
Abstract
Acute myeloid leukemia (AML) is one of the deadliest leukemias for which there is an urgent and unmet need for the development of novel treatment strategies. Multiple drug resistance mechanisms mediate poor drug response and relapse in patients, and a selective Mcl-1 inhibitor has been speculated to be a promising agent in the treatment of AML. Here, we describe that maritoclax, a small molecule Mcl-1 inhibitor, induces Mcl-1 proteasomal degradation without transcriptional downregulation. Maritoclax killed AML cell lines and primary cells with elevated Mcl-1 levels through selective Mcl-1 downregulation, and synergized with ABT-737 to overcome Mcl-1-mediated ABT-737 resistance. Maritoclax was more effective than daunorubicin at inducing leukemic cell death when co-cultured with HS-5 bone marrow stroma cells, while being less toxic than daunorubicin against HS-5 stroma cells, primary mouse bone marrow cells, and hematopoietic progenitor cells. Moreover, maritoclax administration at 20 mg/kg/d intraperitoneally caused significant U937 tumor shrinkage, as well as 36% tumors remission rate in athymic nude mice, without apparent toxicity to healthy tissue or circulating blood cells. In summary, our studies suggest that maritoclax belongs to a novel class of Mcl-1 inhibitors that has the potential to be developed for the treatment of AML.
Introduction
Acute myeloid leukemia (AML) is one of the deadliest forms of leukemia and is expected to cause 10 370 deaths in year 2013 in the United States alone.Citation1 Standard therapy for AML utilizes cytarabine in combination with daunorubicin. However, a lack of response to therapy and frequent patient relapse makes the management of AML difficult.Citation2 As new therapeutics have not been approved for AML in decades, there is an urgent and unmet need for new therapeutic intervention to improve patient outcomes.
Myeloid cell leukemia sequence-1 (Mcl-1), an anti-apoptotic protein in the B-cell lymphoma-2 (Bcl-2) family of proteins, is reported to contribute to the pathogenesis and drug resistance in AML. Relapsed primary human AML samples revealed a marked elevation in Mcl-1 protein levels,Citation3,Citation4 and Mcl-1 was shown to be necessary to sustain translocation-driven murine AML at both induction and relapse.Citation5 The FMS-like tyrosine kinase-3-internal tandem duplications (FLT3-ITD) mutation, a prognostic marker that indicate frequent relapse and poor survival in patients, could drive Mcl-1 upregulation.Citation6 While ABT-737 and its analogs ABT-263 and ABT-199 are clinically promising selective Bcl-2 inhibitors against leukemia that do not inhibit Mcl-1 function,Citation7-Citation9 resistance to these compounds frequently occurred through Mcl-1 upregulation.Citation10 Mcl-1 upregulation was further implicated in multidrug resistance and stroma-mediated drug resistance.Citation11-Citation13 Therefore, a selective pharmacologic inhibitor of Mcl-1 might be effective in treating AML.
However, a body of evidence suggests that Mcl-1 inhibition might impair physiological function. Genetic Mcl-1 knockout caused peri-implantation embryonic lethality in mice,Citation14 and the organ-specific knockout of Mcl-1 resulted in acute failure of hematopoietic stem cells and the heart.Citation15-Citation17 Nonetheless, a number of anti-cancer agents approved for use or in clinical trials to treat cancer could downregulate Mcl-1, such as sorafenib, PKC412, and flavopiridol.Citation18-Citation20 Given that genetic and pharmacologic modulations of a protein are fundamentally different, it is possible that there exists a margin of safety between the anti-cancer and toxic effects of Mcl-1 small molecule inhibitors in the treatment of AML.
We recently discovered that a selective Mcl-1 inhibitor, marinopyrrole A (hereafter referred to as maritoclax), demonstrated an unique mechanism of action to downregulate Mcl-1 through proteasomal degradation.Citation21,Citation22 In this report, we determined that maritoclax potency strongly correlated with elevated levels of Mcl-1 in both AML cell lines and primary human AML samples independent of transcriptional repression. Maritoclax could overcome ABT-737 resistance and synergize with ABT-737 to kill ABT-737-resistant AML cells. Maritoclax also remained potent against U937 cells cultured with stroma, while being less toxic toward the HS-5 human stroma cell line, primary mouse bone marrow, and hematopoietic progenitor cells compared with daunorubicin. Finally, in athymic nude mice bearing U937 xenografts, 20 mg/kg/d intraperitoneal (IP) injection of maritoclax caused significant tumor reduction without apparent toxicity to organs or circulating blood. Overall, we demonstrated that Mcl-1 inhibition through maritoclax can potentially be developed as a viable strategy for the treatment of Mcl-1-overexpressing AML cells.
Results
Maritoclax downregulates Mcl-1 expression through proteasomal degradation without transcriptional repression
Maritoclax is found naturally in a marine-derived strain of Streptomyces as the (−) enantiomer,Citation23 but the racemic compound was used in previous biological assays testing maritoclax as a Mcl-1 inhibitor.Citation21,Citation22 We confirmed that the (+)-, (−)-, and racemic maritoclax demonstrate similar biological activity in viability assays against U937 and Mcl-1-IRES-Bim K562 cells (Fig. S1A and B).
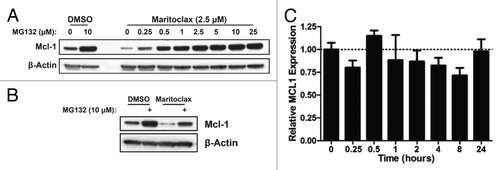
Maritoclax induced endogenous Mcl-1 downregulation in U937 cells that could be completely reversed by MG132 treatment in a concentration-dependent manner (). As prolonged proteasome inhibition may disrupt transcription and synthesis, MG132 was added to U937 cells in the last 3 h of a 12-h incubation with maritoclax. U937 cells not treated with maritoclax demonstrated a 4-fold increase in Mcl-1 protein levels, and maritoclax-treated cells demonstrated a 6.5-fold increase in Mcl-1 protein levels when incubated with MG132 (). We then analyzed Mcl-1 mRNA expression in the presence of maritoclax over 24 h by qRT-PCR, and determined that Mcl-1 mRNA levels following treatment did not significantly differ from basal levels (ANOVA, P > 0.05).
