
Abstract
Restriction of nutrients and oxygen in the tumor microenvironment disrupts ER homeostasis and adaptation to such stress is mediated by the key UPR effector PERK. Given its pro-tumorigenic activity, significant efforts have been made to elucidate the molecular mechanisms that underlie PERK function. Chemical-genetic approaches have recently proven instrumental in pathway mapping and interrogating kinase function. To enable a detailed study of PERK signaling we have generated an analog-sensitive PERK allele that accepts N6-alkylated ATP analogs. We find that this allele can be regulated by bulky ATP-competitive inhibitors, confirming the identity of the PERK gatekeeper residue as methionine 886. Furthermore, this analog-sensitive allele can be used to specifically label substrates with thiophosphate both in vitro and in cells. These data highlight the potential for using chemical-genetic techniques to identify novel PERK substrates, thereby providing an expanded view of PERK function and further definition of its signaling networks.
Introduction
The tumor microenvironment is characterized by limitation of nutrients and oxygen, both of which are necessary for tumor expansion. These conditions arise from the combination of increased demand triggered by rapidly proliferating tumor cells and a compromised vasculature in the surrounding tissue. One consequence of this environmental challenge is a reduced capacity for tumor cells to properly fold secretory proteins. The accumulating misfolded proteins trigger a cell adaptive response termed the unfolded protein response (UPR). The pro-survival nature of this response is co-opted by cancer cells, allowing them to survive the harsh tumor microenvironment.Citation1
The UPR is mediated by three primary signal transducers: ATF6, Ire1, and PERK. These three factors are embedded in the ER membrane and act as ER stress sensors. Under homeostatic conditions, ATF6, Ire1, and PERK are bound by the ER luminal chaperone GRP78/BiP, preventing their activation.Citation2,Citation3 However, under conditions that result in the accumulation of unfolded proteins in the ER, the demand for BiP chaperone function increases, triggering its release from UPR signal transducers. Release from BiP/GRP78 permits the oligomerization and activation of Ire1 and PERK, and migration of ATF6 to the trans-Golgi where it is proteolytically processed.Citation2,Citation3 The initial stages of this response are stress adaptive. To this end, activation of Ire1 and ATF6 upregulate ER chaperones to facilitate protein folding. Activation of PERK triggers eIF2α phosphorylation, which inhibits protein synthesis, thus lowering the protein load.Citation4,Citation5 In addition, eIF2α phosphorylation contributes to cell cycle arrest through inhibiting cyclin D1 translation; cell cycle arrest reduces cellular biosynthetic needs and thereby contributes to ER stress resolution.Citation6 PERK signaling also selectively upregulates certain proteins, like the transcription factor ATF4. ATF4 regulates the expression of pro-survival genes that encode detoxifying enzymes as well as ER chaperones and foldases.Citation7,Citation8 Paradoxically, ATF4 also increases expression of the pro-apoptotic transcription factor GADD153/CHOP after prolonged or acute ER stress, setting the stage for cell death.Citation9
Consistent with the idea that the pro-survival effects of the UPR are important for tumor cell survival, key UPR mediators have been implicated in promoting tumorigenesis.Citation8,Citation10 Among these is the primary UPR effector PERK, which has been characterized for its role in tumor growth, cell migration, metastasis, angiogenesis, survival of ECM-detached cells, and the epithelial–mesenchymal transition.Citation11-Citation17 In light of these pro-tumorigenic effects, there has been significant interest in developing cancer therapeutics that target PERK activity, and in defining the molecular mechanisms that underlie PERK function.Citation18,Citation19
To further interrogate PERK-signaling networks, we have generated an analog-sensitive PERK allele that specifically binds N6-alkylated ATP analogs. This involves mutation of a conserved residue in the ATP-binding pocket known as the “gatekeeper”.Citation20,Citation21 The gatekeeper residue blocks access to a deeper hydrophobic pocket; when mutated to a smaller amino acid, the resultant enlarged pocket specifically binds ATP analogs modified with a bulky alkyl group. Here, we show that analog-sensitive PERK alleles are specifically inhibited by bulky ATP-competitive analogs, and that PERK M886A can utilize N6-alkylated ATPγS to label substrate both in vitro and in the context of permeabilized cells. These data highlight a strong potential for use of PERK M886A in network mapping.
