basal levels of phosphorylated ErbB2-Y1248 were detected in trastuzumab-resistant JIMT1 cells and trastuzumab did not induce ErbB2-Y1248 phosphorylation in these cells (). Moreover, the inability of trastuzumab to induce ErbB2-Y1248 phosphorylation correlated with trastuzumab-resistant phenotype of JIMT1 cells. As shown in , trastuzumab did not inhibit JIMT1 cells growth, consistent with previous reports.Citation10,Citation34 In addition, no interactions between CHK and ErbB2 were detected by Duolink assays in JIMT1 cells that were either treated with trastuzumab or left untreated (Fig. S2) although the expression levels of endogenous CHK in JIMT1 cells were even slightly higher than that in SKBR3 cells (). This suggests that the interaction between CHK and ErbB2 is dependent on the phosphorylation of ErbB2-Y1248. Next, we asked whether overexpression of CHK induces ErbB2-Y1248 phosphorylation in JIMT1 cells. showed that CHK overexpression did not enhance ErbB2-Y1248 phosphorylation in JIMT1 cells. We asked a question of whether overexpression of CHK inhibited JIMT1 cell growth or rendered JIMT1 cells to be sensitive to trastuzumab. demonstrated that overexpression of CHK not only did not inhibit BT474 cell growth, but also was unable to render JIMT1 cells to be sensitive to trastuzumab. Taken together, these data provide evidence that sensitivity of cells to trastuzumab is independent of CHK expression. ErbB2-Y1248 phosphorylation and ErbB2/CHK complex formation induced by trastuzumab are critical for the sensitivity of trastuzumab treatment. The inset in showed CHK expression in JIMT1 cells at the indicated time points.
Figure 6. Trastuzumab is unable to induce ErbB2-Y1248 phosphorylation in JIMT1 cells, but is still capable of inhibiting Akt activity. (A) Analysis of ErbB2-Y1248 phosphorylation in trastuzumab-sensitive and trastuzumab-resistant cells as indicated following treatment with trastuzumab. Cells were plated in 10% serum containing media for 24 h and then treated with trastuzumab (4 μg/mL) for 1 h or left untreated. The levels of ErbB2-pY1248, total ErbB2, and endogenous CHK in WCL were detected by western blot analysis using anti-phospho-ErbB2-Y1248, anti-ErbB2, and anti-CHK antibodies. (B) JIMT1 cells were plated at 2.5 × 104/well in 12-well plates in triplicate in media containing either trastuzumab (10 μg/mL) or no trastuzumab (control). Cells were trypsinized, mixed with trypan blue, and counted at the indicated times using a BioRad TC10 automated cell counter. Data represent the mean ± SEM from one of the two independent experiments, and each was performed in triplicate. (C) JIMT1 cells were electroporated with either empty pCMV6-entry vector or pCMV-entry vector encoding DDK-tagged CHK. WCL were harvested 48 h post-transfection and then subjected to immunoprecipitation using an antibody directed against ErbB2. The levels of ErbB2-pY1248 in immuneprecipitates were evaluated by western blot analysis using an antibody directed against ErbB2-pY1248. Total ErbB2 in the immunoprecipitates was detected using an antibody directed against ErbB2. The levels of DDK-CHK in WCL were detected using an antibody directed against DDK tag. IgG was used as a negative control. (D) JIMT1 cells were electroporated with either empty pCMV6-entry vector or pCMV-entry vector encoding DDK-tagged CHK. Cells then were seeded at 10 × 103/well in 12-well plate in triplicate and treated with trastuzumab (50 μg/mL) or left untreated. Cells were trypsinized, mixed with trypan blue and counted using a BioRad TC10 automated cell counter on the indicated days. Data represent the mean ± SEM from one of the two independent experiments, and each was performed in triplicate. Inset: Levels of DDK-CHK expression in WCL were detected in JIMT1cells by western blot analysis using an antibody directed against DDK at the indicated times. (E) JIMT1 cells were plated in media containing 10% serum for 24 h and then serum-starved for another 24 h. Cells then were treated with trastuzumab at the indicated concentration for 1 h or left untreated. The levels of P-Akt-T308 and total Akt in WCL were detected by western blot analysis. (F) The experimental procedures were essentially the same as described in (E) except that SKBR3 and BT474 cells were also included and the levels of P-Akt-S473 and total Akt in WCL were detected by western blot analysis. (G) MCF7 cells were plated at 2.5 × 104/well in a 12-well plate overnight, and then treated with either trastuzumab (4 and 10 μg/mL) or left untreated for the indicated days. Cells were trypsinized, mixed with trypan blue, and counted using BioRad TC10 automated cell counter on the indicated days. Data represent the mean ± SEM from one of the two independent experiments, and each was performed in triplicate. (H) P-ERK1/2 and total ERK1/2 detected in MCF7 WCL by western blot analysis using antibodies directed against P-ERK1/2 and ERK1/2, respectively.
