
Abstract
We have previously reported that infection with the non-pathogenic, tumor suppressive, wild-type adeno-associated virus type 2 (AAV2) inhibited proliferation of breast cancer-derived lines representing both weakly invasive (MCF-7 and MDA-MB-468), as well as aggressive (MDA-MB-231) cancer types. AAV2-induced death occurred via targeting pathways of apoptosis and necrosis. In contrast, normal human mammary epithelial cells were unaffected upon AAV2 infection. The current study characterizes AAV2 infection and subsequent death of the highly aggressive, triple-negative (ER−/PR−/HER2−) MDA-MB-435 cell line derived from metastatic human breast carcinoma. Monolayer MDA-MB-435 cultures infected with AAV2 underwent complete apoptotic cell death characterized by activation of caspases -7, -8, and -9 and PARP cleavage. Death was further correlated with active AAV2 genome replication and differential expression of viral non-structural proteins Rep78 and Rep52. Cell death coincided with increased entry into S and G2 phases, upregulated expression of the proliferation markers Ki-67 and the monomeric form of c-Myc. Expression of the p16INK4, p27KIP1, p21WAF1, and p53 tumor suppressors was downregulated, indicating marked S phase progression, but sharply contrasted with hypo-phosphorylated pRb. In parallel, MDA-MB-435 breast tumor xenografts which received intratumoral injections of AAV2 were growth retarded, displayed extensive areas of necrosis, and stained positively for c-Myc as well as cleaved caspase-8. Therefore, AAV2 induced death of MDA-MB-435 xenografts was modulated through activation of caspase-regulated death pathways in relation to signals for cell cycle controls. Our findings provide foundational studies for development of novel AAV2 based therapeutics for treating aggressive, triple-negative breast cancer types.
Introduction
Breast cancer is the most common female cancer and the leading cause of female cancer-related deaths.Citation1 Breast cancer also constitutes greater than 25% of cancers in women.Citation2 Despite the development of targeted therapeutics, treatment of breast cancer remains complex due to the involvement of multiple signaling pathways which promote tumor growth, development of resistance to ongoing therapy, and associated cytotoxicity.Citation3-Citation10 Selection of appropriate treatment is patient-specific, guided by the molecular heterogeneity of different disease subtypes characterized by expression of estrogen (ER), progesterone (PR), and Her-2 receptors, and identification of tumor grade.Citation11,Citation12 Differential expression of these hormone and growth factor receptors are utilized as therapeutic targets in different breast cancer subtypes and provide the foundation for individualized treatments.Citation13,Citation14 On the other hand, triple-negative breast cancer (TNBC) is a clinical term for a subgroup of breast cancers characterized by the lack of expression of ER, PR, and Her-2, its aggressiveness and metastatic potential, as well as poor prognosis.Citation11 Unlike patients with hormone and growth factor receptor-positive breast cancer subtypes that respond to targeted therapeutics, patients with TNBC do not benefit from similar therapies. For these patients, chemotherapy is the only option, and is associated with a high chance of relapse and lower survival rates compared with patients exhibiting receptor positive breast cancer subtypes.Citation15 As a result, there is an urgent and ongoing need for the development of novel therapies that efficiently target TNBC subtypes, as well as development of therapeutics which bypass the requirement for personalized patient treatments.
We have reported that infection with the non-pathogenic, tumor suppressive human adeno-associated virus type 2 (AAV2) induced apoptosis in a number of different breast cancer cell lines which cumulatively represent multiple grades of aggressiveness.Citation16 We showed that AAV2 induced caspase-independent apoptosis in MCF-7 (ER+) cells, caspase-dependent apoptosis in MDA-MB-468 (ER−) cells and caspase-dependent apoptosis, mixed with necrosis, in MDA-MB-231 (ER−) cells.Citation16 A 100% decrease in viability of the AAV2-infected breast cancer cell lines was correlated with the ability of the virus to undergo active genome replication and differentially express its non-structural proteins: Rep78, Rep68, and Rep40, but not Rep52. Although our studies failed to identify common AAV2-targeted cell death pathways among the different breast cancer cell lines, our studies did show that cellular demise coincided with increased expression of c-Myc and Ki-67 protein levels, irrespective of the breast cancer cell line studied.Citation16 The upregulated expression of c-Myc was a significant observation since this protein has been shown to have the dual ability to induce cell cycle progression/proliferation as well as apoptosis, in the context of other cellular signals.Citation17,Citation18 In contrast, expression of Ki-67 is a typical marker of proliferation indicative of active S phase cell cycle progression.Citation19,Citation20 We hypothesized that the inherent endonuclease activity of AAV2 Rep78 and Rep68 proteins induces a DNA damage response and activates death signaling pathways, as well as simultaneously mediate conflicting activation of cell proliferation pathways, which culminates in cancer cell death.Citation16 In contrast, normal human mammary epithelial cells infected with AAV2 failed to express Rep proteins or undergo apoptosis.Citation16 Our studies suggest that the mechanistic ability of AAV2 to distinguish between normal and cancer cells to mediate the cell killing could be clinically advantageous.
Our published studies demonstrated the common ability of AAV2 to induce death of a number of breast cancer,Citation16 as well as multiple human papillomavirus (HPV) positive cervical cancer-derived cell lines,Citation21 which not only represented gradations in aggressiveness,Citation16,Citation21 but also represented varying sensitivities related to hormone and growth factor signaling.Citation16 In the current study, we investigated the ability of AAV2 to infect and induce apoptosis in the human MDA-MB-435 breast cancer cell line which was derived from metastatic (pleural effusion), infiltrating ductal breast carcinoma.Citation22 This cell line is categorized as being representative of the triple-negative (ER−/PR−/HER2−) breast disease type.Citation23 Additionally, the MDA-MB-435 cell line has also been shown to form tumors when injected into the mammary fat pads of immuno-compromised mice.Citation22 In the current study, AAV2 infection induced 100% cell death of the MDA-MB-435 cell line and was achieved via activation of caspases which regulate both the intrinsic as well as the extrinsic pathways of apoptosis. Cell death coincided with active AAV2 genome replication and differential expression of AAV2 non-structural Rep proteins: Rep78 and Rep52, but not Rep68 and Rep40. AAV2 induced cell death was characterized by an increase in the number of cells with S phase and G2 phase cell cycle DNA content. Decreased cell viability of AAV2-infected cells was characterized by upregulated expression of Ki-67 and increased expression of the monomeric form of c-Myc. Our corresponding in vivo studies demonstrated that intratumoral injections of AAV2 into tumor xenografts in nude mice bearing MDA-MB-435 cells delayed tumor growth and induced extensive areas of tumor necrosis. These results portray the ability of AAV2 to regulate targeting of distinct cell growth/proliferation, as well as a mixture of cell death pathways in this triple-negative breast cancer derived line. Cumulatively, our current study and previously published dataCitation16 suggest that AAV2 mediated cell killing of multiple breast cancer cell lines representing both low and high grades of cancer, and that virus targeting of cancer cells is independent of hormone and growth factor signaling status. Thus, AAV2 has the potential for derivation of novel therapeutics for TNBC types as well as in other breast cancer subtypes.
Results
AAV2 infection induces complete cell death in the MDA-MB-435 metastatic breast cancer-derived cell line
In the current study, we tested the ability of AAV2 to infect and induce death of the MDA-MB-435 cell line. This cell line is characterized as a highly aggressive, triple-negative cell line, derived from metastatic (pleural effusion), infiltrating ductal breast carcinoma, and was shown to form tumors when injected into the mammary fat pads of immunocompromised mice.Citation22 The MDA-MB-435 cells were synchronized as described in Materials and Methods, by which approximately two-thirds or more of the cells were in the G1 phase of the cell cycle.Citation21,Citation24 Under the growth conditions utilized for our studies, this is the maximum level of synchronization that we are normally able to achieve with or without the addition of exogenous chemicals. Synchronized MDA-MB-435 cultures were infected with AAV2 using an MOI of 0.02. We have previously used an MOI of 0.02 and shown it to be the lowest dose of AAV2 which was effective in killing multiple breast cancer lines as well as human papillomavirus positive cervical cancer lines.Citation16,Citation21 Both AAV2-infected and mock-infected cells were grown to 80% confluence (day 2), at which time the cells were passaged at a ratio of 1:2, followed by a period of further growth and passaging again at a 1:2 ratio on day 5, day 7, day 11, day 13, day 17, and day 20. On day 17, the AAV2-infected cells showed growth retardation, and eventually culminated in 100% cell death as portrayed by the steady-state decrease in cell viability over days 17–21, determined utilizing the trypan blue exclusion method ().
Figure 1. AAV2 infection of MD-MB-435 cells induces steady decrease in cell viability which results from virus induced cell death. (A) Viability of MDA-MB-435 breast cancer cells infected with AAV2. Synchronized MDA-MB-435 cells were infected with AAV2. Both mock and virus-infected cells were grown to 80% confluence (day 2) and passaged 1:2, followed by further growth and passaging again at a 1:2 ratio on days 5, 7, 11, 13, 17, and 20 as indicated by arrows. At each time point, cells were counted using the trypan blue exclusion method. Graph generated is representative of 3 independent experiments. (B) Apoptotic death of AAV2-infected MDA-MB-435 cells was determined using FACS analysis. On the days indicated, both mock and AAV2-infected cells were stained with propidium iodide and the histogram of the cell cycle profile was used to determine the appearance of the sub-G0-G1 fraction that progressively increased with days in culture. FACS analysis is representative of 3 independent experiments.