Results
Methionine 886 is the gatekeeper residue
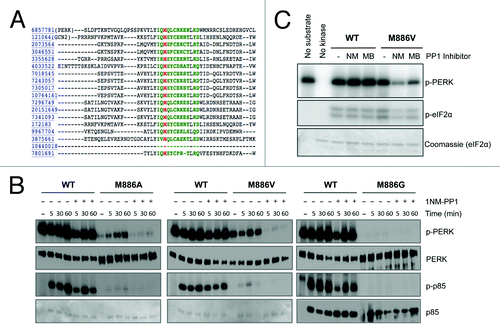
The gatekeeper residue for most protein kinases can be predicted through primary sequence alignment with related family members.Citation22 To identify the gatekeeper residue for PERK, this alignment was performed through the Kinase Sequence Database.Citation23 Conserved residues contacting ATP are represented in green, with the predicted residue for conferring analog sensitivity highlighted in red (). The predicted gatekeeper residue for PERK is methionine 886. To generate an analog-sensitive PERK allele, methionine 886 was mutated to three distinct small hydrophobic residues: glycine, alanine, and valine. To test whether M886 functions as the gatekeeper residue, recombinant wild-type and mutant kinases were purified and used for radiometric in vitro kinase assays (). The p85 subunit of PI3K was used as substrate, as unpublished work from our lab revealed it to be a direct PERK substrate in vitro. We found that the three mutant PERK alleles retained varying degrees of kinase activity. Mutation of M886 to glycine severely crippled PERK activity; therefore, this mutant was not used for further study. M886V retained the highest level of kinase activity, and was thus considered the strongest candidate. In the presence of the bulky ATP-competitive inhibitor 1-NM-PP1, wild-type kinase activity was unaffected, while all mutant alleles were inhibited both in terms of PERK autophosphorylation and phosphorylation of p85 substrate. This suggests that mutation of M886 to a smaller amino acid opened an affinity pocket that allowed the bulky ATP analog to bind, confirming M886 as the gatekeeper residue. These results were verified for M886V by additional radiometric assays in which recombinant PERK was incubated with eIF2α substrate in the presence or absence of 1-NM-PP1 or another bulky ATP-competitive analog, 3-MB-PP1 ().
Figure 1. The gatekeeper residue for PERK is methionine 886. (A) Sequence alignment of the PERK ATP-binding pocket with related kinases predicts the conserved gatekeeper residue, shown in red. (B) PP1 inhibitors inhibit the gatekeeper mutant in vitro. Recombinant WT PERK-ΔN and PERK-ΔN M886V (left) or M886A (right) were pre-incubated with 1-NM-PP1 (NM) or 3-MB-PP1 (MB). Kinase was then incubated with recombinant eIF2α in the presence of γ-32P-ATP. Reactions were run on an SDS-PAGE gel and exposed to film. (C) Recombinant WT PERK-ΔN or PERK-ΔN M886G/A/V were pre-incubated with 1-NM-PP1. Kinase was then incubated alone (−) or with p85 substrate in the presence of γ-32P-ATP. Reactions were run on an SDS-PAGE gel and exposed to film. Western blots were performed against total levels to confirm equal loading (Ponceau stains for p85 shown for M886A, M886V).
PERK M886V and M886A are analog-sensitive in the context of cells
To assess kinase activity and our ability to regulate PERK gatekeeper mutants in vivo, PERK−/− embryonic fibroblasts were transduced with retrovirus encoding wild-type PERK, PERK M886A, or PERK M886V. Stably transduced cell lines were subsequently challenged with thapsigargin to induce ER stress, in the presence or absence of the indicated doses of 3-MB-PP1. Consistent with in vitro results, the bulky ATP analog did not affect wild type PERK autophosphorylation or phosphorylation of eIF2α at any concentration tested (). PERK M886V exhibited sensitivity to 3-MB-PP1 at concentrations between 20 and 40 μM, with maximal inhibition at 100 μM (). Of note, though this concentration did not affect wild-type activity, it is well above the concentration of PP1 inhibitor generally used in cells.Citation24-Citation26 In contrast, PERK M886A exhibited maximal sensitivity at significantly lower concentrations, suggesting that the alanine mutation permits more efficient analog binding (; data not shown).