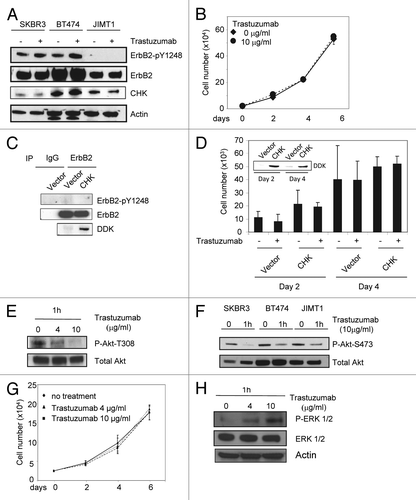
Since trastuzumab was unable to induce ErbB2-Y1248 phosphorylation in JIMT1 cells, we then tested whether the activity of Akt was altered by trastuzumab in these cells. Surprisingly, trastuzumab was still able to downregulate phosphorylation of Akt at two phosphorylation sites (T308 and S473) (). This result indicates that although trastuzumab inhibition of PI3K-Akt signaling is one of major mechanisms to mediate growth inhibition,Citation5,Citation12 resistance to trastuzumab in JIMT1 cells was not due to the inability of trastuzumab to inhibit Akt signaling. These data further demonstrate that the phosphorylation status at ErbB2-Y1248 or inability to induce phosphorylation of ErbB2-Y1248 by trastuzumab might be a potential predictive biomarker for primary resistance to trastuzumab.
MCF7 cells, which are derived from a breast adenocarcinoma, express relatively low levels of ErbB2 as compared with SKBR3 and BT474 cellsCitation7 (also our unpublished data) and are resistant to trastuzumab.Citation18,Citation35 ErbB2-Y1248 phosphorylation was not detectable in MCF7 cell, and trastuzumab also was unable to induce ErbB2-Y1248 phosphorylation in these cells (data not shown). showed that trastuzumab was unable to inhibit MCF7 cell growth. While Akt was detected in MCF7 cells, the phospho-Akt was not detected in MCF7 cells under either trastuzumab-treated or untreated conditions (data not shown). Similar to SKBR3 and BT474 cells, upregulation of ERK1/2 phosphorylation (P-ERK1/2) was readily detectable in MCF7 cells treated with trastuzumab (), suggesting that ERK phosphorylation induced by trastuzumab is likely not associated with inhibitory effects of trastuzumab on cell growth. Taken together, these data provide another line of evidence that resistance to trastuzumab correlated with the loss of ErbB2-Y1248 phosphorylation.
The status of ErbB2-Y1248 phosphorylation correlates with the response to trastuzumab treatment in neoadjuvant settings
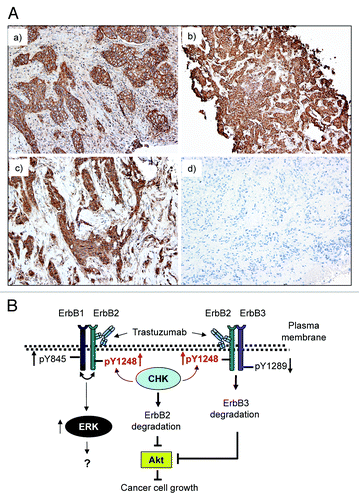
Our data suggested that the phosphorylation status at ErbB2-Y1248 might be an important indicator for sensitivity to trastuzumab. We next examined whether ErbB2-Y1248 phosphorylation was associated with response to trastuzumab in ErbB2-positive breast cancer. Analysis of clinical specimens for the presence of ErbB2-pY1248 staining was performed on tumor samples from the patients who were indicated to receive trastuzumab therapy in the neoadjuvant settings. As shown in , 4 out of 5 patients who progressed or had residual disease after neoadjuvant trastuzumab treatment were negative for ErbB2-pY1248 staining (either 0, +, or +/++), whereas 4 out of 5 patients who either achieved complete or near complete pathological remission after neoadjuvant trastuzumab treatment were positive for ErbB2-pY1248 staining (3+). Representative anti-ErbB2-pY1248 immunohistochemical staining of core breast cancer biopsy samples is shown in . Panels a and b show positive ErbB2-pY1248 staining (3+). Panel c shows negative ErbB2-pY1248 staining (1+) with predominantly cytoplasmic and focal incomplete membrane staining, and panel d shows a negative staining. Cases a and b had a complete pathologic response after neoadjuvant trastuzumab treatment, whereas cases c and d had residual disease after neoadjuvant trastuzumab treatment. These data indicated that ErbB2-pY1248-positive staining correlated with response to trastuzumab treatment in ErbB2-positive breast cancer patients.