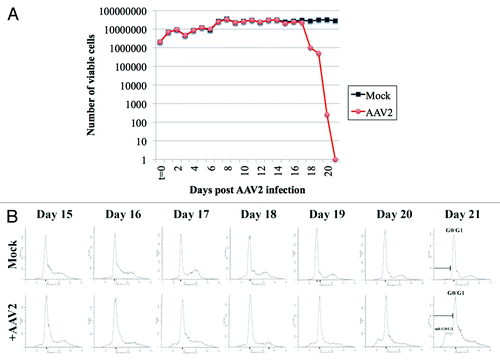
The kinetics of AAV2 mediated death of MDA-MB-435 cells over the 21 d period contrasts sharply with those of MCF-7, MDA-MB-468, and MDA-MB-231 cells, all cell lines which routinely underwent apoptosis upon infection with AAV2 over a 7 d period.Citation16 The disparate timelines of AAV2 regulated cell killing could be a reflection of the aggressive nature of the MDA-MB-435 cell line representing its characteristic metastatic/pro-survival nature and resistance to cell killing. It is also possible that the MDA-MB-435 cells are less permissive in expressing AAV2 death inducing proteins and/or transcripts compared with other cell lines. Using AAV2 MOIs of 10, 20, 30, and 100 to infect the MDA-MB-435 line yielded similar results, therefore, the 21 d time period of cell death was not limited to virus dosage. In the current study, it was 14 d before we observed the initial drop in viability of the AAV2-infected MDA-MB-435 cells, after which cell death increased precipitously till day 21. The abrupt decrease in cell viability could be due to accumulating AAV2 non-structural Rep proteins (discussed below) to a critical threshold needed to activate the cell death machinery. The observed time line of AAV2-induced death in MDA-MB-435 cells is not surprising since we have observed similar kinetics of demise using mesothelioma-derived cell lines (unpublished observations).
It is of note that complete AAV2-mediated cell killing of MCF-7, MDA-MB-468, MDA-MB-231,Citation16 and MDA-MB-435 (current study) was accomplished using an initial MOI of 0.02, which meant that at best, 1 out of 50 cells were infected. Therefore, the initial mechanism of cell killing in all these different breast cancer cell lines is potentially mediated by a “bystander” effect, in which dying cells release cytotoxic metabolites, cellular and/or AAV2 encoded proteins, which are able to affect the neighboring non-infected cells via movement through cellular gap junctions to mediate cell killing, a phenomenon which has been previously described elsewhere.Citation25,Citation26 Subsequent mass cytotoxicity of AAV2-infected cells is potentially due to a downstream amplification step, since we also observed that the AAV2-mediated cell killing was positively associated with viral genome amplification (discussed below). Therefore, low levels of AAV2 infectious particles potentially synthesized in the infected breast cancer cell lines and released into the medium could infect neighboring cells, thereby amplifying the bystander effect. We have not further pursued this possibility. In future studies, the idea of AAV2-regulated bystander effect, whether virus-mediated or otherwise, and potential cell killing could be examined using the supernatant from virus-infected cultures to test its efficacy in killing cancer cells.
We further wanted to demonstrate that the observed decline in viability of the AAV2-infected MDA-MB-435 cells was due to apoptotic cell death. The MDA-MB-435 cells were synchronized, infected with AAV2, and passaged over the 21 d period, as described above. Each day, cells were prepared for flow cytometric analysis of DNA content as described in Materials and Methods. We then analyzed the histogram of the cell cycle profile and determined the appearance of sub-G0/G1 fraction, which is indicative of apoptotic cell death. Since AAV2 induced decrease in cell viability occurred after passaging on day 14 (), we analyzed the histogram of AAV2-infected MDA-MB-435 cells and compared them with control cells at each time point after day 14. We observed that the sub-G0/G1 fraction of AAV2-infected cells progressively increased during the day 15 to day 21 time period (), which is indicative of apoptotic cell death, a property consistent with the tumor suppressive activities of AAV2 our laboratory and work of others have previously reported. Apoptotic death was also correlated with increased entry into S phase of the cell cycle (presented in Fig. S2). Histogram analysis of AAV2-infected cells at earlier time points (day 1–day 14) were similar to non-infected cells (Fig. S1).
AAV2 infection of breast cancer cells results in Rep78 and Rep52 protein expression and genome replication which culminates in cell death
AAV2 encodes four non-structural proteins: Rep78 and Rep68, which regulate multiple viral functions including DNA replication and transcription; and Rep52 and Rep40, which are involved in packaging of viral genomes into capsids.Citation27 Only the Rep78 and Rep52 proteins were found to be clearly expressed in the AAV2-infected MDA-MB-435 cell line, beginning on day 15 and levels of which steadily increased up to day 21 (). On the other hand, expression of the Rep68 and Rep40 proteins could not be clearly determined in the AAV2-infected cells (). As expected, Rep78 and Rep52 protein expression on day 15 () coincided with the clear decrease in cell viability of the AAV2-infected cells compared with mock infected cells (). In contrast to Rep protein expression, AAV2 genome replication was detected in low levels as early as day 6 in the AAV2-infected cells, but was clearly detected on day 13 onwards (). These results suggest that low-level Rep protein expression presumably occurred as early as day 6 but its expression was at the limits of detection using these techniques. In contrast, on day 13, AAV2 genome replication was significantly higher and correlated with increased G2 phase cell cycle entry (Fig. S2C), an indication of active cell division. High levels of the 4.7 kb monomer AAV2 genome was detected from day 18 to day 21 (). The 4.7 kb monomer is indicative of active genome replication. This is also the time period of active cell death accompanied by non-discriminate DNA degradation, both cellular and viral, and is reflected in the quality of the bands in the Southern blots (). For presenting the bands with clarity, we have included a similar Southern blot obtained using the MCF-7 breast cancer cell line infected with AAV2 and protocols described herein (Fig. S4A).
Figure 2. AAV2 induced apoptosis/cell death of MDA-MB-435 cells correlates with (A) Rep protein expression. For detecting Rep proteins in immunoblots, total protein extracts were prepared and 60 μg of total protein extracts from AAV2-infected and mock MDA-MB-435 breast cancer cells were resolved in a 7.5% SDS-PAGE gel and detected with an AAV2 Rep specific antibody. Results are representative of three individual experiments. t, time; +, AAV2-infected; −, mock. Actin is used as a loading control. (B) AAV2 genome replicates in virus infected MDA-MB-435 cells. Southern blot analysis to detect 4.7 kb AAV2 replicative form monomer representing active genome replication in the virus infected breast cancer cell line. A total of 5 μg of total DNA was then detected with AAV2 genomic DNA as probe. One hundred nanograms of total DNA isolated from cells was used as loading control. Results shown are representative of 3 individual experiments. t, time; +, AAV2-infected; −, mock. *Low levels of AAV2 genome replication depicted by the 4.7 kb band on days 6 and 13.
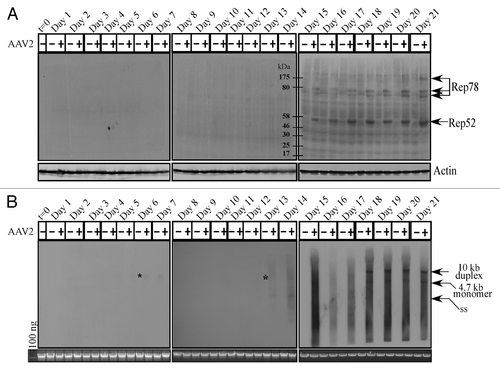
Thus, these experiments show that Rep protein expression, which is necessary for active genome replication, coincides with decreased viability and eventual cell death. This result is similar to our previously published studies which showed that AAV2 induced death of MCF-7, MDA-MB-468, and MDA-MB-231 cell lines coincided with expression of Rep proteins.Citation16 These results suggest the possibility that AAV2 regulated kinetics of death in infected breast cancer cells is in part due to the differential ability of those cells to allow expression of specific Rep proteins. Most likely, growth characteristics which determine aggressiveness potentially determine permissiveness for expressing AAV2 Rep proteins. For comparison, we have included a western blot obtained using the HPV31b positive cervical cancer cell line infected with AAV2, showing expression of all four Rep proteins (Fig. S4B).
We also consistently detected the dimer form of Rep78 in the AAV2-infected MDA-MB-435 cell line (), which is not an uncommon observation since others have also reported finding the high molecular weight Rep78 complexes, which are essentially Rep–Rep protein aggregates containing 2 to 6 Rep proteins which persist in sodium dodecyl sulfate (SDS).Citation28,Citation29 We have also detected such high molecular weight Rep78 complexes in AAV2 co-infected HPV-positive cervical cancer linesCitation21 as well as multiple breast cancer cell lines infected with AAV2 undergoing apoptosis.Citation16
We have previously demonstrated that differential expression of specific Rep proteins determine cell killing in different breast cancer cell lines.Citation16 However, the optimal Rep protein, alone or in combination has been difficult to determine, due to varying sensitivities of the cell lines for transfection. We have reported that only the MCF-7 breast cancer cells transfected with the infectious AAV2 vector undergoes apoptosis,Citation16 but not when transfected alone with Rep78 (Alam and Meyers, unpublished observations), suggesting the need for more than one Rep protein to be expressed for efficient AAV2 regulated cell death.