Figure 2. Analog-sensitive PERK alleles are functional in the context of cells. (A) 3-MB-PP1 inhibits PERK M886V. PERK−/− mouse fibroblasts expressing PERK WT or M886V were pre-treated with the indicated doses of 3-MB-PP1, then challenged with thapsigargin (TG) for 1 h. PERK activation was assessed by western blot for PERK and eIF2α phosphorylation. (B) 3-MB-PP1 inhibits PERK M886A. PERK−/− mouse fibroblasts expressing PERK WT or M886A were treated as described in (A). (C) Cells were pre-treated with the indicated doses of 3-MB-PP1, then challenged with TG for 4 h to induce ATF4 and CHOP expression. Cytoplasmic fractions were probed for PERK and p-eIF2α, with total eIF2α to control for equal loading. Nuclear fractions were probed for ATF4 and CHOP, with lamin B as a loading control.
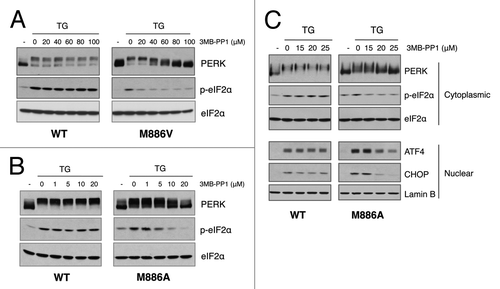
To further characterize PERK M886A, effects on downstream signaling were assessed following treatment of cells with thapsigargin for a more extended period to induce ATF4 and CHOP expression. 3-MB-PP1 inhibited PERK M886A and eIF2α phosphorylation as well as ATF4 and CHOP induction, while having no influence on wild type activity (). Collectively, these data demonstrate that methionine 886 is the PERK gatekeeper residue. Moreover, these studies suggest that mutation of M886 to alanine allows the highest binding affinity for PP1 inhibitors while retaining adequate kinase activity.
3-MB-PP1 inhibits PERK activity, but exhibits previously uncharacterized off-target effects
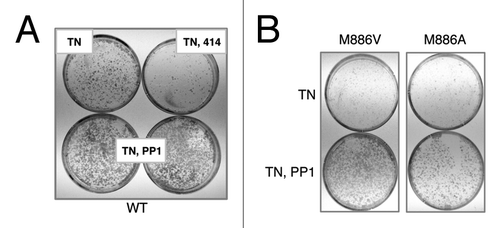
PERK is essential for cell survival following acute ER stress;Citation27 therefore, we asked whether 3-MB-PP1 treatment impaired survival in cells expressing the PERK gatekeeper mutants. To address this, acute tunicamycin treatment was delivered in the presence or absence of 3-MB-PP1. Cells were also treated with the commercially available PERK inhibitor GSK2606414Citation19 as a positive control. The outgrowth of colonies following ER stress was visualized by Giemsa staining. As anticipated, the control PERK inhibitor suppressed colony outgrowth, however, all cells regardless of genotype exhibited increased survival with 3-MB-PP1 treatment (; data not shown). Given that PP1 inhibitors are known to be highly selective,Citation21 this off-target effect was surprising. Though this precludes further study of PERK function with 3-MB-PP1, the in vitro assays served to confirm the gatekeeper mutant and spur an effort to determine whether PERK M886A could be used as a tool for pathway mapping.
Figure 3. 3-MB-PP1 rescues tunicamycin-sensitivity in wildtype cells, demonstrating an off-target effect. (A) Immortalized PERK−/− mouse fibroblasts stably expressing wild-type PERK were pre-treated with 10 μM (bottom, left) or 15 μM (bottom, right) 3-MB-PP1 for 1 h, or with GSK2606414 as a control. Cells were then acutely stressed with tunicamycin for 30 min and allowed to grow for 6 d. Colonies were stained with Giemsa. (B) Cells of the indicated genotypes were pre-treated with 10 μM 3-MB-PP1 for 1 h, then treated as described in (A).