Table 1. Phosphorylation status of ErbB2-Y1248 for trastuzumab non-responders and trastuzumab responders in trastuzumab neoadjuvant settings
Figure 7. Positive ErbB2-pY1248 staining in ErbB2-positive breast cancer biopsies correlates with the increased trastuzumab response in trastuzumab neoadjuvant settings. (A) Immunohistochemical staining using an anti-phospho-ErbB2-Y1248 antibody on representative cases of core biopsies obtained from breast cancer patients before neoadjuvant trastuzumab treatment. (a and b) Positive with 3+ staining; (c) negative (+, with predominantly cytoplasmic and focal incomplete membrane staining); (d) negative staining. Cases (a and b) had complete pathologic response after trastuzumab treatment. Cases (c and d) had residual disease. (B) Model depicting trastuzumab-mediated interaction between CHK and ErbB2 to regulate ErbB2 phosphorylation at Y1248 and degradation. Overexpression of ErbB2 in trastuzumab-sensitive breast cancer cells leads to heterodimer formation among ErbB family members (ErbB1/ErbB2 and ErbB2/ErbB3). This results in the basal phosphorylation of ErbB2 at Y1248 creating docking sites for downstream effectors such as CHK. When binding to the extracellular domain of ErbB2, trastuzumab stimulates kinase activity of ErbB2, resulting in an increase in ErbB2-pY1248. This in turn promotes recruitment of CHK to ErbB2 via ErbB2-pY1248. Upon binding to ErbB2, CHK further enhances the phosphorylation of ErbB2-Y1248 and induces ErbB2 degradation. Binding of trastuzumab to ErbB2 does not interfere with heterodimer formation between ErbB1 and ErbB2, but rather induces ErbB1-Y845 phosphorylation. This may lead to the induction of ERK1/2 phosphorylation. However, the role of ERK1/2 phosphorylation induced by trastuzumab in trastuzumab-mediated growth inhibition remains elusive. Binding of trastuzumab to ErbB2 in ErbB2/ErbB3 heterodimers leads to the inhibition of ErbB3-Y1289 phosphorylation and promotes ErbB3 degradation. Downregulation of both ErbB2 and ErbB3 results in the inhibition of Akt activity and breast cancer cell growth.
Discussion
Unlike cetuximab, which binds to the extracellular domain of EGFR/ErbB1 and blocks ligand-induced EGFR/ErbB1 tyrosine phosphorylation and downstream signaling events,Citation36,Citation37 trastuzumab induces ErbB2 tyrosine phosphorylation upon binding to ErbB2.11,12,17,19. Furthermore, trastuzumab-induced tyrosine phosphorylation of ErbB2 is correlated with its inhibitory effect on tumor growth.Citation16 This effect may be unique to trastuzumab structure, since 4D5, a mouse monoclonal antibody from which epitope regions were used to generate trastuzumab was reported to inhibit phosphorylation of ErbB2.Citation38 This raises the question of how trastuzumab-induced tyrosine phosphorylation of ErbB2 is coordinated with its ability to inhibit tumor growth.
Using EGFR phosphorylation antibody array 1, we compared phosphorylation levels among ErbB family receptors induced by either EGF or trastuzumab in trastuzumab-sensitive SKBR3 and BT474 cells. Trastuzumab-induced phosphorylation of ErbB1-Y845 is eliminated by lapatinib, suggesting that trastuzumab-induced phosphorylation at ErbB1-Y845 is ErbB1 and/or ErbB2 kinase dependent (). However, trastuzumab is still able to induce ErbB2-Y1248 phosphorylation in the presence of lapatinib, which indicates that trastuzumab-induced ErbB2-Y1248 phosphorylation is at least in part independent of the kinase activity of ErbB2 ().