AAV2 Rep protein expression results in activation of both extrinsic and intrinsic pathways of apoptosis induction
It was of interest to determine whether AAV2-induced death of MDA-MB-435 cells could be correlated with activation of specific caspase-dependent pathways. Apoptotic cell death occurs via activation of two signaling pathways: first, the extrinsic or receptor-mediated pathway, and second, the intrinsic or mitochondrial pathway.Citation30 Two groups of caspases characterize these pathways: the initiator (caspase-8, -9, and -10) and executioner (caspase-3, -6, and -7) caspases. Initiator caspases cleave and activate executioner caspases, which in turn process cellular substrates that orchestrate the biochemical execution of cell death.Citation31
To investigate which of the executioner caspases were cleaved upon AAV2 induced cell death in the MDA-MB-435 cells, we examined expression and activation of the executioner caspases -3, -6, and -7 in total protein extracts of mock and AAV2-infected cells (). On day 21, caspase-7 was cleaved to its 30 kDa form in the AAV2-infected cells (, lane 43). In contrast, expression of the full-length caspase-6 protein levels did not display any significant changes and cleavage of caspase-6 was not observed under these conditions (). Caspase-6 expression was more abundant overall during day 15–day 21, compared with earlier time-points, and appears to correlate with extended time of cells in culture. The MDA-MB-435 cells maintained high levels of both full-length caspase-3 and its 17 kDa cleaved form in controls cells (). Although expression of full-length caspase-3 did not change upon AAV2 infection, during day 15–day 21, the steady-state levels of the 17 kDa cleaved form declined in the AAV2-infected cells compared with mock infected cells (), which is potentially attributed to degradation by non-caspase proteases as has been previously suggested.Citation32,Citation33 In addition, we have previously reported that AAV2 infection of MDA-MB-231 cells resulted in simultaneous decline of both the full-length and cleaved forms of multiple executioner caspasesCitation16 which potentially indicated activation of cell death pathways also by non-caspase proteases.Citation32 On day 15–day 21, AAV2-infected MDA-MB-435 cells additionally displayed the 89 kDa cleaved form of PARP (), a DNA repair enzyme which is generally inactivated by cleavage from one of the excecutioner caspases.Citation34 It is noteworthy that the 89 kDa cleaved form of PARP appeared in AAV2-infected cells as early as day 16 (, lane 33), whereas caspase-7 cleavage was not detected in AAV2-infected cells until day 21 (, lane 43). These observations suggest the possibility that AAV2 targeting of non-caspase proteases regulate PARP cleavage during early time points (day 15–day 20) (), but at later times (day 21) act in concert with activated caspase-7 (, lane 43). Our data showed that both caspase 8 and caspase 9 are activated well before day 21, indicating that downstream activation of mitochondrial functions, such as disruption of the mitochondrial membrane potential, membrane permeabilization, and cytochrome c release, are likely initiated earlier than day 21. Since our in vivo results suggest activation of necrosis as a pathway of cell death (discussed below), detecting activation of an executioner caspase, in this case caspase 7, is likely to be difficult earlier than day 21. However, identification of a specific executioner caspase may not be significant. Our results potentially suggest PARP-1 cleavage and cell death, earlier than day 21, was potentially caused by caspase independent pathways. Active AAV2 protein synthesis and active genome replication could increase intracellular ROS levels by placing a greater energy demand on a cancer cell which is already under a certain level of oxidative stress. Caspase-independent pathways, such as increased intracellular ROS, and its induction of double-strand breaks in genomic DNA, are also known to regulate PARP-1 activation, and apoptotic as well as necrotic forms of cell death.Citation35-Citation39 Additionally, increased levels of intracellular ROS are necessary for dissipation of the mitochondrial membrane potential, and subsequent PARP-1-dependent AIF translocation from the mitochondria to the nucleus, where AIF functions to mediate nuclear condensation, chromatinolysis, and cell death.Citation40 A similar mechanism may be implemented by AAV2 to induce death of the MDA-MB-435 cells in the current study.
Figure 3. AAV2 induction of apoptosis/cell death in the MDA-MB-435 cells results in activation of caspases of both the intrinsic and extrinsic pathways, ultimately resulting in PARP cleavage. Monolayer cell cultures were synchronized in G1, followed by infection with AAV2. Cell pellets were collected each day over a 21 d period as described in Materials and Methods. Detection of caspases and their cleavage/activation was performed by western blotting. Total protein extracts were prepared as described. Sixty micrograms of total protein extracts from AAV2-infected and mock infected cells were resolved in SDS-polyacrylamide (SDS-PAGE) gel electrophoresis. To detect the 35 kDa pro-caspase form of caspase-3, proteins were resolved in a 10% SDS-PAGE gel and detected with caspase-3 rabbit monoclonal antibody (Cell Signaling Technology). To detect the 17 kDa cleaved caspase-3 form, proteins were resolved in a 15% SDS-PAGE gel and detected with a rabbit polyclonal antibody against cleaved caspase-3 (Cell Signaling Technology). To detect the 35 kDa pro-caspase form of caspase-6, proteins were resolved in a 10% SDS-PAGE gel and to detect the 15 kDa cleaved form of caspase-6, proteins were resolved in a 15% SDS-PGE gel and detected with a rabbit polyclonal antibody (Cell Signaling Technology). To detect both the pro- and cleaved- forms of caspase-7, caspase-8, and caspase-9, proteins were resolved in a 10% SDS-PAGE gel. The 35 kDa pro-caspase form and the 30 kDa/20 kDa cleaved form of caspase-7 was detected with a mouse monoclonal antibody (Cell Signaling). The pro-caspase and cleaved 28 kDa form of caspase-8 was detected with a mouse monoclonal antibody (Alexis Biochemicals). The 47 kDa pro-caspase and 37 kDa/35 kDa cleaved forms of caspase-9 were detected with a rabbit polyclonal antibody (Cell Signaling). To detect the pro- (116 kDa) form of PARP, proteins were resolved in a 7.5% SDS-PAGE gel and detected with a rabbit monoclonal antibody (Cell Signaling). t, time; +, AAV2-infected; −, mock. Actin was used as a loading control. Results shown are representative of three individual experiments. t, time; +, AAV2-infected; −, mock. Bottom panel: caspase-7 cleavage on day 21, enlarged for clarity.
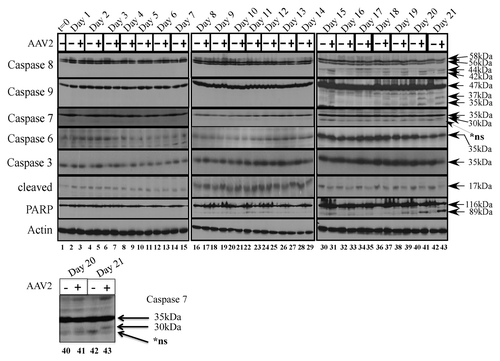
In contrast to the executioner caspases, during the day 15–day 21 time period, decreased viability of AAV2-infected MDA-MB-435 cells was correlated with cleavage of both the initiator caspase-8 to its 44 kDa and 42 kDa, and caspase-9 to its 37 kDa and 35 kDa proteolytic species (). The AAV2-regulated cleavage of caspase-9 implicated disruption of mitochondrial functions and release of cytochrome c, which is characteristic of the intrinsic pathway of apoptosis.Citation30 On the other hand, simultaneous cleavage of caspase-8 in the AAV2-infected cells also indicated the activation of the extrinsic pathways of apoptosis.Citation30 Cumulatively, our results suggest AAV2 mediated co-activation of the extrinsic and the intrinsic pathways of apoptotic cell death in the MDA-MB-435 cells. In addition, our studies cannot rule out simultaneous triggering of non-caspase proteases that act in concert and/or amplify induction of these cell death pathways.
AAV2-mediated manipulation of cell cycle check-points is related to changes in c-Myc, Ki-67, and PCNA protein levels
Our results also show that AAV2 induction of apoptosis in the MDA-MB-435 cells coincided with increased breach of the G1/S check-point and movement of cells into S phase (Figs. S2 and S3), which is indicative of implementation of cellular proliferation signals.Citation41 Likewise, AAV2-mediated increase in G2 phase progression signals readiness of the cells to enter mitosis.Citation42 AAV2 targeted interference with mechanisms that control cell cycle check-points in cells undergoing DNA damage could be a trigger for apoptosis induction. Therefore, we reasoned that one or more signaling cascades upstream of cell cycle controls could mechanistically couple increased S and G2 phase progression, proliferation, and apoptosis induction.
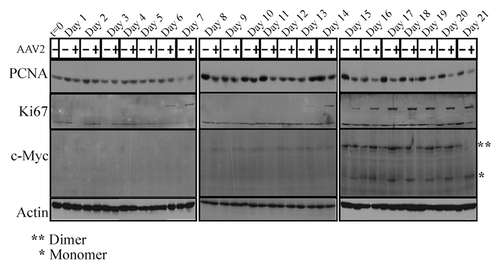
In normal cells, entry of cells into the G1 phase and execution of the G1/S cell cycle check-point is dependent on multiple signaling mechanisms, a key regulator of which is c-Myc.Citation41 On the other hand, proliferating cell nuclear antigen (PCNA) and Ki-67 are markers of S phase proliferation.Citation19,Citation20 We performed western blot analysis to detect expression of PCNA, Ki-67, and c-Myc in AAV2-infected MDA-MB-435 cell line. PCNA expression was found to be decreased in apoptotic AAV2-infected cells compared with mock infected cells (, day 15–day 21). On the other hand, Ki-67 expression was increased in the AAV2-infected cells (, day 6–day 7; day 13–day 14; day 15–day 21), which is indicative of signals for proliferation but which also coincide with decreased cell viability (). Expression of the monomeric form of c-Myc was greatly upregulated in the AAV2-infected breast cancer cells starting on day 15 whereas the dimeric form of c-Myc was detected equally in both the control and AAV2-infected cells (). In the current study, selectively increased expression of the monomeric form of c-Myc in AAV2-infected cells and its relation to proliferation and cell death signals is an interesting observation. We have previously reported that AAV2 infection upregulated expression of both the dimer- and monomer forms of c-Myc in multiple breast cancer lines tested,Citation16 which suggested that AAV2-regulated expression and/or stabilization of c-Myc could serve to amplify proliferation signals which allow the breast cancer cells to bypass cell cycle checkpoint controls in the presence of damaged cellular DNA.Citation16 On the other hand, c-Myc is also a protein which limits growth by sensitizing cells to apoptosis,Citation43 and which has the added potential to regulate DNA damage response upon imposition of genotoxic stressCitation44 mediated via the inherent endonuclease activity of AAV2 Rep78 and Rep68 proteins.Citation45 We have previously proposed that in the breast cancer cells, AAV2 regulation of c-Myc activation could potentially mediate simultaneous growth stimulatory (G1/S, S, and G2 phase targeting), and growth inhibitory (apoptosis associated DNA damage) signals which could represent a central unifying mechanism of AAV2-induced cell death in multiple breast cancer types.Citation16 In the current study, since the kinetics of cell death of the AAV2-infected MDA-MB-435 cells was much slower than the time frame we reported for other breast cancer cell linesCitation16 (21 d compared with 7 d), it is possible that increased appearance of the c-Myc monomers result from AAV2-mediated de novo synthesis. Such an increase would essentially add to pre-existing pools of c-Myc in order to achieve the threshold required for apoptotic induction. In contrast to the monomer form of c-Myc, the dimer form was more abundant overall during day 15–day 21, compared with earlier time-points, and appears to correlate with extended time of cells in culture.