PERK M886A can utilize bulky ATP analogs to label substrates in vitro
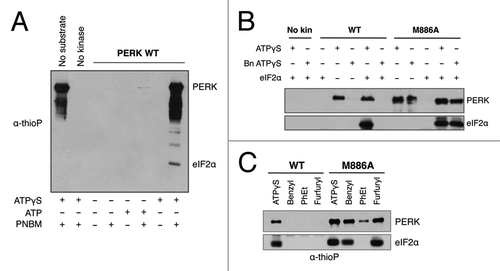
Chemical-genetic techniques have also been employed to map signaling networks through use of an analog-sensitive kinase that can specifically label substrates.Citation28-Citation31 With this approach, the analog-sensitive kinase uses N6-alkylated ATPγS to transfer a thiophosphoryl group to its substrates. The bulky group again confers specificity for the analog-sensitive kinase, and the thiophosphoryl group acts as a label for distinguishing direct substrates from all other phosphorylated proteins in the cell. To determine whether PERK was able to use ATPγS as a phosphodonor, recombinant wild-type PERK was incubated with eIF2α in the presence or absence of ATPγS. Reactions were alkylated and probed by western blot with an anti-thiophosphate ester antibody that recognizes alkylated, thiophosphorylated substrate ().Citation29,Citation30 PERK and eIF2α phosphorylation could be detected only in the presence of the ATPγS and the alkylating agent, PNBM, demonstrating not only that PERK can use ATPγS but that the thioP antibody is specific in this system. We then asked whether the M886A mutant could specifically use a bulky ATPγS analog to thiosphorylate substrate. In vitro kinase assays demonstrated that indeed, the gatekeeper mutant could use bulky and non-bulky ATPγS to label eIF2α, while wild type PERK could only use the non-bulky analog (). Furthermore, a comparison of various bulky analogs established N6-furfuryl ATPγS as the preferred phosphodonor ().
Figure 4. PERK M886A can utilize bulky ATPγS to thiophosphorylate substrate in vitro. (A) The thioP antibody specifically recognizes alkylated, thiophosphorylated PERK substrate. Recombinant WT PERK-ΔN was incubated with eIF2α in the presence or absence of 1mM ATPγS or ATP. Samples were alkylated (PNBM) and assessed by western blot for thiophosphorylated protein using an anti-thiophosphate ester antibody (α-thioP). (B) Only PERK M886A can use bulky ATPγS to thiophosphorylate substrate in vitro. Recombinant WT or M886A PERK-ΔN was incubated with eIF2α in the presence of ATPγS or benzyl (Bn) ATPγS. Reactions were alkylated, then and probed by western blot with the thioP antibody. (C) PERK M886A prefers N6-furfuryl ATPγS. Recombinant WT or M886A PERK-ΔN were used in kinase assays, as described in (B) with the inclusion of the indicated bulky ATPγS analogs.
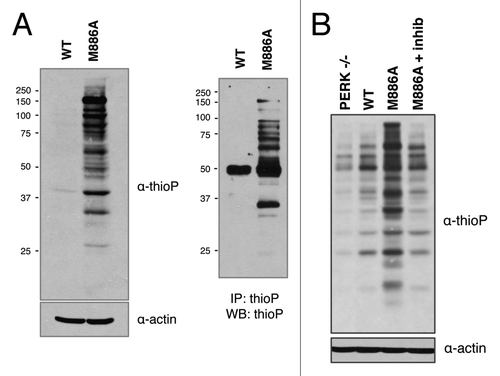
M886A thiophosphorylates substrate in permeabilized cells
Maintaining a kinase in its appropriate subcellular compartment is arguably the most desirable context for substrate labeling and identification. Therefore, we asked whether PERK could thiophosphorylate substrate under near-physiological conditions. This was addressed by gently permeabilizing cells expressing PERK wild-type or M886A with a low concentration of digitonin. Cells were then incubated with thapsigargin in the presence of N6-furfuryl ATPγS for in vivo substrate labeling. Analysis of whole cell lysates by western blot revealed increased thiosphorylation in the M886A mutant as compared with wild type (). Moreover, when labeled lysates were subjected to immunoprecipitation with the thioP antibody, increased labeling was again detected for M886A in enriched samples. To verify that the increase in signal was in fact dependent upon PERK kinase activity, cells were pre-treated with GSK2606414 prior to labeling. PERK inhibition decreased thiophosphorylation in the M886A-labeled sample to background levels, as seen in the PERK−/− and wild-type lanes (). Together, these data reveal a strong potential for PERK M886A in screening for novel substrates.