Here, we describe a previously unappreciated relationship between trastuzumab and the non-receptor kinase CHK. Based on a number of lines of evidence presented in this study, we propose a novel molecular mechanism by which trastuzumab promotes recruitment of CHK to ErbB2 to regulate ErbB2 phosphorylation at Y1248 and ErbB2 degradation. As shown in the proposed model presented in , the basal phosphorylation at ErbB2-Y1248 allows CHK to bind to ErbB2. Upon binding to the extracellular domain of ErbB2, trastuzumab activates the ErbB2 kinase. This further induces ErbB2 phosphorylation at Y1248 and promotes recruitment of CHK to the ErbB2. Upon binding to ErbB2, CHK enhances ErbB2-Y1248 phosphorylation, leading to an increase in trastuzumab-induced ErbB2 degradation, the reduction in Akt signaling and inhibition of cell growth.
Additionally, our data suggest a novel mechanism by which trastuzumab acts differently on existing heterodimers (ErbB1/ErbB2 vs. ErbB2/ErbB3) in both SKBR3 and BT474 cells. As shown in , binding of trastuzumab to ErbB2 does not interfere with heterodimer formation between ErbB1 and ErbB2, but rather induces ErbB1-Y845 phosphorylation, which may account for the induction of ERK1/2 phosphorylation.Citation31 In contrast, binding of trastuzumab to ErbB2 in ErbB2/ErbB3 heterodimers leads to the inhibition of ErbB3-Y1289 phosphorylation, resulting in ErbB3 degradation and downregulation of Akt activity.
Collectively, this study highlights an important role for ErbB2-Y1248 phosphorylation in mediating trastuzumab-induced growth inhibition. Data indicate that the inhibitory effect of trastuzumab on breast cancer cells may result not only from inhibiting receptor-coupled signaling but also from activating its kinase activity to mediate signaling pathways that negatively regulate cell growth. Trastuzumab-induced interactions between CHK and ErbB2-pY1248 are identified as a novel mechanism of action that mediates growth inhibition. The novel mechanistic insights into trastuzumab action revealed by this study may impact the design of next generation of therapeutic monoclonal antibodies targeting receptor tyrosine kinases for the cancer therapy. For example, modulation of the kinase activity of a receptor, together with its associated downstream signaling pathways, may be incorporated in the screening strategies for the development of therapeutic monoclonal antibodies.
About two-thirds of ErbB2-positive breast cancers are primarily resistant to trastuzumab treatment.Citation39,Citation40 While downregulation of Akt is believed to be one of the major mechanisms of action of trastuzumab, trastuzumab is still capable of inhibiting Akt activity in trastuzumab-resistant JIMT1 breast cancer cells. This finding indicates that resistance to trastuzumab may not be due to the inability of trastuzumab to inhibit Akt activity. This also suggests that reduction in Akt phosphorylation induced by trastuzumab may not be a reliable indicator for the response to the trastuzumab treatment. Based on our data, we propose that the phosphorylation status of ErbB2-Y1248 might be an important biomarker for the response to the trastuzumab treatment. This observation is supported by our clinical data that positive staining of ErbB2-pY1248 correlated with increased response to trastuzumab treatment in neoadjuvant settings (; ). Prospective clinical investigations of the relationship between ErbB2-Y1248 phosphorylation and response to trastuzumab treatment are warranted for the development of ErbB2-Y1248 phosphorylation as a predictive biomarker for the response to trastuzumab treatment.
Methods/Materials
Antibodies and reagents
Antibodies against ErbB2 (29D8), phospho-ErbB2 (pY1248), phospho-ErbB1 (pY845), ERK1/2 (clone L34F12), phospho-ERK1/2 (202T/204Y), Akt (clone 11E7), phospho-Akt (S473, T308), were obtained from Cell Signaling Technology. Antibodies against actin, CHK and immunohistochemistry quality anti-phospho ErbB2 (pY1248) were obtained from Sigma-Aldrich. Antibody against ErbB1 was obtained from BD Transduction Laboratories. Antibody against DDK was obtained from Origene. Anti-DYKDDDDK tag (L5) affinity gel was obtained from BioLegend. Trastuzumab was purchased from the pharmacy at the National Institutes of Health (NIH). Recombinant human EGF was obtained from Invitrogen. pCMV6-entry (Myc/DDK) tagged vector encoding CHK and empty vector were obtained from Origene. The Duolink In Situ kit was obtained from Olink Bioscience.