Figure 1. Maritoclax induces Mcl-1 proteasomal degradation but not transcriptional repression. (A) U937 cells were treated with DMSO or 2.5 μM maritoclax with the indicated concentrations of MG132 for 12 h, and protein expression was analyzed by immunoblotting. (B) U937 cells were treated with DMSO or 2.5 μM maritoclax for 9 h before adding 10 µM MG132 for 3 h, and protein expression was analyzed by immunoblotting. (C) U937 cells were treated with 2.5 µM maritoclax for the indicated times, and MCL1 mRNA expression was analyzed by qRT-PCR.
Maritoclax kills primary human AML cells overexpressing Mcl-1 through Mcl-1 downregulation
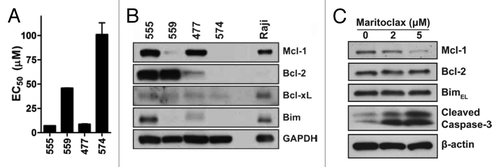
We therefore surveyed the potency of maritoclax treatment in four primary human AML patient samples with varying prognoses (; Table S1). AML samples 555 and 477 were sensitive to maritoclax treatment (EC50 = 7.2 µM, 8.8 µM respectively), while samples 559 and 574 were resistant at EC50’s above 40 µM. Interestingly, when we probed for Bcl-2 family expression in the primary patient samples, maritoclax-sensitive samples 555 and 477 expressed elevated Mcl-1 levels while samples 559 and 574 contained markedly lower Mcl-1 protein levels (). Sensitivity to maritoclax in primary patient samples correlated with the protein levels of Mcl-1, but not with the levels of Bcl-2 or Bcl-xL. We further observed that maritoclax caused the downregulation of Mcl-1, but not that of Bcl-2 or Bim, in a concentration-dependent manner in patient sample 555 leading to induction of caspase-3 cleavage ().
Figure 2. Maritoclax potency correlates with Mcl-1 expression in primary human AML. (A) The EC50 of maritoclax in 4 primary human AML samples were assayed by treating samples with maritoclax over 48 h. Error bars = SD (n = 3). (B) The expression of Bcl-2 family proteins were detected for the same 4 primary human AML samples through immunoblotting, with the Raji Burkitt lymphoma cell line as positive control. (C) Primary human AML case #555 was treated with the indicated concentrations of maritoclax for 24 h, and protein expression was analyzed by immunoblotting.
Maritoclax overcomes Mcl-1-mediated drug resistance in AML cells
Given that maritoclax potency correlated with Mcl-1 protein levels in primary AML patient cells, we surveyed the potency of maritoclax at 48 h in a panel of AML cell lines (). We further observed that parental AML cell lines HL60 and Kasumi-1, which express elevated Mcl-1, were sensitive to maritoclax (EC50 = 2.0 µM, 1.7 µM respectively). On the other hand, parental KG-1 and KG-1a cell lines expressing lower Mcl-1 protein levels were more resistant to maritoclax treatment (EC50 = 6.1 µM, 5.5 µM respectively). The U937 cell line expressed the highest levels of Mcl-1 among tested cell lines, and demonstrated the highest sensitivity to maritoclax treatment (EC50 = 1.4 µM).
Figure 3. Maritoclax induces apoptosis through Mcl-1 degradation in Mcl-1-dependent AML cell lines. (A) The Bcl-2 family protein expression for a number of parental and drug-resistant AML cell lines. (B) The effective concentration for 50% viability (EC50) of parental and drug-resistant AML cell lines in response to ABT-737 and maritoclax treatment. (C) Detection of Mcl-1 degradation and caspase activation by immunoblotting in the HL60/ABTR cell line with 2 µM maritoclax over the indicated time. (D) HL60/ABTR (top) and KG1a/ABTR (bottom) cell lines were treated with a single concentration of maritoclax (2 and 1 µM, respectively) and the indicated concentrations of ABT-737 to measure viability. Error bars = SD (n = 3).
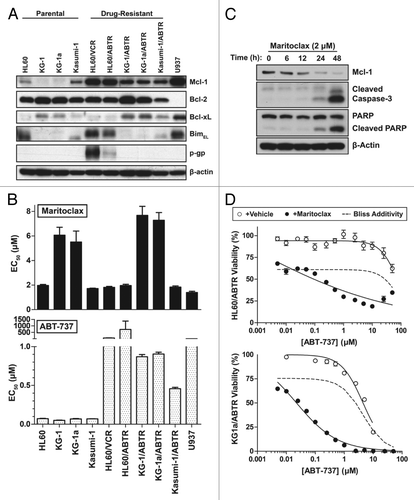
Previous studies indicated that Mcl-1 upregulation is a major mechanism of resistance to selective Bcl-2 inhibitor ABT-737 in cancer cells.Citation10 Therefore, we generated a panel of ABT-737-resistant (ABTR) cell lines through prolonged culture with ABT-737. While parental AML cell lines were very sensitive to ABT-737 treatment, ABTR cell lines demonstrated markedly increased resistance to ABT-737 (). Remarkably, all ABTR phenotypes in the tested AML cell lines demonstrated elevated Mcl-1 levels (). Both HL60/ABTR and Kasumi-1/ABTR cell lines remained sensitive to maritoclax treatment (EC50 = 1.7 µM, 1.8 µM respectively). The KG-1/ABTR and KG-1a/ABTR cell lines demonstrated an increase in resistance to maritoclax (EC50 = 7.7 µM, 7.3 µM respectively), where we also observed Bcl-xL upregulation compared with parental cell lines. The multi-drug resistant HL60/VCR cell line, expressing high levels of p-gp, was resistant to ABT-737 treatment (EC50 > 50 µM), but was sensitive to maritoclax treatment (EC50 = 1.8 µM). We confirmed that maritoclax caused a time-dependent downregulation of Mcl-1 in the HL60/ABTR cells, leading to caspase-3 activation and PARP cleavage, a marker for the activation of caspase-dependent apoptosis (). A number of previous approaches demonstrated that the inhibition of Mcl-1, both pharmacologically by kinase inhibitors and genetically by siRNA approaches, synergized with ABT-737 to sensitize ABTR cells.Citation24,Citation25 Indeed, combining escalating concentrations of ABT-737 with a sub-optimal concentration of maritoclax resulted in significant synergistic interactions, sensitizing both HL60/ABTR and KG1a/ABTR cell lines to cell death (). At higher ABT-737 concentrations in the HL60/ABTR cell line, the addition of maritoclax tended to be additive, which was likely a result of off-target effects.