Figure 4. AAV2 infection of MDA-MB-435 cells and apoptosis induction results in altered expression of proliferation markers PCNA, Ki-67, and c-Myc. Cells were synchronized by trypsinization of 80% confluent cultures and plating at a density of 1 × 106 cells in E medium. The cells were incubated for 10 to 12 h at which point at least two-thirds of the cells are maximally synchronized in the G1 phase. This time point was designated time zero (t = 0). Cells were then infected with AAV2. Cells were further cultured and passaged as described in Materials and Methods and pellets were collected each day over the 21 d period. Detection of cell cycle proliferation proteins were performed by western blotting. Total protein extracts were prepared and 60 μg of total protein extracts from AAV2-infected and mock cells were resolved in 7.5% SDS-PAGE gels. The PCNA protein was detected using a rabbit polyclonal antibody (Santa Cruz). The Ki67 protein was detected using a rabbit polyclonal antibody (Santa Cruz). The c-Myc protein was detected using a rabbit polyclonal antibody (Santa Cruz). Results shown are representative of three individual experiments. t, time; +, AAV2-infected; −, mock.
AAV2 infection of MDA-MB-435 breast tumor xenografts inhibits tumor growth
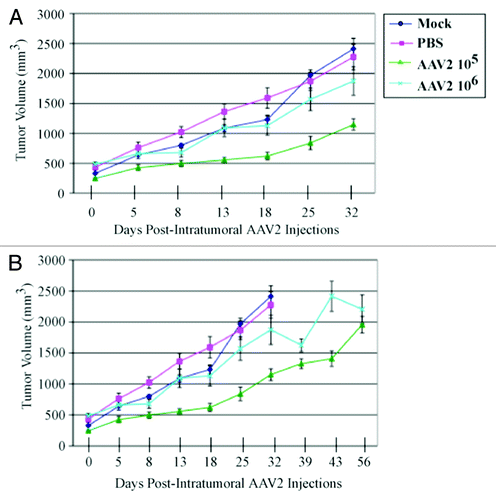
Our in vitro data indicate that infecting MDA-MB-435 monolayer cultures with AAV2 resulted in apoptosis induction via activation of caspase-8- and caspase-9-dependent pathways, demonstrating that in this breast cancer line AAV2 targeted cell death via regulating mitochondrial dysfunction () and targeting of multiple cell cycle phases and associated check-points (Figs. S2 and S3). These studies suggest the potential use of AAV2 to target growth/proliferation pathways in tumor tissues of breast cancer patients. It is well known that potential agents for use as chemotherapeutics which are active in vitro could be restricted in their activity in vivo, due to low bioavailability, delivery failure, or a fast rate of removal.Citation46 To address this issue, it was then of interest to investigate the efficacy of AAV2-mediated tumor suppression in vivo using the human breast cancer model established in female nude mice. Athymic mice were xenografted with the MDA-MB-435 cell line as described in Materials and Methods. All mice developed palpable tumors within two weeks after implantation, and reached an average volume of 300 mm3. We then determined whether infecting the human breast cancer xenografts with the wild-type AAV2 could regulate growth of those tumors. We tested the dose dependence of AAV2 infection on tumor growth by administering intratumoral injections of 105 and 106 infectious AAV2 particles, whereas the control tumors were injected with phosphate buffered saline (PBS) or left untreated (Mock). We observed significant reduction and/or growth delay of tumors infected with AAV2 compared with both mock and PBS-treated tumors (). In contrast, both mock and PBS-treated tumors grew to high tumor volumes (). On day 32, all the mice in the mock and PBS control groups were euthanized due to observed morbidity resulting from the high tumor burden, which was further associated with the failure of the mice to move or feed normally. In contrast, at the day 32 time point, all the mice in both 105 and 106 AAV2-treated mice showed no ill effects and morbidity. The AAV2-treated mice were further maintained and observed for changes in tumor volumes. Over the time course of the experiment, the large fluctuations in the tumor volumes were observed, which was especially evident in mice infected with 106 AAV2 particles, which was attributed to growth characteristics related to tumor heterogeneity (). On day 56, the tumor volumes of both AAV2 treatment groups were comparably similar (). It is notable that the two AAV2 dosages did not show a linear relationship with the observed reduction in tumor volumes, since tumors receiving 106 virus particles were larger compared with tumors receiving the lower dosage. This could potentially be due to virus clumping prior to injecting the tumors, thereby impairing efficient delivery. Another possibility is that viral receptors within in the tumors could be a limiting factor for AAV2 binding prior to infection. The experiment was terminated on day 56 and all the mice were euthanized. A general post-mortem examination of all animals revealed no obvious organ-specific toxicity in any of the AAV2-treated animals.
Figure 5. AAV2 infection reduced MDA-MB-435 tumor growth and increased survival in nude mice. (A) MDA-MB-435 cells (5 × 106) were implanted subcutaneously in the shoulder of mice. Tumor size was measured every other day. When xenografts reached volumes of ~300 mm3 (approximately 2 wk), the mice were randomly assigned to control and AAV2 treated groups (n = 5). Two sets of 5 mice each received a single AAV2 dosage of 105 and 106 infectious units per tumor administered via intratumoral injections. The respective dosage of AAV2 virus was diluted in 200 μL PBS and used for injecting multiple sites of the tumor. Control tumors either received intratumoral injections of 200 μL PBS or not manipulated (Mock). When the control tumors reached ~2400 mm3 (on day 32), the mock and PBS-treated mice were sacrificed due to their grossly restricted ability to move and to feed arising from the tumor burden. (B) The mice receiving intratumoral injections of AAV2 at both 105 and 106 dosages survived longer than the control mice and did not display similar restrictions in movements and feeding habits as the controls, and were eventually euthanized on day 56. Statistical analyses are expressed as means (SD).
AAV2-mediated inhibition of tumor growth in nude mice results from necrotic cell death within tumors
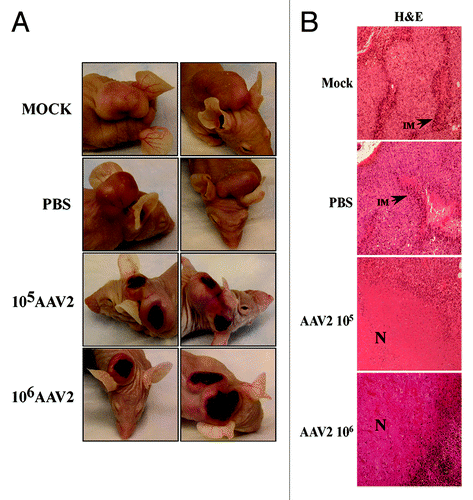
A visual difference observed between the control and the AAV2-infected tumors was the appearance of dry, dark, necrotic patches, on the skin over the tumors of mice receiving intratumoral AAV2 injections (, left panel). Since the appearance of necrotic areas was specific to tumors infected with AAV2, it was then of interest to determine morphological features of the virus infected as well as the control tumors. We performed hematoxylin and eosin (H&E) staining of the tumor sections and observed that the mock and PBS-treated tumors were packed with nucleated cells and presented areas of strong infiltrate of inflammatory macrophages (, right panel). This observed pattern of infiltrating macrophages is phenotypically similar to studies reported elsewhere.Citation47 It has been previously reported that breast tumors containing increased numbers of macrophages are significantly more vascularized and metastatic than tumors with low number of macrophage infiltrates.Citation47,Citation48 In contrast, AAV2-infected tumors showed noticeable decrease in the number of tumor-surrounding inflammatory cells (, right panel). In contrast to the mock and PBS-treated tumors, AAV2-infected tumors from both 105 and 106 treatment groups, presented large areas of central necrosis (, right panel). The effective tumor cell death appears to be evident in both treatment groups as determined from the appearance of the necrotic areas within the tumors (, right panel). These results are significant, since tumor necrosis in response to therapy is also used as the measure of an effective chemotherapeutic,Citation49 reduced viable tissue mass within the tumor and consequently a reduced tumor burden.Citation50 Therefore, our results suggest that AAV2 infection of human breast cancer xenografts displays the hallmarks of an effective chemotherapeutic.
Figure 6. AAV2 infection of MDA-MB-435 tumors induced tumor necrosis which correlates with growth inhibition. (A) AAV2 infection of MDA-MB-435 xenografts induced dark patches of necrotic areas on the skin surrounding the tumors. (B) Tumors were examined using H&E staining. Mock and PBS-injected tumors were compared with AAV2-injected tumors (magnification, 100×). Both mock and PBS-injected tumors show areas of infiltrating macrophages (IM) as indicated with arrows, whereas AAV2-treated tumors show large areas of tumor necrosis (N) that correlate with the appearance of darkly staining patches on the external surface.
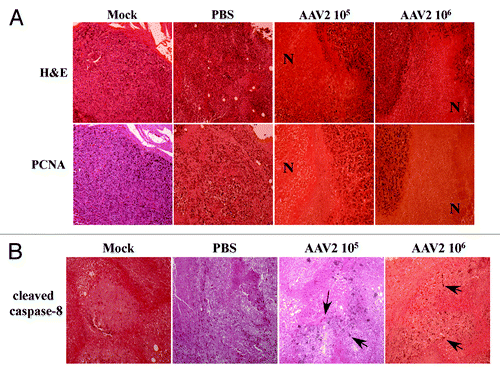
To further confirm that the necrotic areas observed in the tumors were indeed devoid of live cells and not the result of an artifact due to incomplete H&E staining, we performed immunohistochemical analysis of identical tumor sections using a rabbit polyclonal antibody against PCNA, which stains active nuclei. We observed that the pattern of nuclei expressing PCNA coincided with nuclei observed in the H&E-stained tumor sections, and PCNA staining further failed to stain nuclei within the necrotic areas (). These results further confirm the expected tumor morphology of the AAV2-infected tumors. In addition, the observed AAV2-mediated tissue necrosis further correlated with virus-regulated tumor growth inhibition and increased survival of mice () compared with tumors in the control groups.
Figure 7. AAV2-induced necrotic death of MDA-MB-435 xenografts correlated with lack of PCNA nuclear staining. (A) Tumor sections were examined with H&E staining and further stained with PCNA antibody. Identical areas in both types of stained tumor sections showed lack of PCNA staining in areas of necrosis induced upon AAV2 infection. AAV2-treated tumors show large areas of necrosis (N). (B) AAV2-induced necrotic death of MDA-MB-435 xenografts correlated with increased staining with antibody against cleaved caspase-8. Tumor sections were examined with H&E staining and further stained with an antibody against cleaved caspase-8. AAV2-infected tumors show increased focal areas of staining which correlates with tumor cell death compared with mock and PBS injected controls.