Cell culture and transfection
SKBR3, BT474, and MCF7 cells were obtained from American Type Culture Collection (ATCC). SKBR3 and BT474 cells were grown in DMEM (Lonza) containing 10% fetal bovine serum (FBS) and 1% antibiotic/antimycotic (Invitrogen). The JIMT-1 cell line was purchased from DSMZ (German Collection of Microorganisms and Cell Cultures) and was grown in DMEM supplemented with 10% FBS. Transient expression of CHK in cells was done by Nucleofector technology (Lonza).
RayBio human EGFR phosphorylation antibody array 1
The array was obtained from RayBiotech, Inc. and the assays were performed according to manufacturer’s instructions. RayBio Human EGFR phosphorylation antibody array 1 can detect 17 phosphorylation sites of ErbB family of receptor tyrosine kinases.
ErbB2 kinase assay
ErbB2 tyrosine kinase activity was detected using a commercially available sandwich ELISA universal tyrosine kinase assay detection kit (Takara Bio, Inc.). This assay monitors the transfer of γ-phosphate residue from ATP to peptide substrates immobilized on plate. In brief, whole cell lysate (WCL) was harvested and subjected to immunoprecipitation with either control human IgG or anti-ErbB2 antibody (trastuzumab). Immunoprecipitates were incubated with tyrosine kinase substrate immobilized on the ELISA plate. Detection of tyrosine phosphorylation of substrate was performed by colorimetric reaction following incubation with the anti-phosphotyrosine (pY20) antibody conjugated to horse radish peroxidase that was measured at OD450. In each reaction, a set of tyrosine kinase activity standards provided by the manufacturer was also included and used to calculate tyrosine kinase activity of the respective samples. Normalization of the ErbB2 tyrosine kinase activity was performed by subtracting background (nonspecific human IgG) from the ErbB2 tyrosine kinase values, including trastuzumab-treated or untreated and EGF-treated samples. The resulting numbers were used to calculate the fold changes in ErbB2 kinase activity using the values obtained from cells untreated with trastuzumab as the reference.
Heterodimer (ErbB1/ErbB2) detection and immunoprecipitation
For the analysis of ErbB2/ErbB1 complexes in BT474 cells following trastuzumab treatment, the cells were cross-linked using DTSSP, according to the protocol described in Junttila et al.Citation5 Immunoprecipitation reactions were performed by the standard procedures.Citation41
Duolink proximity ligation assay
Duolink proximity ligation assays (PLA) were performed according to manufacturer’s instructions (Olink Bioscience). The detailed protocol for this assay is available at http://www.olink.com/products/duolink/downloads/duolink-manuals-and-guidelines. Briefly, cells were fixed and permeabilized, and then incubated overnight with two different primary antibodies raised in different species (mouse anti-ErbB2 and rabbit anti-CHK). The conditions with either no primary antibody or one primary antibody were used as controls. After washing, cells were incubated with secondary antibodies conjugated with the oligonucleotide probes (PLA probe plus and PLA probe minus) for 1 h at 37 °C followed by addition of ligase into the slides for another 30 min incubation at 37 °C. In the amplification step, nucleotides and fluorescently labeled oligonucleotides were added together with polymerase. After hybridization, signals from fluorescently-labeled oligonucleotides in the concatemeric products were visualized by fluorescent microscopy. Distinct single fluorescent spots indicate that there was an interaction between ErbB2 and CHK.
Cell growth assays
Cell growth assays were performed as previously described.Citation41 At the indicated times, cells were trypsinized, mixed with trypan blue and counted using a BioRad TC10 automated cell counter.
Analysis of clinical samples
Tissue sections were deparaffinized and rehydrated. Nonspecific binding was blocked after which sections were incubated with primary anti-phospho-ErbB2 (pY1248) antibody (1:50 dilution, Sigma). Slides were incubated with ready-to-use secondary antibody (Vectastain Elite ABC Universal Kit; Vector laboratories), then with the avidin-biotin peroxidase complex (1:100 Vectastain Elite ABC kit), after which visualization was conducted with chromogenic 3,3′-diaminobenzedine (Dako).
| Abbreviations: | ||
| ErbB | = | erythroblastosis protein B |
| HER-2 | = | human epidermal growth factor receptor 2 |
| CHK | = | Csk-homologous kinase |
| MATK | = | megakaryocyte-associated tyrosine kinase |
| EGF | = | epidermal growth factor |
| ERK | = | extracellular signal-regulated kinase |
| MAPK | = | mitogen activated protein kinase |
| PI3K | = | phosphatidylinositol 3-kinase |
| ADCC | = | antibody dependent cellular cytotoxicity |
| GFP | = | green fluorescent protein |
| DMEM | = | Dulbecco’s modified essential medium |
| RPMI | = | Roswell Park Memorial Institute |
| CMV | = | cytomegalovirus |
| ELISA | = | enzyme-linked immunosorbent assay |
| WCL | = | whole cell lysates |
| PLA | = | proximity ligation assay |
Additional material
Download Zip (188.8 KB)Disclosure of Potential Conflicts of Interest
The authors declare that they have no conflict of interests.