Maritoclax overcomes stroma-mediated drug resistance in AML cells
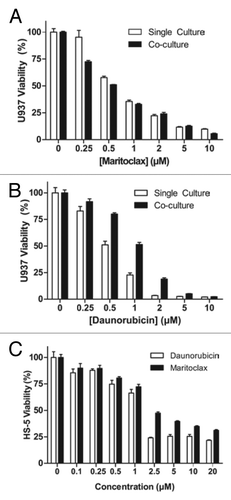
The culture of AML cells with bone marrow stroma mimics the tumor microenvironment, resulting in decreased leukemic proliferation and drug resistance.Citation26 A study has suggested Mcl-1 upregulation as a mechanism of resistance in AML cells cultured with stroma.Citation12 We therefore assayed the effects of maritoclax on leukemic cells co-cultured with bone marrow stroma in a novel system. After allowing U937 cells expressing luciferase (U937-luc) to attach to cultured human stroma cell line HS-5, we treated the cells with maritoclax or daunorubicin. After treatment, we measured luciferase activity to specifically assay the viability of the leukemic cells. While the single-cultured U937-luc cells were sensitive to maritoclax at the EC50 of 0.75 µM, co-cultured cells were slightly more susceptible at the EC50 of 0.56 µM (). On the other hand, co-cultured U937-luc cells were 2-fold more resistant to daunorubicin at the EC50 of 1.01 µM, compared with single-cultured cells at the EC50 of 0.52 µM (). We confirmed that the susceptibility of co-cultured U937-luc cells was not due to toxicity to HS-5 cells, as maritoclax was in fact less toxic for the HS-5 cells compared with daunorubicin at all concentrations tested ().
Figure 4. Maritoclax induces leukemic cell death in co-culture with HS-5 stroma while sparing stroma cells. U937-luc cells were cultured with or without HS-5 stroma cells and treated with maritoclax (A) or daunorubicin (B) for 48 h, and luciferase activity was measured. (C) HS-5 cells were treated with daunorubicin or maritoclax for 48 h, and viability was assayed. Error bars = SD (n = 3).
Maritoclax kills mouse AML cell line C1498 while being less toxic than daunorubicin to primary mouse bone marrow cells
We examined the potency of maritoclax compared with ABT-737 and daunorubicin in the mouse AML cell line C1498. The C1498 cell line was sensitive to daunorubicin and maritoclax treatment at the respective EC50 of 1.86 µM and 2.26 µM, while being more resistant to ABT-737 with an EC50 of 4.66 µM (). We further confirmed that maritoclax induced cell death in the C1498 cell line through Mcl-1 degradation (). Maritoclax was able to induce a concentration-dependent Mcl-1 degradation in C1498 cells to induce caspase-3 cleavage and subsequent PARP cleavage ().
Figure 5. Maritoclax spares bone marrow while inducing apoptosis in mouse AML C1498 through Mcl-1 degradation. (A) Mouse AML cell line C1498 was treated with the indicated concentrations of maritoclax, ABT-737, or daunorubicin to measure cell viability. Error bars = SD (n = 3). (B) C1498 cells were treated with the indicated concentrations of maritoclax or ABT-737 for 24 h, and protein expression was analyzed by immunoblotting. (C) An in vitro culture of primary mouse bone marrow was treated with the indicated concentrations of maritoclax, ABT-737, or daunorubicin for 48 h to measure cell viability. Error bars = SD (n = 3). (D) Primary mouse bone marrow was seeded in methylcellulose medium to allow for progenitor cell growth, and treated with the indicated concentrations of maritoclax or daunorubicin for 7 d, and the CFU-GEMM, CFU-GM, BFU-E, and CFU-E colonies were counted.
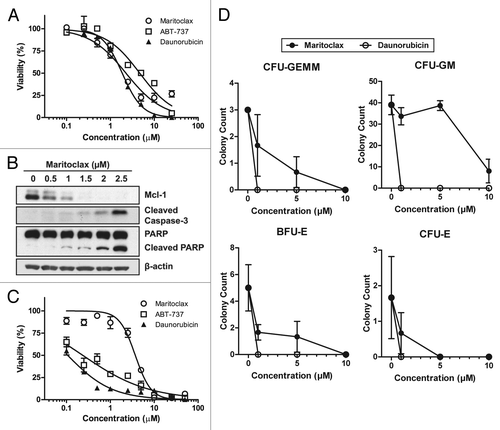
As Mcl-1 has been reported to be necessary for hematopoietic stem and progenitor cell survival,Citation15 we tested whether maritoclax was toxic to mouse bone marrow cells. After treating freshly isolated mouse bone marrow cells with maritoclax, ABT-737, and daunorubicin, we observed that maritoclax was the least toxic to the bone marrow cells in the panel of 3 drugs (). Maritoclax demonstrated more than 10-fold higher EC50 against the primary bone marrow cells at 3.70 µM, compared with ABT-737 and daunorubicin’s EC50 at 0.24 µM and 0.11 µM respectively.