AAV2-mediated necrosis coincides with the appearance of cleaved caspase-8 in tumors
AAV2 induced tumor necrosis is indicative of the terminal stages of activating one, or possibly more, cell death pathways. In our current study we have shown that AAV2 induced death of the MDA-MB-435 cells in culture results from activation and cleavage of both caspase-8 and caspase-9 and downstream induction of apoptosis (). Since activation of caspase-8 occurs upstream of caspase-9, we wanted to determine whether AAV2 also induced cleavage of caspase-8 in the MDA-MB-435 xenografted tumors. We performed immunohistochemical analysis of tumor sections using a mouse monoclonal antibody against cleaved caspase-8, which specifically recognizes the cleaved form of the protease. We observed scattered patches of cells within the AAV2-infected tumors which stained positively for the cleaved form of caspase-8 (). In contrast, a similar pattern of staining was not observed in the mock and PBS-treated tumors (). These results suggest that the tumor necrosis observed within the AAV2-infected tumors in nude mice () resulted from virus targeted activation of the extrinsic pathway of apoptosis (), although other pathways of death may also be active under these conditions. Thus, AAV2 activation of the extrinsic pathway of apoptosis via caspase-8 cleavage observed in vitro () also positively correlated with the detection of the cleaved form caspase-8 in AAV2-infected tumors in vivo (). Further, AAV2-specific staining of cell clusters corresponding to activation of caspase-8 () correlated with virus regulated inhibition of tumor growth and increased survival of mice with tumors treated with the virus compared with mice with tumors in the control groups ().
AAV2-mediated inhibition of tumor growth coincides with xenografts positively staining with c-Myc
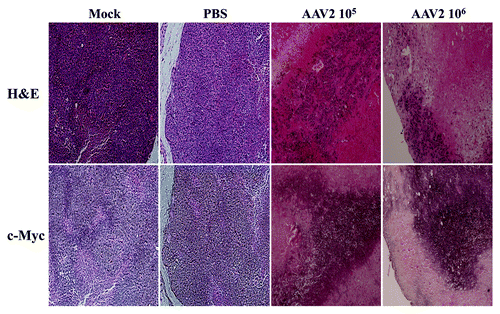
In the current study, our in vitro results showed that MDA-MB-435 monolayer cultures infected with AAV2 underwent apoptotic cell death, which further coincided with increased expression of proliferation markers including the monomeric form of the c-Myc protein (). We then determined whether the AAV2-infected tumors in nude mice also exhibited altered c-Myc expression in vivo, along with other hallmarks of cell death, including tumor necrosis and the appearance of cleaved caspase-8. We performed immunohistochemical analysis of tumor sections using a mouse monoclonal antibody against c-Myc, which was also used for detecting the same protein in western blot analysis (). We observed that the AAV2-infected tumors stained strongly for c-Myc, whereas in contrast, the necrotic areas were devoid of this staining (). In contrast, similar staining was not observed in the mock and PBS-treated tumors (). These results suggested that the AAV2-mediated inhibition of tumor growth in nude mice () resulted from virus-targeted activation of the cell proliferation pathways in conjunction with activation of cell death pathways in vivo (), which was also observed in vitro (). Thus, AAV2 regulated increased in the total levels of c-Myc observed in the current study in vitro () also correlated positively with the detection of strongly stained areas in AAV2-infected tumors in vivo (). Our results further suggest that detection of the cleaved form of caspase-8 as well as c-Myc, in addition to visual determination of tumor necrosis, could be used as diagnostic markers for determining the efficacy of AAV2 as an anti-tumor agent in the treatment of breast tumors in the nude mouse model. Cumulatively, our results in this study positively correlate our observations in vitro with our in vivo observations and suggest the potential for developing wild-type AAV2-based therapeutics for breast cancer.
Figure 8. AAV2-induced necrotic death of MDA-MB-435 xenografts correlated with enhanced c-Myc staining. Tumor sections were examined with H&E staining and further stained with c-Myc antibody. Identical areas in both types of stained tumor sections showed darker areas of c-Myc staining in cells surrounding areas of necrosis induced upon AAV2 infection.
Discussion
Triple-negative breast cancer is characterized by rapid growth, high recurrence rate and poor prognosis. Chemotherapy is the only systemic treatment available for TNBC, in conjunction with surgery and radiation.Citation51 Treatment of breast cancer involves determination of individual patient tumor profiles comprising both hormone and growth factor status for delivering personalized therapy.Citation52 Recent studies have further established the classification of breast cancer into ten novel subgroups with distinct clinical outcomes,Citation53 further fine-tuning future opportunities for providing tailored therapy. In the interim, development of therapies for novel targets in TNBC continues on an evolutionary path, rendered difficult due to multiple mechanisms of death evasion/survival culminating in metastasis of cancer cells. Multiple studies, including published studies from our laboratory,Citation16,Citation21 have documented the anti-proliferative nature of AAV2. In particular, we have recently reported that AAV2 infection suppressed growth and proliferation, via apoptosis induction, of multiple breast cancer-derived cell lines.Citation16 We tested a range of breast cancer cell lines representing both low and high grades of aggressiveness, including MCF-7 (ER+), MDA-MB-468 (ER−), and MDA-MB-231 (ER−) cells.Citation16 In contrast, normal human breast epithelial cells infected with AAV2 were unaffected.Citation16 Together with these findings, our current study strongly suggests the potential for developing wild-type AAV2 as a novel therapeutic for TNBC, regardless of identified subgroups, as well as other grades of breast cancer, and establishes the foundation for further exploration of this possibility.
AAV2 is a potential inducer of DNA damage and abrogates cell cycle check-points
In the current study, we have shown that AAV2 infection induces apoptotic death of the human MDA-MB-435 triple-negative breast cancer cell line, in culture resulting in 100% loss of viability, as well as in breast tumor xenografts. The MDA-MB-435 cell line has been derived from metastatic (pleural effusion), infiltrating ductal breast carcinomaCitation22 and is widely used as a model of metastatic breast cancer.Citation54 The molecular mechanism of death potentially involves the ability of the AAV2 Rep proteins to propel the cancer cells toward a proliferative phenotype, as suggested by the increased movement of cells into S and G2 phases. Increased entry into S phase also suggests a breach of the G1/S cell cycle check-point, within which Cyclin/CDK complexes could be a target of AAV2. Alternatively, upstream signaling mediated by MAP kinase-regulated pathways could also be a target of AAV2, which could explain the observed increase in S phase entry in AAV2-infected cells. AAV2-mediated breach of cell cycle controls to mediate enhanced S phase progression is suggestive of AAV2 overcoming mechanisms of DNA damage checkpoint regulatory controls. In normal cells, DNA damage signals activate checkpoint kinases (Chk), which play a critical role in determining cellular responses to DNA damage by initiating cell cycle arrest, thereby allowing for DNA repair.Citation55 The S, intra-S, and the G2-M phase checkpoints are predominantly regulated by Chk1, whereas Chk2 regulates the G1 phase checkpoint.Citation55 In the current study, addition of DNA damage via AAV2 genomic inverted terminal repeats,Citation56 or inherent Rep protein endonuclease functions,Citation45 could induce a significant level of cellular stress, particularly in cancer cells which are already compromised for DNA damage checkpoint functions.Citation55 These observations suggest that AAV2 promotes infected breast cancer cell line to increasingly enter S and G2 phases, and potentially indicates the ability of AAV2 to interfere with Chk1 kinase functions. One possibility is that AAV2 action on the breast cancer cells is somewhat parallel to functions of DNA damage checkpoint kinase inhibitors which sensitize cancer cells to genotoxic chemotherapeutics, while non-cancerous cells are more resistant to such therapeutics due to the presence of intact DNA damage checkpoint pathways.
Specific combinations of the AAV2 Rep protein potentially determine the kinetics of tumor cell death
Our current studies are not only significant from the viewpoint of potentially developing AAV2 as a therapeutic for TNBC, but also for development of AAV2 as a common therapeutic for less aggressive breast cancer types. We have previously reported that MCF-7, MDA-MB-468, and MDA-MB-231 human breast cancer-derived cell lines, representing both low and high grade types, also underwent cell death in response to AAV2, whereas primary breast epithelial cells were unaffected upon AAV2 infection.Citation16 These results further suggested the property of AAV2 to specifically target cancer cells, which in itself is an attribute of an ideal chemotherapeutic. Although there did not appear to be a “universal” mechanism of cell death among the three cell types,Citation16 the ability of AAV2 to push these cancer cells into S phase also coincides with the ability of the virus to initiate a more proliferative phenotype in the MDA-MB-435 cells (current study), and appears to be a common mechanism of death induction and a specific hallmark of AAV2 regulated tumor suppression. A difference relating to the kinetics of AAV2 regulated cell death was the gradual demise of the virus infected MDA-MB-435 cells over a period of 21 d (current study), which contrasts sharply with the 7 d period characteristic of the other breast cancer cell lines.Citation16 The observed difference could be due to the differential expression of the four Rep proteins expressed in the different breast cancer cell lines. Whereas death of AAV2-infected MDA-MB-435 cells was correlated with the expression of Rep78 and Rep52 but death of MCF-7, MDA-MB-468, and MDA-MB-231 cells was correlated with expression of Rep78, Rep68, and Rep40. This is potentially due to some Rep proteins being expressed below the level of detection under these conditions. On the other hand, it is also possible that biochemical environment of the different breast cancer cell lines determine differential expression of specific combinations of Rep proteins, which enables their targeting of cell death. Highly metastatic breast cancer cells, similar to growth characteristics of the MDA-MB-435 cells, are generally more resistant to chemotherapeutics, which may translate to the longer kinetics for the observed 100% cell killing observed in the current study.