Figure 5. PERK M886A thiophosphorylates substrate in permeabilized cells. (A) PERK−/− cells expressing either PERK WT or M886A were permeabilized, then incubated with thapsigargin and NCitation6-furfuryl ATPγS for substrate labeling. Cells were lysed, and lysates alkylated. Lysates were probed by western blot for total thiophosphorylated substrate (left). Alkylated lysates were then immunoprecipitated with α-thioP (right). (B) Cells expressing M886A were pre-treated with GSK2606414 inhibitor as a control for PERK-dependent activity. The indicated cell types were then treated as described in (A).
Discussion
Protein kinases are important regulators of normal and tumor cell biology. Analysis of their functional properties in the context of disease is of vital importance. From a therapeutic standpoint, this is reflected in an intense focus on kinase inhibitor development, and in the array of kinase inhibitors currently in clinical trial.Citation32 Despite the demand for kinome profiling and kinase characterization, this area of study has proven challenging. When employing the use of small molecule inhibitors to study kinase function, the fact that most inhibitors often target the conserved ATP-binding pocket presents a specificity issue. On the other hand, genetic manipulations (i.e., kinase deletion) achieve specificity in that only the kinase of interest is targeted. This method, however, has the drawback of depleting the kinase for an extended period, during which compensatory mechanisms and indirect downstream effects are often confounding. This method also cannot distinguish between effects due to loss of kinase activity and those due to loss of the entire protein. Chemical-genetic techniques have recently enabled significant advances in this field. Using a system whereby the kinase of interest is genetically altered to selectively bind a bulky inhibitor analog has offered benefits of both transience and specificity. As testament to the appeal of this method, analog-sensitive alleles have been generated for at least 85 kinases to date.Citation33
To assess PERK function and identify novel signaling branches, we report an analog-sensitive PERK allele, M886A, that is inhibited by the bulky ATP-competitive analog 3-MB-PP1 both in vitro and in vivo. These results confirm methionine 886 as the gatekeeper residue for PERK. It is interesting to note, however, that our characterization of cellular responses to 3-MB-PP1 suggest an off-target activity of pyrazolo(3,4-d)pyrimidine (PP) inhibitors that has not been previously reported. Not only did the PP1 inhibitor enhance survival of the M886V and M886A mutants, which was the exact opposite of its anticipated effect, it also increased survival of cells expressing wild type PERK. Subsequent experiments performed by pulsing in 3-MB-PP1 at an even lower dose (5 μM) recapitulated this result (data not shown). This could reflect inhibition of a stress induced pro-apoptotic pathway, or possibly activation of another UPR branch. Although we have not explored alternative inhibitors for use with M886A, several possibilities do exist for reducing off-target effects. Recent work from the Shokat lab proposed mutating the gatekeeper residue to cysteine for use with electrophilic inhibitors, a panel of which were screened for potency and specificity.Citation34 These, in addition to a new set of inhibitors with even higher potency and selectivityCitation33 would provide a solid platform from which to launch such a study.
In terms of using the analog-sensitive version of PERK for novel substrate identification, the work presented here will be instrumental. To date, PERK substrate mapping has only been attempted via yeast two-hybrid screening.Citation35 This study identified the antioxidant response factor Nrf2 as a direct PERK substrate, one of only three that have been identified thus far. We have shown that PERK M886A can label substrates with a unique thiophosphoryl tag, both in vitro and in permeabilized cells. This tag can subsequently be used for purification and mass spectrometry-based substrate identification. Moreover, the conditions have been optimized to allow substrate labeling in gently permeabilized cells, which should maintain PERK in its native subcellular compartment. For screening purposes, this should reduce the number of false positives. In vivo labeling techniques have recently been used to successfully identify novel substrates of Erk2 and AMPK,Citation28,Citation30 giving us confidence in identifying PERK substrates in a similar fashion. Such a screen would provide significant information for understanding PERK signaling from a purely biological standpoint, and also may provide additional mechanistic data to consider for PERK inhibitor development in the treatment of cancer.