Acknowledgments
We thank Drs Avin Lalmansingh and Ying-Xin Fan for the critical review of the manuscript. US Food and Drug Administration Critical Path Funding for FY2011.
Disclaimer
The information presented in this article reflects the views of the authors and does not represent the policy of the US Food and Drug Administration.
References
- Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med 2007; 357:39 - 51; http://dx.doi.org/10.1056/NEJMra043186; PMID: 17611206
- Wu WJ, Dokmanovic M. Trastuzumab. In Encyclopedia of Cancer, M. Schwab, ed. Berlin, Heidelberg, Germany: SpringerReference; Springer-Verlag; 2009. http://dx.doi.org/10.1007/SpringerReference_177618.
- Molina MA, Codony-Servat J, Albanell J, Rojo F, Arribas J, Baselga J. Trastuzumab (herceptin), a humanized anti-Her2 receptor monoclonal antibody, inhibits basal and activated Her2 ectodomain cleavage in breast cancer cells. Cancer Res 2001; 61:4744 - 9; PMID: 11406546
- Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol 2007; 18:977 - 84; http://dx.doi.org/10.1093/annonc/mdl475; PMID: 17229773
- Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, Sampath D, Sliwkowski MX. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009; 15:429 - 40; http://dx.doi.org/10.1016/j.ccr.2009.03.020; PMID: 19411071
- Sarup JC, Johnson RM, King KL, Fendly BM, Lipari MT, Napier MA, Ullrich A, Shepard HM. Characterization of an anti-p185HER2 monoclonal antibody that stimulates receptor function and inhibits tumor cell growth. Growth Regul 1991; 1:72 - 82; PMID: 1688187
- Cuello M, Ettenberg SA, Clark AS, Keane MM, Posner RH, Nau MM, Dennis PA, Lipkowitz S. Down-regulation of the erbB-2 receptor by trastuzumab (herceptin) enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in breast and ovarian cancer cell lines that overexpress erbB-2. Cancer Res 2001; 61:4892 - 900; PMID: 11406568
- Valabrega G, Montemurro F, Sarotto I, Petrelli A, Rubini P, Tacchetti C, Aglietta M, Comoglio PM, Giordano S. TGFalpha expression impairs Trastuzumab-induced HER2 downregulation. Oncogene 2005; 24:3002 - 10; http://dx.doi.org/10.1038/sj.onc.1208478; PMID: 15735715
- Austin CD, De Mazière AM, Pisacane PI, van Dijk SM, Eigenbrot C, Sliwkowski MX, Klumperman J, Scheller RH. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol Biol Cell 2004; 15:5268 - 82; http://dx.doi.org/10.1091/mbc.E04-07-0591; PMID: 15385631
- Dokmanovic M, Hirsch DS, Shen Y, Wu WJ. Rac1 contributes to trastuzumab resistance of breast cancer cells: Rac1 as a potential therapeutic target for the treatment of trastuzumab-resistant breast cancer. Mol Cancer Ther 2009; 8:1557 - 69; http://dx.doi.org/10.1158/1535-7163.MCT-09-0140; PMID: 19509242
- Dokmanovic M, Shen Y, Bonacci TM, Hirsch DS, Wu WJ. Trastuzumab regulates IGFBP-2 and IGFBP-3 to mediate growth inhibition: implications for the development of predictive biomarkers for trastuzumab resistance. Mol Cancer Ther 2011; 10:917 - 28; http://dx.doi.org/10.1158/1535-7163.MCT-10-0980; PMID: 21487052
- Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, Klos KS, Li P, Monia BP, Nguyen NT, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004; 6:117 - 27; http://dx.doi.org/10.1016/j.ccr.2004.06.022; PMID: 15324695
- Esteva FJ, Guo H, Zhang S, Santa-Maria C, Stone S, Lanchbury JS, Sahin AA, Hortobagyi GN, Yu D. PTEN, PIK3CA, p-AKT, and p-p70S6K status: association with trastuzumab response and survival in patients with HER2-positive metastatic breast cancer. Am J Pathol 2010; 177:1647 - 56; http://dx.doi.org/10.2353/ajpath.2010.090885; PMID: 20813970