Next, we specifically assayed the relative toxicity of maritoclax and daunorubicin to mouse hematopoietic progenitor cells by seeding mouse primary bone marrow cells to methylcellulose medium supplemented with growth factors and cytokines. In vehicle-treated plates, we could detect the multipotential CFU-GEMM colonies, the more differentiated monocytic/granulocytic CFU-GM colonies, as well as erythroid CFU-E and BFU-E colonies (Fig. S2). After incubating the bone marrow cells with daunorubicin for 7 d, we could not detect any colonies at any of the tested concentrations (). On the other hand, maritoclax was significantly less toxic to these progenitor colonies than daunorubicin. Maritoclax was the least toxic to CFU-GM colonies.
Maritoclax significantly reduces U937 xenograft tumor burden in female athymic nude mice
As maritoclax was less toxic than daunorubicin to primary bone marrow and hematopoietic progenitor cells in vitro, we next determined the toxicity of maritoclax in mice. The MTD and LD50 of maritoclax in female athymic nude mice were 20 mg/kg and 25 mg/kg respectively as once-daily intraperitoneal (IP) injections. At MTD, we occasionally observed temporary somnolence in treated mice lasting 1–2 h.
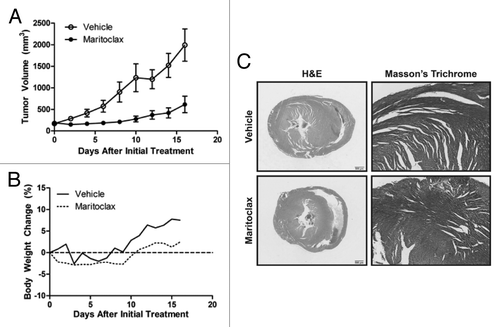
We engrafted U937 tumors to female athymic nude mice, and treated the animals daily with a single IP injection of vehicle or 20 mg/kg maritoclax following tumor staging. A statistically significant decrease in tumor volumes for maritoclax-treated animals was observed starting one day after treatment initiation (Student t test, P < 0.05) (). Maritoclax significantly decreased the U937 xenograft tumor size for the duration of treatment (ANOVA, P < 0.0001). We observed a significant decrease in the weights of the nude mice treated with maritoclax (ANOVA, P < 0.001), which however was not more than 5% of the initial weights of the animals (). We speculate that the decrease in weights of maritoclax-treated animals compared with vehicle control is a result of the corresponding decrease in tumor volumes.
Figure 6. Maritoclax is effective in vivo against U937 xenografts in athymic nude mice. (A) The tumor volumes of nude mice bearing U937 xenograft through daily treatment with vehicle or 20 mg/kg maritoclax. Error bars = SD. (B) The body weight of the same nude mice throughout the course of treatment, expressed as percent change relative to initial treatment. (C) Histological analyses of the heart sections by H&E and Masson trichrome from a representative nude mouse from each treatment arm 16 d after initial treatment.
We continued to observe the tumor burden for 17 d after terminating treatment, allowing the classification of the responses as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). In vehicle-treated mice, 80% of mice were classified as PD, as tumor volumes increased more than 50% compared with tumor volume at staging, and 20% as SD. On the other hand, we observed a 36.4% CR rate with non-palpable tumors and a 22.7% PR rate, demonstrating more than 50% decrease in tumor volume compared with that at staging, for maritoclax-treated animals. Cumulatively, 59.1% of treated tumors were responsive to maritoclax and continued to demonstrate diminished tumor size at 45 d following initial treatment ().
Table 1. Classification of tumor response to maritoclax 17 d following treatment termination
We assayed the effects of maritoclax treatment on peripheral blood counts in the same athymic nude mice bearing U937 xenografts at day 16 of treatment to confirm if maritoclax was toxic to the hematopoietic system. Maritoclax treated animals did not demonstrate significantly depleted blood cell populations as detected through CBC compared to vehicle treated animals (). Two recent reports showed that Mcl-1 depletion leads to rapid and acute cardiac failure.Citation16,Citation17 We examined the heart sections from both treatment arms with Masson trichrome staining in addition to HE. We could not observe any histopathological abnormalities in hearts of maritoclax-treated animals to suggest heart dysfunction (). We further did a complete toxicological assessment of other major organs of the tumor-bearing athymic nude mice from both maritoclax and vehicle treatments at 16 d (n = 3). We were unable to detect any histopathological abnormalities in the liver, kidneys, brain, or spleen (data not shown).
Table 2. The complete blood count (mean ± SEM) from mice treated with vehicle or maritoclax for 16 d
Discussion
The Bcl-2 family proteins are central to the regulation of apoptosis, the dysregulation of which is a hallmark of cancer. ABT-737 and its analogs are extremely potent against Bcl-2-dependent cancers, but lack efficacy toward Mcl-1-overexpressing cancer cells.Citation4 As studies have indicated that Mcl-1 can be necessary for the survival of AML cell populations, it became likely that the small molecule inhibition of Mcl-1 would be a promising strategy for the treatment of AML.Citation5,Citation27
Maritoclax was identified in a natural compound library screen as a novel class of Bcl-2 family inhibitors that selectively induced the proteasomal degradation of Mcl-1.Citation21,Citation22 In this report, we demonstrated that maritoclax induces the proteasomal degradation of Mcl-1 without interfering with its transcription (), unlike previously identified Mcl-1 inhibitors such as flavopiridol or sorafenib.Citation18,Citation20 Maritoclax potency highly correlated with Mcl-1 protein levels in both AML cell lines and primary patient samples, where we observed time- and concentration-dependent Mcl-1 downregulation ( and ). Caspase-3 and PARP cleavage occurred subsequent to apparent Mcl-1 downregulation, suggesting that caspase-dependent apoptosis was activated in response to Mcl-1 degradation (). Indeed, inhibition of maritoclax-induced Mcl-1 degradation by proteasome inhibitor MG132 suppressed caspase activation and apoptosis.Citation21 Therefore, the mechanism of action of maritoclax is distinct from canonical BH3 mimetics or kinase inhibitors. Instead, maritoclax appears to cause the proteasomal degradation of Mcl-1, which in turn may release previously sequestered BH3-only and multi-domain pro-apoptotic Bcl-2 family proteins to activate intrinsic apoptotic pathways.