Materials and Methods
Cell culture and treatments
Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 4 mM l-glutamine, 55 mM β-mercaptoethanol, and nonessential amino acids. To induce ER stress, cells were treated with 500 nM thapsigargin (Sigma T9033) or 2.5 ug/ml tunicamycin (Sigma T7765) for the indicated times. For PERK inhibition, cells were pre-treated with 1 μM GSK2606414Citation19 or the indicated concentrations of 3-MB-PP1 (EMD Millipore 529582) for 1 h.
Retroviral vectors and stable cell lines
Retroviral vectors for expression of PERK M886A and M886V were generated by QuikChange site-directed mutagenesis (Agilent 200521) of Myc-tagged full-length mPERK in pBabe-puro. Stable cell lines were generated by transducing PERK−/− mouse embryonic fibroblastsCitation36 with retrovirus carrying these constructs. Cells were selected with and maintained in 5 ug/ml puromycin.
Antibodies and immunoblotting
Cells were lysed in EBC buffer (50 mM Tris pH 8.0, 120 mM NaCl, 0.5% NP-40) or RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1.0% NP-40, 0.1% SDS) supplemented with protease and phosphatase inhibitors. The following antibodies were used for immunoblotting: anti-PERK (Cell Signaling 3192), anti-phosphoserine 51 of eIF2α (Cell Signaling 3597), anti-CHOP (Cell Signaling 2895), anti-eIF2α (Invitrogen AHO0802), anti-ATF4 (Santa Cruz Biotechnology sc-200), anti-lamin B (Santa Cruz Biotechnology sc-6216), and anti-β actin (Sigma A5441). The thioP antibody has been previously describedCitation29,Citation30 and was available from Epitomics/Abcam (ab92570).
Radiometric in vitro kinase assay
PERK M886G, M886A, and M886V were generated by QuikChange site-directed mutagenesis (Agilent 200521) of GST-tagged PERK-ΔN in pGEX.Citation37 Recombinant PERK was expressed in BL21 bacterial cells and purified using GST Purification columns (Clontech 635619). HIS-tagged eIF2α in pET15b was expressed in BL21(DE3) cells and purified on HisTALON gravity columns (Clontech 635654). Recombinant p85 was from SignalChem (P31–30H). PERK was pre-treated with indicated concentrations of 1-NM-PP1 (EMD Millipore 529581) or 3-MB-PP1 (EMD Millipore 529582) for 30 min. PERK was then incubated with recombinant substrate in the presence of γ-32P-ATP for an additional 30 min at 30 °C. Reactions were run on an SDS-PAGE gel, Coomassie-stained for loading, and exposed to film.
ATPγS in vitro kinase assay
Recombinant GST-tagged PERK-ΔN (described above) was incubated with recombinant eIF2α in the presence of 1 mM ATPγS (Biolog A060) or N6-substituted ATPγS (Biolog F008, B072, P026) for 30 min at 30 °C.Citation38 Following thiophosphorylation, reactions were alkylated by adding p-nitrobenzyl mesylate (Abcam ab138910) to 2.5 mM for 1 h at room temperature. Samples were boiled in SDS sample buffer, run on an SDS-PAGE gel, and probed by western blot for thiophosphorylated protein.
Fractionation
Cells were lysed in 6 pellet volumes Harvest Buffer (10 mM HEPES pH 7.9, 50 mM NaCl, 0.5 M sucrose, 0.1 mM EDTA, 0.5% Triton X 100) supplemented with 1 mM DTT, and protease and phosphatase inhibitors. Nuclei were pelleted, washed in Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA), and lysed in 4 volumes Buffer C (10 mM HEPES pH 7.9, 500 mM NaCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.1% NP-40). Nuclei were vortexed for 15 min at 4 °C and clarified. Cytoplasmic and nuclear extracts were analyzed by western blot.