AAV2 regulates increased expression of monomeric c-Myc in late-stage apoptotic cells
AAV2-mediated induction of apoptosis in the MDA-MB-435 breast cancer line coincided with upregulated expression of monomeric form of the c-Myc protein. The c-Myc protein is a transcription factor which is both a regulator of cell proliferation as well as apoptosis.Citation17 Under normal conditions, c-Myc controls growth regulatory signals in early G1 and at the G1/S cell cell cycle check-points.Citation18 Ectopic expression of c-Myc has been shown to induce S phase entry in quiescent cells, observations which first suggested that c-Myc could play a role in G1/S transition.Citation18 In addition, c-Myc expression also targets cell cycle regulatory proteins in the G2 phase and could mediate a G2/M arrest and downstream apoptosis induction.Citation57 We have reported that AAV2 infection of multiple breast cancer cell lines and ensuing apoptotic death was correlated with upregulated expression levels of the c-Myc protein.Citation16 Taken together, our studies suggest that AAV2 modulation of c-Myc expression in breast cancer cells, either through enhancing specific expression of the monomer form (current study), or via upregulating total protein levels,Citation16 is correlated with apoptosis induction. In the current study, AAV2-mediated increase of c-Myc specifically of the monomer form in the infected MDA-MB-435 cells. Under normal conditions, c-Myc binding to E box DNA recognition sequences and functional regulation of its target genes requires binding and heterodimerization with its partner Max.Citation58 Myc function in transcriptional activation, transformation and apoptosis is dependent on the herterodimeration with MaxCitation59 as a minimal functioning unit.Citation58 Since c-Myc needs to heterodimerize with Max in order to be transcriptionally active, increased expression of the monomeric species of c-Myc in the AAV2-infected cells could be in response to the need to shift the balance in favor of its heterodimerization with Max or other partners, perhaps necessary for reaching the threshold for favoring the induction of apoptosis.
AAV2 infection of MDA-MB-435 xenografts inhibits tumor growth and induces central necrosis
AAV2 infection of MDA-MB-435 xenografts inhibited tumor growth. Two different AAV2 dosages successfully inflicted extensive areas of tumor necrosis and virus infected tumors, and both dosages showed a trend toward decreased size. Additionally, induction of necrotic death within tumors is used as the index of an effective therapeuticCitation49 as necrosis is further correlated with decreased angiogenesis and metastasis.Citation47 The visualization of pronounced necrotic areas, tumor growth inhibition, and 100% survival of mice with AAV2-infected tumors compared with controls all point to the effectiveness of the wild-type AAV2 as a novel therapeutic for TNBC.
TNBC-derived tumors are difficult to treat as they are devoid of typical hormone and growth factor targets, which are common to less aggressive cancer types.Citation11,Citation12 The need for identification of novel targets for TNBC is crucial. In the current study, we determined the visualization of cleaved caspase-8 as an AAV2-specific index of apoptosis in the MDA-MB-435 three dimensional xenografts, as well as in cultured cells, which provides the potential for the consideration of a novel target for therapeutic development. Cell signaling pathways in aggressive/metastatic cancers have found ways to activate multiple pathways of survival by inhibiting pathways of cell death.Citation60 Our studies suggest that AAV2 potentially removed blocked signals which allowed for sensitization of caspase-8-dependent apoptosis, further disallowing death evasion signals. These data also suggest the possibility that CD95, a member of the death receptor family, and downstream signaling molecules in that pathway, are potential targets of AAV2. Under normal conditions, stimulation of CD95 mediated signaling ultimately mediates activation/cleavage of procaspase-8 via downstream formation of DISC.Citation61 The ability of AAV2 to activate the caspase-8-dependent pathway of apoptosis also suggests the ability of the virus to mediate mitochondrial dysfunction in the infected tumors. Our current study demonstrates proof of principle for the use of AAV2 as a therapeutic for tumors of the breast.
In the current study, we demonstrated that AAV2 induction of tumor necrosis was accompanied by a reduction in the number of inflammatory macrophages compared with control tumors. Others have reported that breast tumors containing increased numbers of macrophages are significantly more vascularized and metastatic than tumors with a low number of macrophage infiltrates.Citation48 Therefore, our data also suggests the potential for AAV2 to target pathways of angiogenesis in TNBC.
Conclusions
Our data presented in the current report suggests the potential for developing the wild-type AAV2 as a novel and effective anti-cancer therapeutic agent for treatment of TNBC. Our current study clearly demonstrates the ability of AAV2 to sensitize pathways which regulate caspase-8 activation for mediating tumor cell death, although activation of other pathways potentially remain to be explored. Developing the wild-type AAV2 in its entirety, as well as identifying active protein domains and derivation of peptides of one or more Rep proteins as small molecule therapeutics, are possibilities for future consideration. Our studies suggest that very low dosages of AAV2 are required for inducing cell death and promoting tumor regression. Future studies would necessitate testing the wild-type AAV2 in an immune-competent mouse model. Overall, our studies provide proof of principle for developing wild-type AAV2 as a novel therapy for TNBC as well as other grades of breast cancers.
Methods
Cell lines
The MDA-MB-435 cell line has been previously described.Citation22 Cells were maintained in monolayer culture with E medium containing 5% fetal bovine serum.
Viral stocks
AAV2 stocks were prepared in our laboratory using the whole-cell lysate method, by banding twice on CsCl gradients and determination of infectious titers as we have described previously.Citation21 The AAV2 virus stocks were tested for residual contamination with helper adenovirus using the following methods. A 2 μL aliquot from 0.5 μL CsCl gradient fractions was used for infecting 293 cells in 24-well dishes. The AAV2 positive fractions exhibiting low cytopathic effects were pooled, and heated at 56 °C for 30 min. The AAV2 stocks were then serially diluted and further used to infect 293 cells in 60-mm dishes and the presence of residual contaminating adenovirus was visualized using plaque assays.
Cell synchronization and infection of cells
The MDA-MB-435 cell line was grown to approximately 80% confluence, trypsinized, and plated at a density of 1 × 106 cells in 100-mm plates in E medium. After plating, cells were incubated between 10 and 12 h, at which time about two-thirds or more of the cells are synchronized at the G1 phase (without the addition of exogenous chemicals) as previously described.Citation21 This time point was designated time zero. The medium was aspirated from the plates, and infections were performed using AAV2 at a multiplicity of infection (MOI) of 0.02 (using AAV2 MOIs of 10, 20, 30, and 100 yielded similar results). The infections were performed by diluting the AAV2 stocks into 1 mL of E medium without serum and used for infection of cells. Mock infections were performed using 1 mL of E medium without serum. Plates were incubated at 37 °C for 2 h with intermittent swirling. At the end of the incubation, residual medium was aspirated from the plates and replaced with 10 mL of fresh E medium supplemented with serum. Virus infected and mock infected cells were collected at t = 0 and day 1 through day 21 post-infection with AAV2. Both mock-infected and AAV2-infected cell samples were trypsinized, inactivated with the addition of serum, pelleted, and stored at −70 °C until further manipulations. Both floating cells and adherent cells were collectively pooled before pelleting, followed by storage at −70 °C. Additionally, on day 2, day 5, day 9, day 12, and day 16 (when cells were approximately 80% confluent), both mock and AAV2-infected cells were passaged at a ratio of 1:2 and plated in fresh E medium.
Southern blot analysis of AAV2 DNA replication
A 5 μg aliquot of the isolated low molecular weight DNA was separated by electrophoresis in a 0.8% TAE agarose gel followed by transferring to GeneScreen Plus membranes (New England Nuclear Research Products), as previously reported.Citation62 Samples were probed with 32P-labeled total AAV2 genomic DNA probe generated by random primer extension using methods previously described.Citation62
Preparation of total cellular protein extracts and western blotting
Total protein extracts were prepared from the monolayer cultures and quantitated using the Lowry Method as previously described.Citation21,Citation24 For detecting cell cycle proteins, a total of 60 μg whole cell extract was used in western blots to determine expression of p21WAF1, p27KIP1, and p16INK4. To detect pRb and p53 protein expression, 30 μg of whole cell extracts were used. Protein extracts were applied to sodium-dodecyl sulfate (SDS)-polyacrylamide (SDS-PAGE) gel (acrylamide/bisacrylamide ratio, 30:0.8). Gel compositions for resolving various proteins are as follows. A 12% gel was used to detect p21WAF1, 15% gel for p16INK4, 10% gel for p27KIP1, and 7.5% SDS-PAGE gel for pRb and p53. Polyclonal antibodies against p21WAF1, p27KIP1, p16INK4, and pRb (Santa Cruz) were each used at a dilution of 1:2000 to detect the respective proteins, and have been described previously.Citation21,Citation24 A monoclonal antibody against p53 (Oncogene) was used at a dilution of 1:1000 for detecting the protein and has been described previously.Citation21,Citation24
To detect c-Myc and Ki-67 protein expression, 60 μg of whole cell extracts were used. Monoclonal antibodies against c-Myc (Santa Cruz) were used at a dilution of 1:500 and polyclonal antibodies against Ki-67 (Santa Cruz) were used at a dilution of 1:2000.
To detect apoptosis regulatory proteins, 60 μg of whole cell extracts were used. To detect the pro-caspase form of caspase-3, proteins were resolved in a 10% SDS-PAGE gel and detected with caspase-3 rabbit monoclonal antibody (Cell Signaling Technology) and used at a dilution of 1:1000. To detect the 19 kDa and 17 kDa cleaved caspase-3 forms, proteins were resolved in a 15% SDS-PAGE gel and detected with rabbit polyclonal antibody against cleaved caspase-3 (Cell Signaling Technology) and used at a dilution of 1:1000. To detect the pro-caspase form of caspase-6, proteins were resolved in a 10% SDS-PAGE gel and to detect the cleaved form of caspase-6, proteins were resolved in a 15% SDS-PAGE gel and detected with a rabbit polyclonal antibody (Cell Signaling Technology) used at a dilution of 1:1000. To detect both the pro- and cleaved forms of caspase-7, caspase-8, and caspase-9, proteins were resolved in a 10% SDS-PAGE gel. Caspase-7 was detected with a mouse monoclonal antibody (Cell Signaling) and used at a dilution of 1:1000. Caspase-8 was detected with a mouse monoclonal antibody (Alexis Biochemicals) and used at a dilution of 1:2000. Caspase-9 was detected with a rabbit polyclonal antibody (Cell Signaling) and used at a dilution of 1:1000. To detect the pro- and cleaved forms of PARP, proteins were resolved in a 7.5% SDS-PAGE gel and detected with a rabbit monoclonal antibody (Cell Signaling) and used at a dilution of 1:1000. Proteins were detected using enhanced chemiluminescence (ECL) method (Perkin Elmer) as per manufacturer’s instructions.