Eichhorn et al. recently questioned maritoclax as a selective Mcl-1 inhibitor, as the compound did not apparently demonstrate selectivity between HeLa and RS4;11 cells.Citation28 We also observed that maritoclax was not effective in HeLa, HEK293, or MEF cells (Table S2), suggesting that maritoclax sensitivity is cell type specific. Proteasomal Mcl-1 degradation through maritoclax might be mediated by other factors, such as the recruitment of specific E3 ubiquitin ligases. While the overexpression of Mcl-1 in HeLa cells might not have altered sensitivity to maritoclax,Citation28 the co-expression of Mcl-1 and Bim sensitized K562 cells to maritoclax,Citation21 suggesting that Mcl-1 in complex with BH3-only proteins such as Bim might facilitate maritoclax to induce Mcl-1 degradation. Therefore, cells lacking such factors required for maritoclax-mediated proteasomal degradation of Mcl-1 could be resistant to maritoclax treatment. As we have previously determined that maritoclax does not modify the Ser159 phosphorylation status of Mcl-1, phosphorylation-dependent E3 ubiquitin ligases of Mcl-1 such as β-TRCP or FBW7 may not be involved in maritoclax-induced Mcl-1 degradation.Citation29,Citation30 However, the precise mechanism by which maritoclax recruits Mcl-1 to the proteasome remains to be elucidated.
The feasibility of selective Mcl-1 inhibition has been challenged by genetic Mcl-1 knockout models, which demonstrated that Mcl-1 is necessary for hematopoietic stem cell survival and cardiac muscle function.Citation15-Citation17 However, our data indicate that a margin of safety can potentially exist for Mcl-1 inhibition through maritoclax between leukemic cell death and hematopoietic cell toxicity. We observed that maritoclax and daunorubicin were similarly efficacious against the mouse AML cell line C1498 in vitro, but maritoclax was less toxic to an in vitro culture of bone marrow (). Furthermore, maritoclax was significantly less toxic to hematopoietic progenitor cells compared with daunorubicin in the colony formation assay for hematopoietic stem cells (). Given that daunorubicin was approved by the FDA for the treatment of AML, this in vitro evidence suggests that a therapeutic window for maritoclax might exist. Interestingly, maritoclax was the least toxic to CFU-GM, the lineage from which AML originates. As maritoclax may be more selective toward AML than bone marrow cells, AML may indeed become Mcl-1-dependent as they transform.Citation5 In our study in nude mice with U937 xenografts, 20 mg/kg/d maritoclax can reduce leukemic tumor volume without apparent depletion of peripheral blood counts or toxicity to the heart in histopathological analyses (; ). We did not detect a statistically significant depletion of any hematopoietic cell populations in the CBC, suggesting that maritoclax concentrations in the bone marrow did not reach the toxic dose that would affect hematopoietic progenitor cell survival. Therefore, a margin of safety could exist for Mcl-1 inhibition through maritoclax, at least in athymic nude mice.
We demonstrated that maritoclax could overcome drug resistance in AML cells. All ABTR AML cell lines expressed markedly elevated Mcl-1 protein levels compared with their respective parental cell lines (). Maritoclax sensitized these ABTR cells to ABT-737 treatment, synergistically killing resistant AML cell lines (). We also observed that maritoclax potency was not affected by p-gp expression, suggesting that maritoclax is not a substrate for p-gp mediated drug efflux (). Furthermore, we demonstrated that while U937-luc cells cultured with HS-5 stroma are more resistant to daunorubicin, they nonetheless remained sensitive to maritoclax treatment (). U937-luc cells cultured with stroma demonstrated decreased proliferation, which may have caused increased resistance to the chemotherapeutic daunorubicin. However, maritoclax induces Mcl-1 downregulation to activate apoptosis, and may not depend on cell-cycle entry. As a subset of leukemic stem cells are quiescent and resistant to chemotherapy,Citation31 Mcl-1 inhibitors may better target these leukemic stem cells than current chemotherapeutic drugs.
In summary, we showed that maritoclax belongs to a novel class of Bcl-2 family inhibitors that induces the selective degradation of Mcl-1 through the proteasome to kill AML cells that express elevated levels of Mcl-1. We found that maritoclax can overcome stroma-mediated drug resistance and ABT-737 resistance while sparing bone marrow and hematopoietic progenitor cells. We also showed that maritoclax can significantly shrink U937 xenograft tumors without causing apparent toxicity to normal blood cells and the heart. Our studies collectively demonstrated that maritoclax has the potential to be developed as an exciting and promising new therapeutic for the treatment of AML.
Materials and Methods
Antibodies and compounds
Antibodies were obtained from the following sources: hMcl-1Citation32; mMcl-1 (Rockland 600-401-394S); Bcl-2Citation32; Bcl-xL (Sigma B9429); Bim (Sigma B7929); β-actin (Sigma A5441); PARP (Cell Signaling 9542); cleaved caspase-3 (Cell Signaling 9661); GAPDH (Imgenex IMG5019A); p-glycoprotein (Millipore MAB4120). Maritoclax was synthesized as enantiomers or racemic mixture as previously described (maritoclax refers to racemic mixture unless otherwise indicated).Citation33 ABT-737 and daunorubicin hydrochloride were obtained from Abbott Laboratories and Sigma (D8809), respectively.