- Nahta R, Esteva FJ. HER2 therapy: molecular mechanisms of trastuzumab resistance. Breast Cancer Res 2006; 8:215; http://dx.doi.org/10.1186/bcr1612; PMID: 17096862
- Garrett TP, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Kofler M, Jorissen RN, Nice EC, Burgess AW, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell 2003; 11:495 - 505; http://dx.doi.org/10.1016/S1097-2765(03)00048-0; PMID: 12620236
- Scott GK, Dodson JM, Montgomery PA, Johnson RM, Sarup JC, Wong WL, Ullrich A, Shepard HM, Benz CC. p185HER2 signal transduction in breast cancer cells. J Biol Chem 1991; 266:14300 - 5; PMID: 1677643
- Diermeier S, Horváth G, Knuechel-Clarke R, Hofstaedter F, Szöllosi J, Brockhoff G. Epidermal growth factor receptor coexpression modulates susceptibility to Herceptin in HER2/neu overexpressing breast cancer cells via specific erbB-receptor interaction and activation. Exp Cell Res 2005; 304:604 - 19; http://dx.doi.org/10.1016/j.yexcr.2004.12.008; PMID: 15748904
- Ginestier C, Adélaïde J, Gonçalvès A, Repellini L, Sircoulomb F, Letessier A, Finetti P, Geneix J, Charafe-Jauffret E, Bertucci F, et al. ERBB2 phosphorylation and trastuzumab sensitivity of breast cancer cell lines. Oncogene 2007; 26:7163 - 9; http://dx.doi.org/10.1038/sj.onc.1210528; PMID: 17525746
- Gijsen M, King P, Perera T, Parker PJ, Harris AL, Larijani B, Kong A. HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer. PLoS Biol 2010; 8:e1000563; http://dx.doi.org/10.1371/journal.pbio.1000563; PMID: 21203579
- Hudelist G, Köstler WJ, Czerwenka K, Kubista E, Attems J, Müller R, Gschwantler-Kaulich D, Manavi M, Huber I, Hoschützky H, et al. Her-2/neu and EGFR tyrosine kinase activation predict the efficacy of trastuzumab-based therapy in patients with metastatic breast cancer. Int J Cancer 2006; 118:1126 - 34; http://dx.doi.org/10.1002/ijc.21492; PMID: 16161043
- Dankort D, Jeyabalan N, Jones N, Dumont DJ, Muller WJ. Multiple ErbB-2/Neu Phosphorylation Sites Mediate Transformation through Distinct Effector Proteins. J Biol Chem 2001; 276:38921 - 8; http://dx.doi.org/10.1074/jbc.M106239200; PMID: 11500516
- Dankort DL, Wang Z, Blackmore V, Moran MF, Muller WJ. Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol Cell Biol 1997; 17:5410 - 25; PMID: 9271418
- Akiyama T, Matsuda S, Namba Y, Saito T, Toyoshima K, Yamamoto T. The transforming potential of the c-erbB-2 protein is regulated by its autophosphorylation at the carboxyl-terminal domain. Mol Cell Biol 1991; 11:833 - 42; PMID: 1671296
- Kim S, Zagozdzon R, Meisler A, Baleja JD, Fu Y, Avraham S, Avraham H. Csk homologous kinase (CHK) and ErbB-2 interactions are directly coupled with CHK negative growth regulatory function in breast cancer. J Biol Chem 2002; 277:36465 - 70; http://dx.doi.org/10.1074/jbc.M206018200; PMID: 12122014
- Chong YP, Chan AS, Chan KC, Williamson NA, Lerner EC, Smithgall TE, Bjorge JD, Fujita DJ, Purcell AW, Scholz G, et al. C-terminal Src kinase-homologous kinase (CHK), a unique inhibitor inactivating multiple active conformations of Src family tyrosine kinases. J Biol Chem 2006; 281:32988 - 99; http://dx.doi.org/10.1074/jbc.M602951200; PMID: 16959780
- Zrihan-Licht S, Deng B, Yarden Y, McShan G, Keydar I, Avraham H. Csk homologous kinase, a novel signaling molecule, directly associates with the activated ErbB-2 receptor in breast cancer cells and inhibits their proliferation. J Biol Chem 1998; 273:4065 - 72; http://dx.doi.org/10.1074/jbc.273.7.4065; PMID: 9461599