In vivo substrate labeling and immunoprecipitation
Samples were prepared as previously describedCitation30 with slight modifications. In brief, cells were trypsinized, counted, and resuspended to 3 × 106 cells/mL in cold kinase buffer (25 mM Tris pH 7.5, 10 mM MgCl2 in PBS) and 50 ug/mL digitonin to permeabilize. Cells were incubated on ice for 5 min, pelleted gently, then resuspended in kinase buffer containing 500 nM thapsigargin, 100 μM N6-furfuryl ATPγS (Biolog F008), and 1 mM GTP (Sigma G8877) for substrate labeling. Cells were incubated for 1 h at 30 °C with gentle shaking, then lysed in RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 1.0% NP-40, 0.1% SDS) containing 25 mM EDTA to quench. Lysates were alkylated by adding p-nitrobenzyl mesylate (Abcam ab138910) to 2.5 mM for 1 h at room temperature with nutation. For immunoprecipitation, lysates were exchanged to RIPA buffer on PD Miditrap G-25 columns (GE Healthcare 28-9180-08) to remove PNBM. Lysates were then pre-cleared with rProtein G agarose (Invitrogen 15920-010) and subjected to immunoprecipitation with thioP antibody bound to rProtein G agarose beads.
Cell survival after ER stress
Cells were seeded at 20 000 per 60 mm dish and pre-treated with indicated doses of 3-MB-PP1 for 1 h. Cells were then challenged with an acute dose of tunicamycin (2.5 ug/mL) for 30 min. Colony outgrowth was assessed by Giemsa staining after 6 d.
| Abbreviations: | ||
| UPR | = | unfolded protein response |
| ER | = | endoplasmic reticulum |
| PERK | = | PKR-like endoplasmic reticulum kinase |
| ATF4 | = | activating transcription factor 4 |
| CHOP | = | C/EBP-homologous protein |
| TG | = | thapsigargin |
| PNBM | = | p-nitrobenzyl mesylate |
| α-thioP | = | anti-thiophosphate ester |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by National Institutes of Health grants P01 CA104838 (J.A.D.).
References
- Luo B, Lee AS. The critical roles of endoplasmic reticulum chaperones and unfolded protein response in tumorigenesis and anticancer therapies. Oncogene 2013; 32:805 - 18; http://dx.doi.org/10.1038/onc.2012.130; PMID: 22508478
- Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2000; 2:326 - 32; http://dx.doi.org/10.1038/35014014; PMID: 10854322
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell 2002; 3:99 - 111; http://dx.doi.org/10.1016/S1534-5807(02)00203-4; PMID: 12110171
- Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, Wek RC. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol 1998; 18:7499 - 509; PMID: 9819435
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999; 397:271 - 4; http://dx.doi.org/10.1038/16729; PMID: 9930704
- Brewer JW, Hendershot LM, Sherr CJ, Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci U S A 1999; 96:8505 - 10; http://dx.doi.org/10.1073/pnas.96.15.8505; PMID: 10411905
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 2003; 11:619 - 33; http://dx.doi.org/10.1016/S1097-2765(03)00105-9; PMID: 12667446
- Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov 2013; 12:703 - 19; http://dx.doi.org/10.1038/nrd3976; PMID: 23989796
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13:184 - 90; http://dx.doi.org/10.1038/ncb0311-184; PMID: 21364565
- Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe?. Nat Rev Cancer 2004; 4:966 - 77; http://dx.doi.org/10.1038/nrc1505; PMID: 15573118
- Feng Y, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2a and sensitizes cells to endoplasmic reticulum stress. Cancer Discov 2014; http://dx.doi.org/10.1158/2159-8290.CD-13-0945; PMID: 24705811
- Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, et al. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 2005; 24:3470 - 81; http://dx.doi.org/10.1038/sj.emboj.7600777; PMID: 16148948
- Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, Cavener D, Diehl JA. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010; 29:3881 - 95; http://dx.doi.org/10.1038/onc.2010.153; PMID: 20453876