Cell culture and transfection
All cell lines were obtained from ATCC and maintained per the manufacturer’s recommendations in complete medium with 1% antibiotic/antimycotic solution (Cellgro 30-004-CI) at 37 °C and 5% CO2 unless otherwise specified. Kasumi-1 cells were cultured in RPMI 1640 with 10% FBS for treatment studies. Indicated compounds were added to cells with <1% DMSO. The HL60/ABTR, KG-1/ABTR, KG-1a/ABTR, and Kasumi-1/ABTR cell lines were created through prolonged incubation of the parental cell lines in maintenance medium with escalating concentrations of ABT-737, up to 50 µM. The Mcl-1-IRES-Bim K562 cell line was generated as previously described.Citation21 The MSCV-luciferase-IRES-YFP vector was obtained from Dr Gerard Grosveld (St. Jude Children’s Research Hospital) and transduced to U937 cells as described previouslyCitation34 to generate U937-luc clones. Immunoblotting was done as previously described after cells were lysed in 1% CHAPS or RIPA lysis buffer.Citation21
Real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total mRNA was extracted from 1–5 × 106 U937 cells with TRIzol reagent (Ambion), and a cDNA library was created using the SuperScript III First-Strand Synthesis System (Life Technologies) with Oligo(dT)20 per manufacturers’ recommendations. Real-time polymerase chain reaction (qPCR) was then performed on the Bio-Rad CFX96 Real-time PCR Detection System with the Quantitect SYBR Green PCR Kit (Qiagen 204141) according to the manufacturer’s recommendations in triplicate with specific human MCL1, GAPDH, and ACTB primers, and relative MCL1 mRNA expression was normalized to the geometric mean of GAPDH and ACTB expression. The following specific primers were used: sense 5′-AGAAAGCTGC ATCGAACCAT-3′ and antisense 5′-CCAGCTCCTA CTCCAGCAAC-3′ for human MCL1, sense 5′-GACCCCTTCA TTGACCTCAA CTACATG and antisense 5′-GTCCACCACC CTGTTGCTGT AGCC-3′ for human GAPDH, and sense 5′-CCACCATGTA CCCAGGCATT-3′ and antisense 5′-AGGGTGTAAA ACGCAGCTCA-3′ for human ACTB.
Cell viability assay
Cell viability was determined following 48 h of treatment unless otherwise specified by the indicated compounds by measuring intracellular ATP levels with the CellTiter Glo Luminescent Cell Viability Assay kit (Promega G7571) according to the manufacturer’s recommendations, unless otherwise specified.
Primary human AML
Peripheral blood from AML patients with >60% circulating malignant cells were separated on ficoll-hypaque (specific gravity 1.077). After centrifugation, mononuclear cells were collected from the interface and washed twice by resuspension in PBS. Fresh samples were used for viability assays except case 477, which was frozen in 10% DMSO and 90% FBS prior to rapid thawing at 37 °C followed by resuspension in IMDM media with 10% FBS prior to pelleting, washing, and seeding at 1 × 106 cells/mL in the same media. Frozen cells were used for immunoblot analysis.
In vitro culture of U937 with stroma cells
HS-5 cells were seeded at 4 × 105 cells/mL in maintenance media. After 8 h, U937-luc cells were seeded at 5 × 105 cells/mL, and the medium was changed to 45% DMEM, 45% RPMI 1640, and 10% FBS. After 8 h, cells were treated with the indicated compounds for 48 h. To measure viability, d-luciferin (Gold Biotechnology, LUCK-100) was added, and luminescence was measured according to the manufacturer’s recommendations.
In vitro culture of primary mouse bone marrow
The bone marrow was collected from the femur and tibia of male C57BL/6J mice, and washed twice in IMDM medium. Viable cells by trypan blue staining were seeded at 1 × 106 cells/mL in IMDM medium with 10% FBS, and treated immediately for cell viability assay.
Primary mouse bone marrow colony formation assay
The bone marrow was collected from the femur and tibia of female C57BL/6J mice, washed twice in IMDM medium, and viable cells by trypan blue staining were seeded at 2 × 105 cells/mL in methylcellulose medium with recombinant cytokines and EPO (MethoCult GF M3434, StemCell Technologies) according to the manufacturer’s recommendations in the presence of DMSO, maritoclax, or daunorubicin (<0.25% DMSO) for 7 d before counting and classifying colony forming unit (CFU) and blast forming unit (BFU) lineages as granulocyte/erythroid/megakaryocyte/macrophage (CFU-GEMM), granulocyte/macrophage (CFU-GM), blast-forming erythroid (BFU-E), and colony-forming erythroid (CFU-E).
Animal models
Female athymic nude (NCI Athymic NCr-nu/nu 01B74) mice were obtained from Jackson Laboratories. Four 6-wk-old mice were injected with PTDCitation7 with <3% DMSO with or without maritoclax intraperitoneally (IP) to determine the maximum tolerated dose (MTD), defined as the maximum dose of maritoclax that the animals received without causing mortality or greater than 10% loss in body weight.