Cell viability assay and flow cytometric analysis of DNA content
MDA-MB-435 cultures were synchronized and infected with AAV2 as described above. At each time point, cell samples were collected at t = 0 and days 1 through 21. On day 2, day 5, day 9, day 12, and day 16, both mock and AAV2-infected cells were passaged 1:2 as described above. Both mock and AAV2-infected cell samples were trypsinized, inactivated with the addition of serum, and counted using trypan blue exclusion using standard protocols.
For performing flow cytometry, mock and AAV2-infected cells were prepared for analysis as previously described.Citation24 Briefly, MDA-MB-435 cells were synchronized and infected with AAV2 as described above. Mock and AAV2-infected cells were sampled at t = 0 and from day 1 through day 21, with passaging on day 2, day 5, day 9, day 12, and day 16, as described. On each day, cells were harvested by trypsinization, washed with PBS, fixed in 70% ethanol, and stored at −20 °C. The fixed cells were washed in PBS and then resuspended in PBS containing 0.1% Triton X-100 (Sigma), 200 μg/mL DNase-free RNase A (Boehringer Mannheim), and 100 μg/mL of propidium iodide (Sigma) for 30 min at 37 °C. Flow cytometric analysis of 106 cells was performed in a fluorescence-activated cell sorter (FACS), and the percentages of cells in the G1, S, and G2/M phases of the cell cycle were determined using the Cell Quest program of Becton Dickinson. Data were analyzed with the Mod Fit LT program.
Human breast tumor xenograft experiments
Five- to six-week-old female athymic nude mice were purchased from Harlan and housed under pathogen-free conditions according to Penn State University College of Medicine animal care guidelines. The protocols of animal experiments were reviewed and approved by Institutional Animal Care and Use Committee of Penn State University College of Medicine. MDA-MB-435 cells (5 × 106) were resuspended in 100 μL PBS and implanted subcutaneously in the shoulder of mice. Tumor size was measured every other day. Since the xenograft tumors develop volumetric shapes which are roughly elliptical, tumor volume (V) was determined by the equation: V = π(w + l)Citation3 / 48, where l is the length and w is the width of the tumor, measured with calipers without applying compression, as published elsewhere.Citation63 When xenografts reached volumes of ~300 mm3 (approximately 2 wk), the mice were randomly assigned to control and AAV2 treated groups (n = 5). Two sets of 5 mice each received a single AAV2 dosage of 105 and 106 infectious units per tumor administered via intratumoral injections. The respective dosage of AAV2 virus was diluted in 200 μL PBS and used for injecting multiple sites of the tumor. Control tumors either received intratumoral injections of PBS or not manipulated (mock). When the control tumors reached ~2400 mm3 (on day 32), the mock and PBS-treated mice were sacrificed due to their grossly restricted ability to move and to feed arising from the tumor burden. The mice receiving intratumoral injections of AAV2 at both 105 and 106 dosages did not display similar restrictions in movements and feeding habits as the control mice, survived longer than the control mice and were eventually euthanized on day 56.
Histochemical analysis
At the end of the experiment, animals were euthanized and tumors were removed, fixed in 10% buffered formalin (for 24 h), paraffin embedded, and 4-μm cross-sections were prepared as we have previously described.Citation64 Sections were stained with hematoxylin and eosin as previously described.Citation65
For immunohistochemical analysis, the c-Myc antibody (Santa Cruz) was used at a dilution of 1:100 using protocols previously described.Citation66 The PCNA antibody (Santa Cruz) was used as we have described previously.Citation67 The cleaved caspase-8 (Cell Signaling) antibodies were used as recommended by the manufacturers.
| Abbreviations: | ||
| AAV2 | = | adeno-associated virus type 2 |
| TNBC | = | triple negative breast cancer |
Additional material
Download Zip (617.5 KB)Disclosure of Potential Conflicts of Interest
The work described in this manuscript is part of US Patent Application No. 8 080 240 applied for by The Pennsylvania State University Intellectual Property Office.
Authors’ Contributions
S.A. and C.M. conceptualized the experimental design and drafted the manuscript. S.A. performed all experiments. B.S.B. performed the western blots for cell cycle analysis. M.I. performed the immuno-histochemical staining of the tumor tissues. M.J.C. performed statistical analysis of the tumor growth curves.
Acknowledgments
We thank D. Welch and A. Mastro for kindly supplying the MDA-MB-435 cell line. We thank Lynn Budgeon for excellent technical assistance with studies of tumor histology. This work was supported by a grant from the Pennsylvania Breast Cancer Coalition to C.M.
References
- DeSantis C, Siegel R, Bandi P, Jemal A. Breast cancer statistics, 2011. CA Cancer J Clin 2011; 61:409 - 18; http://dx.doi.org/10.3322/caac.20134; PMID: 21969133
- Chalasani P, Downey L, Stopeck AT. Caring for the breast cancer survivor: a guide for primary care physicians. Am J Med 2010; 123:489 - 95; http://dx.doi.org/10.1016/j.amjmed.2009.09.042; PMID: 20569749
- Hamilton A, Hortobagyi G. Chemotherapy: what progress in the last 5 years?. J Clin Oncol 2005; 23:1760 - 75; http://dx.doi.org/10.1200/JCO.2005.10.034; PMID: 15755984
- Colditz GA. Relationship between estrogen levels, use of hormone replacement therapy, and breast cancer. J Natl Cancer Inst 1998; 90:814 - 23; http://dx.doi.org/10.1093/jnci/90.11.814; PMID: 9625169
- Pegram MD, Pietras R, Bajamonde A, Klein P, Fyfe G. Targeted therapy: wave of the future. J Clin Oncol 2005; 23:1776 - 81; http://dx.doi.org/10.1200/JCO.2005.11.029; PMID: 15755985
- Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem 2001; 276:9817 - 24; http://dx.doi.org/10.1074/jbc.M010840200; PMID: 11139588
- Simstein R, Burow M, Parker A, Weldon C, Beckman B. Apoptosis, chemoresistance, and breast cancer: insights from the MCF-7 cell model system. Exp Biol Med (Maywood) 2003; 228:995 - 1003; PMID: 14530507
- Rahman KM, Sarkar FH. Steroid hormone mimics: molecular mechanisms of cell growth and apoptosis in normal and malignant mammary epithelial cells. J Steroid Biochem Mol Biol 2002; 80:191 - 201; http://dx.doi.org/10.1016/S0960-0760(01)00186-8; PMID: 11897503
- Lewis JS, Cheng D, Jordan VC. Targeting oestrogen to kill the cancer but not the patient. Br J Cancer 2004; 90:944 - 9; http://dx.doi.org/10.1038/sj.bjc.6601627; PMID: 14997187
- Gelber RD, Bonetti M, Cole BF, Gelber S, Goldhirsch A. Quality of life assessment in the adjuvant setting: is it relevant? International Breast Cancer Study Group. Recent Results Cancer Res 1998; 152:373 - 89; http://dx.doi.org/10.1007/978-3-642-45769-2_36; PMID: 9928573
- Minami CA, Chung DU, Chang HR. Management options in triple-negative breast cancer. Breast Cancer (Auckl) 2011; 5:175 - 99; PMID: 21863131
- Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature 2000; 406:747 - 52; http://dx.doi.org/10.1038/35021093; PMID: 10963602
- Ahmad N, Kumar R. Steroid hormone receptors in cancer development: a target for cancer therapeutics. Cancer Lett 2011; 300:1 - 9; http://dx.doi.org/10.1016/j.canlet.2010.09.008; PMID: 20926181
- Orlando L, Schiavone P, Fedele P, Calvani N, Nacci A, Rizzo P, Marino A, D’Amico M, Sponziello F, Mazzoni E, et al. Molecularly targeted endocrine therapies for breast cancer. Cancer Treat Rev 2010; 36:Suppl 3 S67 - 71; http://dx.doi.org/10.1016/S0305-7372(10)70023-2; PMID: 21129614
- Hudis CA, Gianni L. Triple-negative breast cancer: an unmet medical need. Oncologist 2011; 16:Suppl 1 1 - 11; http://dx.doi.org/10.1634/theoncologist.2011-S1-01; PMID: 21278435
- Alam S, Bowser BS, Conway MJ, Israr M, Tandon A, Meyers C. Adeno-associated virus type 2 infection activates caspase dependent and independent apoptosis in multiple breast cancer lines but not in normal mammary epithelial cells. Mol Cancer 2011; 10:97; http://dx.doi.org/10.1186/1476-4598-10-97; PMID: 21827643
- Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 2000; 16:653 - 99; http://dx.doi.org/10.1146/annurev.cellbio.16.1.653; PMID: 11031250
- Matsumura I, Tanaka H, Kanakura Y. E2F1 and c-Myc in cell growth and death. Cell Cycle 2003; 2:333 - 8; http://dx.doi.org/10.4161/cc.2.4.428; PMID: 12851485
- Iatropoulos MJ, Williams GM. Proliferation markers. Exp Toxicol Pathol 1996; 48:175 - 81; http://dx.doi.org/10.1016/S0940-2993(96)80039-X; PMID: 8672872
- Yu CC, Woods AL, Levison DA. The assessment of cellular proliferation by immunohistochemistry: a review of currently available methods and their applications. Histochem J 1992; 24:121 - 31; http://dx.doi.org/10.1007/BF01047461; PMID: 1349881
- Alam S, Meyers C. Adeno-associated virus type 2 induces apoptosis in human papillomavirus-infected cell lines but not in normal keratinocytes. J Virol 2009; 83:10286 - 92; http://dx.doi.org/10.1128/JVI.00343-09; PMID: 19625406
- Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res 2000; 60:2764 - 9; PMID: 10850410