- Bougeret C, Jiang S, Keydar I, Avraham H. Functional analysis of Csk and CHK kinases in breast cancer cells. J Biol Chem 2001; 276:33711 - 20; http://dx.doi.org/10.1074/jbc.M104209200; PMID: 11445575
- Boerner JL, Biscardi JS, Silva CM, Parsons SJ. Transactivating agonists of the EGF receptor require Tyr 845 phosphorylation for induction of DNA synthesis. Mol Carcinog 2005; 44:262 - 73; http://dx.doi.org/10.1002/mc.20138; PMID: 16167350
- Okabayashi Y, Kido Y, Okutani T, Sugimoto Y, Sakaguchi K, Kasuga M. Tyrosines 1148 and 1173 of activated human epidermal growth factor receptors are binding sites of Shc in intact cells. J Biol Chem 1994; 269:18674 - 8; PMID: 8034616
- Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res 2000; 60:3384 - 8; PMID: 10910043
- Mueller KL, Powell K, Madden JM, Eblen ST, Boerner JL. EGFR tyrosine 845 phosphorylation–dependent proliferation and transformation of breast cancer cells require activation of p38 MAPK. Transl Oncol 2012; 5:327 - 34; http://dx.doi.org/10.1593/tlo.12163; PMID: 23066441
- Radhakrishnan Y, Shen X, Maile LA, Xi G, Clemmons DR. IGF-I stimulates cooperative interaction between the IGF-I receptor and CSK homologous kinase that regulates SHPS-1 phosphorylation in vascular smooth muscle cells. Mol Endocrinol 2011; 25:1636 - 49; http://dx.doi.org/10.1210/me.2011-0035; PMID: 21799000
- Thymiakou E, Episkopou V. Detection of signaling effector-complexes downstream of bmp4 using PLA, a proximity ligation assay. J Vis Exp 2011; 49:e2631; PMID: 21403637
- Tanner M, Kapanen AI, Junttila T, Raheem O, Grenman S, Elo J, Elenius K, Isola J. Characterization of a novel cell line established from a patient with Herceptin-resistant breast cancer. Mol Cancer Ther 2004; 3:1585 - 92; PMID: 15634652
- Lai HW, Chien SY, Kuo SJ, Tseng LM, Lin HY, Chi CW, Chen DR. The potential utility of curcumin in the treatment of HER-2-overexpressed breast cancer: an in vitro and In vivo comparison study with herceptin. Evid Based Complement Alternat Med 2012; 2012:486568; http://dx.doi.org/10.1155/2012/486568; PMID: 21876713
- Graham J, Muhsin M, Kirkpatrick P. Cetuximab. Nat Rev Drug Discov 2004; 3:549 - 50; http://dx.doi.org/10.1038/nrd1445; PMID: 15272498
- Doody JF, Wang Y, Patel SN, Joynes C, Lee SP, Gerlak J, Rolser RL, Li Y, Steiner P, Bassi R, et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol Cancer Ther 2007; 6:2642 - 51; http://dx.doi.org/10.1158/1535-7163.MCT-06-0506; PMID: 17913857
- Kumar R, Shepard HM, Mendelsohn J. Regulation of phosphorylation of the c-erbB-2/HER2 gene product by a monoclonal antibody and serum growth factor(s) in human mammary carcinoma cells. Mol Cell Biol 1991; 11:979 - 86; PMID: 1671297
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF, Burchmore M, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol 2002; 20:719 - 26; http://dx.doi.org/10.1200/JCO.20.3.719; PMID: 11821453
- Dokmanovic M, Wu WJ. Trastuzumab-resistance and breast cancer. In Breast Cancer-carcinogenesis, cell growth and signalling pathways. Prof.M. Gunduz, ed. Rijeka, Croatia: In Tech; 2011. 171-204p.
- Hirsch DS, Shen Y, Wu WJ. Growth and motility inhibition of breast cancer cells by epidermal growth factor receptor degradation is correlated with inactivation of Cdc42. Cancer Res 2006; 66:3523 - 30; http://dx.doi.org/10.1158/0008-5472.CAN-05-1547; PMID: 16585176
- Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, et al, American Society of Clinical Oncology/College of American Pathologists. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med 2007; 131:18 - 43; PMID: 19548375