For the U937 xenograft model, 6-wk-old female athymic nude mice were used. In total, 31 mice were subcutaneously transplanted with 5 × 106 U937 cells in a 200 µL solution of PBS with 50% BD Matrigel (BD Biosciences) on both flanks. Tumor volumes were measured by electronic caliper and calculated by the formula: volume = length × width2 / 2. Mice bearing tumors at 150–200mm3 volume were randomly assigned to control (100 µL PTD) or maritoclax (20 mg/kg in 100 µL PTD) treatment, with treatment beginning the same day of staging. The weight and tumor sizes were measured each day immediately prior to treatment. Sixteen days following initial treatment, 6 tumor-bearing mice from maritoclax-treated and 7 tumor-bearing mice from vehicle-treated mice were sacrificed for whole blood collection under CO2 anesthesia for complete blood count (CBC) analysis, and major organs (brain, heart, lungs, liver, kidneys, and spleen) were fixed in 10% formalin for 48 h and transferred to 70% ethanol. Assessment of mice toxicology was performed in hematoxylin and eosin (HE) and Masson trichrome-stained sections of these organs by an experienced animal pathologist, and pictures of histological sections were taken with the Olympus BX51 microscope and DP71 digital camera using cellSens Standard 1.6 imaging software (Olympus America). Treatment continued for the remaining 7 control and 11 maritoclax-treated mice until 28 d after initial treatment, when treatment stopped and tumor volumes were observed until 45 d after initial treatment. Tumors were then classified: complete remission (CR) as not palpable, partial remission (PR) as more than 50% decrease in tumor size from initial volume, stable disease (SD) as tumor sizes between PR and progressive disease (PD), and PD as greater than 50% increase in tumor size from initial volume.
Statistics
All statistical analyses were performed using GraphPad Prism version 5.00 for Windows (GraphPad Software, www.graphpad.com). EC50 calculations for viability were calculated through nonlinear regression with normalized data assuming variable slope. The Bliss model of independence was used for the determination of synergy, where the expected additive effect at a given concentrations of drug A and B (EAB) was the sum of the individual effects minus the product of their effect (EAB = EA + EB − EAEB).Citation35
| Abbreviations: | ||
| ABTR | = | ABT-737 resistant |
| AML | = | acute myeloid leukemia |
| ANOVA | = | analysis of variance |
| Bcl-2 | = | B-cell lymphoma-2 |
| BFU-E | = | blast forming unit-erythroid |
| CBC | = | complete blood count |
| CFU-E | = | colony forming unit-erythroid |
| CFU-GEMM | = | colony forming unit-granulocytic/erythroid/monocytic/megakaryocytic |
| CFU-GM | = | colony forming unit-granulocytic/monocytic |
| EC50 | = | half maximal effective concentration |
| FLT3-ITD | = | FMS-like tyrosine kinase-3-internal tandem duplications |
| HCT | = | hematocrit |
| HE | = | hematoxylin and eosin |
| Hgb | = | hemoglobin |
| IP | = | intraperitoneal |
| LD50 | = | median lethal dose |
| MCH | = | mean corpuscular hemoglobin |
| MCHC | = | mean corpuscular hemoglobin concentration |
| Mcl-1 | = | myeloid cell leukemia sequence-1 |
| MCV | = | mean corpuscular volume |
| MTD | = | maximum tolerated dose |
| PARP | = | poly ADP ribose polymerase |
| p-gp | = | p-glycoprotein |
| qRT-PCR | = | quantitative real-time polymerase chain reaction |
| RBC | = | red blood cells |
| WBC | = | white blood cells |
Additional material
Download Zip (287.5 KB)Disclosures of Conflict of Interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Su-Fern Tan and Dan Zhang from Dr Thomas Loughran’s lab for graciously providing reagents, cell lines, and antibodies. We thank Dr Timothy K Cooper for the complete histological examination of experimental mice. We also thank Dr Gerard Grosveld for graciously providing the MSCV-Luciferase-IRES-YFP construct. This work was supported by the Lois High Berstler Endowment Fund and the Four Diamonds Fund of the Pennsylvania State University College of Medicine.
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11 - 30; http://dx.doi.org/10.3322/caac.21166; PMID: 23335087
- O’Donnell MR, Abboud CN, Altman J, Appelbaum FR, Arber DA, Attar E, Borate U, Coutre SE, Damon LE, Goorha S, et al. Acute myeloid leukemia. J Natl Compr Canc Netw 2012; 10:984 - 1021; PMID: 22878824
- Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, Reed JC. Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse. Blood 1998; 91:991 - 1000; PMID: 9446661
- Liu Q, Wang H-G. Anti-cancer drug discovery and development: Bcl-2 family small molecule inhibitors. Commun Integr Biol 2012; 5:557 - 65; http://dx.doi.org/10.4161/cib.21554; PMID: 23336025
- Glaser SP, Lee EF, Trounson E, Bouillet P, Wei A, Fairlie WD, Izon DJ, Zuber J, Rappaport AR, Herold MJ, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev 2012; 26:120 - 5; http://dx.doi.org/10.1101/gad.182980.111; PMID: 22279045
- Yoshimoto G, Miyamoto T, Jabbarzadeh-Tabrizi S, Iino T, Rocnik JL, Kikushige Y, Mori Y, Shima T, Iwasaki H, Takenaka K, et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009; 114:5034 - 43; http://dx.doi.org/10.1182/blood-2008-12-196055; PMID: 19808698
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005; 435:677 - 81; http://dx.doi.org/10.1038/nature03579; PMID: 15902208
- Gandhi L, Camidge DR, Ribeiro de Oliveira M, Bonomi P, Gandara D, Khaira D, Hann CL, McKeegan EM, Litvinovich E, Hemken PM, et al. Phase I study of Navitoclax (ABT-263), a novel Bcl-2 family inhibitor, in patients with small-cell lung cancer and other solid tumors. J Clin Oncol 2011; 29:909 - 16; http://dx.doi.org/10.1200/JCO.2010.31.6208; PMID: 21282543