- Girgert R, Emons G, Gründker C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res Treat 2012; 134:199 - 205; http://dx.doi.org/10.1007/s10549-012-1968-x; PMID: 22290080
- Alam S, Sen E, Brashear H, Meyers C. Adeno-associated virus type 2 increases proteosome-dependent degradation of p21WAF1 in a human papillomavirus type 31b-positive cervical carcinoma line. J Virol 2006; 80:4927 - 39; http://dx.doi.org/10.1128/JVI.80.10.4927-4939.2006; PMID: 16641284
- Kim YG, Bi W, Feliciano ES, Drake RR, Stambrook PJ. Ganciclovir-mediated cell killing and bystander effect is enhanced in cells with two copies of the herpes simplex virus thymidine kinase gene. Cancer Gene Ther 2000; 7:240 - 6; http://dx.doi.org/10.1038/sj.cgt.7700113; PMID: 10770632
- Trepel M, Stoneham CA, Eleftherohorinou H, Mazarakis ND, Pasqualini R, Arap W, Hajitou A. A heterotypic bystander effect for tumor cell killing after adeno-associated virus/phage-mediated, vascular-targeted suicide gene transfer. Mol Cancer Ther 2009; 8:2383 - 91; http://dx.doi.org/10.1158/1535-7163.MCT-09-0110; PMID: 19671758
- Berns KI, Giraud C. Biology of adeno-associated virus. Curr Top Microbiol Immunol 1996; 218:1 - 23; http://dx.doi.org/10.1007/978-3-642-80207-2_1; PMID: 8794242
- Leonard CJ, Berns KI. Cloning, expression, and partial purification of Rep78: an adeno-associated virus replication protein. Virology 1994; 200:566 - 73; http://dx.doi.org/10.1006/viro.1994.1219; PMID: 8178443
- Nash K, Chen W, Salganik M, Muzyczka N. Identification of cellular proteins that interact with the adeno-associated virus rep protein. J Virol 2009; 83:454 - 69; http://dx.doi.org/10.1128/JVI.01939-08; PMID: 18971280
- Duprez L, Wirawan E, Vanden Berghe T, Vandenabeele P. Major cell death pathways at a glance. Microbes Infect 2009; 11:1050 - 62; http://dx.doi.org/10.1016/j.micinf.2009.08.013; PMID: 19733681
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281:1309 - 12; http://dx.doi.org/10.1126/science.281.5381.1309; PMID: 9721092
- Lan L, Gorke S, Rau SJ, Zeisel MB, Hildt E, Himmelsbach K, Carvajal-Yepes M, Huber R, Wakita T, Schmitt-Graeff A, et al. Hepatitis C virus infection sensitizes human hepatocytes to TRAIL-induced apoptosis in a caspase 9-dependent manner. J Immunol 2008; 181:4926 - 35; http://dx.doi.org/10.4049/jimmunol.181.7.4926; PMID: 18802096
- Bröker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: a review. Clin Cancer Res 2005; 11:3155 - 62; http://dx.doi.org/10.1158/1078-0432.CCR-04-2223; PMID: 15867207
- Agarwal A, Mahfouz RZ, Sharma RK, Sarkar O, Mangrola D, Mathur PP. Potential biological role of poly (ADP-ribose) polymerase (PARP) in male gametes. Reprod Biol Endocrinol 2009; 7:143; http://dx.doi.org/10.1186/1477-7827-7-143; PMID: 19961617
- Althaus FR, Kleczkowska HE, Malanga M, Müntener CR, Pleschke JM, Ebner M, Auer B. Poly ADP-ribosylation: a DNA break signal mechanism. Mol Cell Biochem 1999; 193:5 - 11; http://dx.doi.org/10.1023/A:1006975002262; PMID: 10331631
- Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297:259 - 63; http://dx.doi.org/10.1126/science.1072221; PMID: 12114629
- Ha HC, Snyder SH. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci U S A 1999; 96:13978 - 82; http://dx.doi.org/10.1073/pnas.96.24.13978; PMID: 10570184
- Szabó C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 1998; 19:287 - 98; http://dx.doi.org/10.1016/S0165-6147(98)01193-6; PMID: 9703762
- Alano CC, Ying W, Swanson RA. Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 2004; 279:18895 - 902; http://dx.doi.org/10.1074/jbc.M313329200; PMID: 14960594
- Delavallée L, Cabon L, Galán-Malo P, Lorenzo HK, Susin SA. AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life 2011; 63:221 - 32; http://dx.doi.org/10.1002/iub.432; PMID: 21438113
- Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat 2008; 213:30 - 44; http://dx.doi.org/10.1111/j.1469-7580.2008.00931.x; PMID: 18638068
- Kuntz K, O’Connell MJ. The G(2) DNA damage checkpoint: could this ancient regulator be the Achilles heel of cancer?. Cancer Biol Ther 2009; 8:1433 - 9; http://dx.doi.org/10.4161/cbt.8.15.9081; PMID: 19574738
- Hoffman B, Liebermann DA. Apoptotic signaling by c-MYC. Oncogene 2008; 27:6462 - 72; http://dx.doi.org/10.1038/onc.2008.312; PMID: 18955973
- Guerra L, Albihn A, Tronnersjö S, Yan Q, Guidi R, Stenerlöw B, Sterzenbach T, Josenhans C, Fox JG, Schauer DB, et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 2010; 5:e8924; http://dx.doi.org/10.1371/journal.pone.0008924; PMID: 20111719
- Berthet C, Raj K, Saudan P, Beard P. How adeno-associated virus Rep78 protein arrests cells completely in S phase. Proc Natl Acad Sci U S A 2005; 102:13634 - 9; http://dx.doi.org/10.1073/pnas.0504583102; PMID: 16157891
- Cozzi P. The discovery of a new potential anticancer drug: a case history. Farmaco 2003; 58:213 - 20; http://dx.doi.org/10.1016/S0014-827X(03)00014-4; PMID: 12620417
- Monteiro R, Calhau C, Silva AO, Pinheiro-Silva S, Guerreiro S, Gärtner F, Azevedo I, Soares R. Xanthohumol inhibits inflammatory factor production and angiogenesis in breast cancer xenografts. J Cell Biochem 2008; 104:1699 - 707; http://dx.doi.org/10.1002/jcb.21738; PMID: 18348194
- Bingle L, Lewis CE, Corke KP, Reed MW, Brown NJ. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br J Cancer 2006; 94:101 - 7; http://dx.doi.org/10.1038/sj.bjc.6602901; PMID: 16404363
- Zheng J, Koblinski JE, Dutson LV, Feeney YB, Clevenger CV. Prolyl isomerase cyclophilin A regulation of Janus-activated kinase 2 and the progression of human breast cancer. Cancer Res 2008; 68:7769 - 78; http://dx.doi.org/10.1158/0008-5472.CAN-08-0639; PMID: 18829531
- Basu GD, Pathangey LB, Tinder TL, Gendler SJ, Mukherjee P. Mechanisms underlying the growth inhibitory effects of the cyclo-oxygenase-2 inhibitor celecoxib in human breast cancer cells. Breast Cancer Res 2005; 7:R422 - 35; http://dx.doi.org/10.1186/bcr1019; PMID: 15987447
- Yagata H, Kajiura Y, Yamauchi H. Current strategy for triple-negative breast cancer: appropriate combination of surgery, radiation, and chemotherapy. Breast Cancer 2011; 18:165 - 73; http://dx.doi.org/10.1007/s12282-011-0254-9; PMID: 21290263
- Manson JE. The role of personalized medicine in identifying appropriate candidates for menopausal estrogen therapy. Metabolism 2013; 62:Suppl 1 S15 - 9; PMID: 23018143
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al, METABRIC Group. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012; 486:346 - 52; PMID: 22522925
- Shevde LA, Samant RS, Paik JC, Metge BJ, Chambers AF, Casey G, Frost AR, Welch DR. Osteopontin knockdown suppresses tumorigenicity of human metastatic breast carcinoma, MDA-MB-435. Clin Exp Metastasis 2006; 23:123 - 33; http://dx.doi.org/10.1007/s10585-006-9013-2; PMID: 16830223
- Ashwell S, Zabludoff S. DNA damage detection and repair pathways--recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin Cancer Res 2008; 14:4032 - 7; http://dx.doi.org/10.1158/1078-0432.CCR-07-5138; PMID: 18593978
- Raj K, Ogston P, Beard P. Virus-mediated killing of cells that lack p53 activity. Nature 2001; 412:914 - 7; http://dx.doi.org/10.1038/35091082; PMID: 11528480
- Elliott MJ, Dong YB, Yang H, McMasters KM. E2F-1 up-regulates c-Myc and p14(ARF) and induces apoptosis in colon cancer cells. Clin Cancer Res 2001; 7:3590 - 7; PMID: 11705881
- Lüscher B, Larsson LG. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene 1999; 18:2955 - 66; http://dx.doi.org/10.1038/sj.onc.1202750; PMID: 10378692
- Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature 1992; 359:423 - 6; http://dx.doi.org/10.1038/359423a0; PMID: 1406955
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat 2007; 10:13 - 29; http://dx.doi.org/10.1016/j.drup.2007.01.003; PMID: 17303468
- Lavrik IN, Krammer PH. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ 2012; 19:36 - 41; http://dx.doi.org/10.1038/cdd.2011.155; PMID: 22075988
- Ozbun MA, Meyers C. Human papillomavirus type 31b E1 and E2 transcript expression correlates with vegetative viral genome amplification. Virology 1998; 248:218 - 30; http://dx.doi.org/10.1006/viro.1998.9285; PMID: 9721231
- Song C, Appleyard V, Murray K, Frank T, Sibbett W, Cuschieri A, Thompson A. Thermographic assessment of tumor growth in mouse xenografts. Int J Cancer 2007; 121:1055 - 8; http://dx.doi.org/10.1002/ijc.22808; PMID: 17487841
- Alam S, Conway MJ, Chen HS, Meyers C. The cigarette smoke carcinogen benzo[a]pyrene enhances human papillomavirus synthesis. J Virol 2008; 82:1053 - 8; http://dx.doi.org/10.1128/JVI.01813-07; PMID: 17989183
- Meyers C, Frattini MG, Hudson JB, Laimins LA. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 1992; 257:971 - 3; http://dx.doi.org/10.1126/science.1323879; PMID: 1323879
- Ozbun MA, Meyers C. Transforming growth factor beta1 induces differentiation in human papillomavirus-positive keratinocytes. J Virol 1996; 70:5437 - 46; PMID: 8764055
- Alam S, Bowser BS, Conway MJ, Israr M, Ryndock EJ, Xi LF, Meyers C. Downregulation of Cdc2/CDK1 kinase activity induces the synthesis of noninfectious human papillomavirus type 31b virions in organotypic tissues exposed to benzo[a]pyrene. J Virol 2010; 84:4630 - 45; http://dx.doi.org/10.1128/JVI.02431-09; PMID: 20181698