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 2013; 19:202 - 8; http://dx.doi.org/10.1038/nm.3048; PMID: 23291630
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi Y-X, Sneed T, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 2006; 10:375 - 88; http://dx.doi.org/10.1016/j.ccr.2006.10.006; PMID: 17097560
- Hermanson DL, Das SG, Li Y, Xing C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIβ downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Mol Pharmacol 2013; 84:236 - 43; http://dx.doi.org/10.1124/mol.113.086140; PMID: 23696245
- Balakrishnan K, Burger JA, Wierda WG, Gandhi V. AT-101 induces apoptosis in CLL B cells and overcomes stromal cell-mediated Mcl-1 induction and drug resistance. Blood 2009; 113:149 - 53; http://dx.doi.org/10.1182/blood-2008-02-138560; PMID: 18836097
- Aliabadi HM, Mahdipoor P, Uludağ H. Polymeric delivery of siRNA for dual silencing of Mcl-1 and P-glycoprotein and apoptosis induction in drug-resistant breast cancer cells. Cancer Gene Ther 2013; 20:169 - 77; http://dx.doi.org/10.1038/cgt.2013.8; PMID: 23449477
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev 2000; 14:23 - 7; PMID: 10640272
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307:1101 - 4; http://dx.doi.org/10.1126/science.1106114; PMID: 15718471
- Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, Schuetz JD, Rehg JE, Opferman JT. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 2013; 27:1351 - 64; http://dx.doi.org/10.1101/gad.215855.113; PMID: 23788622
- Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, Fischer KM, Sussman MA, Miyamoto S, Gustafsson AB. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes Dev 2013; 27:1365 - 77; http://dx.doi.org/10.1101/gad.215871.113; PMID: 23788623
- Rahmani M, Davis EM, Bauer C, Dent P, Grant S. Apoptosis induced by the kinase inhibitor BAY 43-9006 in human leukemia cells involves down-regulation of Mcl-1 through inhibition of translation. J Biol Chem 2005; 280:35217 - 27; http://dx.doi.org/10.1074/jbc.M506551200; PMID: 16109713
- Kasper S, Breitenbuecher F, Heidel F, Hoffarth S, Markova B, Schuler M, Fischer T. Targeting MCL-1 sensitizes FLT3-ITD-positive leukemias to cytotoxic therapies. Blood Cancer J 2012; 2:e60; http://dx.doi.org/10.1038/bcj.2012.5; PMID: 22829255
- Ma Y, Cress WD, Haura EB. Flavopiridol-induced apoptosis is mediated through up-regulation of E2F1 and repression of Mcl-1. Mol Cancer Ther 2003; 2:73 - 81; PMID: 12533675
- Doi K, Li R, Sung S-S, Wu H, Liu Y, Manieri W, Krishnegowda G, Awwad A, Dewey A, Liu X, et al. Discovery of marinopyrrole A (maritoclax) as a selective Mcl-1 antagonist that overcomes ABT-737 resistance by binding to and targeting Mcl-1 for proteasomal degradation. J Biol Chem 2012; 287:10224 - 35; http://dx.doi.org/10.1074/jbc.M111.334532; PMID: 22311987
- Pandey MK, Gowda K, Doi K, Sharma AK, Wang H-G, Amin S. Proteasomal degradation of Mcl-1 by maritoclax induces apoptosis and enhances the efficacy of ABT-737 in melanoma cells. PLoS One 2013; 8:e78570; http://dx.doi.org/10.1371/journal.pone.0078570; PMID: 24223823
- Hughes CC, Prieto-Davo A, Jensen PR, Fenical W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org Lett 2008; 10:629 - 31; http://dx.doi.org/10.1021/ol702952n; PMID: 18205372
- Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res 2007; 67:782 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-06-3964; PMID: 17234790
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010; 115:3304 - 13; http://dx.doi.org/10.1182/blood-2009-07-233304; PMID: 20197552
- Garrido SM, Appelbaum FR, Willman CL, Banker DE. Acute myeloid leukemia cells are protected from spontaneous and drug-induced apoptosis by direct contact with a human bone marrow stromal cell line (HS-5). Exp Hematol 2001; 29:448 - 57; http://dx.doi.org/10.1016/S0301-472X(01)00612-9; PMID: 11301185
- Gores GJ, Kaufmann SH. Selectively targeting Mcl-1 for the treatment of acute myelogenous leukemia and solid tumors. Genes Dev 2012; 26:305 - 11; http://dx.doi.org/10.1101/gad.186189.111; PMID: 22345513
- Eichhorn JM, Alford SE, Hughes CC, Fenical W, Chambers TC. Purported Mcl-1 inhibitor marinopyrrole A fails to show selective cytotoxicity for Mcl-1-dependent cell lines. Cell Death Dis 2013; 4:e880; http://dx.doi.org/10.1038/cddis.2013.411; PMID: 24157874
- Ding Q, He X, Hsu J-M, Xia W, Chen C-T, Li L-Y, Lee D-F, Liu J-C, Zhong Q, Wang X, et al. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007; 27:4006 - 17; http://dx.doi.org/10.1128/MCB.00620-06; PMID: 17387146
- Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, et al. SCF(FBW7) regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011; 471:104 - 9; http://dx.doi.org/10.1038/nature09732; PMID: 21368833
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol 2007; 25:1315 - 21; http://dx.doi.org/10.1038/nbt1350; PMID: 17952057
- Krajewski S, Bodrug S, Gascoyne R, Berean K, Krajewska M, Reed JC. Immunohistochemical analysis of Mcl-1 and Bcl-2 proteins in normal and neoplastic lymph nodes. Am J Pathol 1994; 145:515 - 25; PMID: 8080035
- Cheng C, Pan L, Chen Y, Song H, Qin Y, Li R. Total synthesis of (+/-)-marinopyrrole A and its library as potential antibiotic and anticancer agents. J Comb Chem 2010; 12:541 - 7; http://dx.doi.org/10.1021/cc100052j; PMID: 20429575
- Woods NT, Yamaguchi H, Lee FY, Bhalla KN, Wang H-G. Anoikis, initiated by Mcl-1 degradation and Bim induction, is deregulated during oncogenesis. Cancer Res 2007; 67:10744 - 52; http://dx.doi.org/10.1158/0008-5472.CAN-07-3148; PMID: 18006817
- Bliss CI. The calculation of microbial assays. Bacteriol Rev 1956; 20:243 - 58; PMID: 13403845