- Nagelkerke A, Bussink J, Mujcic H, Wouters BG, Lehmann S, Sweep FC, Span PN. Hypoxia stimulates migration of breast cancer cells via the PERK/ATF4/LAMP3-arm of the unfolded protein response. Breast Cancer Res 2013; 15:R2; http://dx.doi.org/10.1186/bcr3373; PMID: 23294542
- Mujcic H, Nagelkerke A, Rouschop KM, Chung S, Chaudary N, Span PN, Clarke B, Milosevic M, Sykes J, Hill RP, et al. Hypoxic activation of the PERK/eIF2α arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin Cancer Res 2013; 19:6126 - 37; http://dx.doi.org/10.1158/1078-0432.CCR-13-0526; PMID: 24045183
- Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol 2011; 31:3616 - 29; http://dx.doi.org/10.1128/MCB.05164-11; PMID: 21709020
- Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, Koumenis C, Harding HP, Ron D, Holcik M, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol 2006; 26:9517 - 32; http://dx.doi.org/10.1128/MCB.01145-06; PMID: 17030613
- Pytel D, Seyb K, Liu M, Ray SS, Concannon J, Huang M, Cuny GD, Diehl JA, Glicksman MA. Enzymatic Characterization of ER Stress-Dependent Kinase, PERK, and Development of a High-Throughput Assay for Identification of PERK Inhibitors. J Biomol Screen 2014; Forthcoming http://dx.doi.org/10.1177/1087057114525853; PMID: 24598103
- Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, Li WH, Heerding DA, Minthorn E, Mencken T, et al. Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem 2012; 55:7193 - 207; http://dx.doi.org/10.1021/jm300713s; PMID: 22827572
- Bishop AC, Shah K, Liu Y, Witucki L, Kung C, Shokat KM. Design of allele-specific inhibitors to probe protein kinase signaling. Curr Biol 1998; 8:257 - 66; http://dx.doi.org/10.1016/S0960-9822(98)70198-8; PMID: 9501066
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000; 407:395 - 401; http://dx.doi.org/10.1038/35030148; PMID: 11014197
- Shokat K, Velleca M. Novel chemical genetic approaches to the discovery of signal transduction inhibitors. Drug Discov Today 2002; 7:872 - 9; http://dx.doi.org/10.1016/S1359-6446(02)02391-7; PMID: 12546954
- Buzko O, Shokat KM. A kinase sequence database: sequence alignments and family assignment. Bioinformatics 2002; 18:1274 - 5; http://dx.doi.org/10.1093/bioinformatics/18.9.1274; PMID: 12217924
- Au-Yeung BB, Levin SE, Zhang C, Hsu LY, Cheng DA, Killeen N, Shokat KM, Weiss A. A genetically selective inhibitor demonstrates a function for the kinase Zap70 in regulatory T cells independent of its catalytic activity. Nat Immunol 2010; 11:1085 - 92; http://dx.doi.org/10.1038/ni.1955; PMID: 21037577
- Liu Y, Warfield L, Zhang C, Luo J, Allen J, Lang WH, Ranish J, Shokat KM, Hahn S. Phosphorylation of the transcription elongation factor Spt5 by yeast Bur1 kinase stimulates recruitment of the PAF complex. Mol Cell Biol 2009; 29:4852 - 63; http://dx.doi.org/10.1128/MCB.00609-09; PMID: 19581288
- Levin SE, Zhang C, Kadlecek TA, Shokat KM, Weiss A. Inhibition of ZAP-70 kinase activity via an analog-sensitive allele blocks T cell receptor and CD28 superagonist signaling. J Biol Chem 2008; 283:15419 - 30; http://dx.doi.org/10.1074/jbc.M709000200; PMID: 18378687
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 2000; 5:897 - 904; http://dx.doi.org/10.1016/S1097-2765(00)80330-5; PMID: 10882126
- Banko MR, Allen JJ, Schaffer BE, Wilker EW, Tsou P, White JL, Villén J, Wang B, Kim SR, Sakamoto K, et al. Chemical genetic screen for AMPKα2 substrates uncovers a network of proteins involved in mitosis. Mol Cell 2011; 44:878 - 92; http://dx.doi.org/10.1016/j.molcel.2011.11.005; PMID: 22137581
- Allen JJ, Lazerwith SE, Shokat KM. Bio-orthogonal affinity purification of direct kinase substrates. J Am Chem Soc 2005; 127:5288 - 9; http://dx.doi.org/10.1021/ja050727t; PMID: 15826144
- Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hübner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, et al. A semisynthetic epitope for kinase substrates. Nat Methods 2007; 4:511 - 6; http://dx.doi.org/10.1038/nmeth1048; PMID: 17486086
- Blethrow JD, Glavy JS, Morgan DO, Shokat KM. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc Natl Acad Sci U S A 2008; 105:1442 - 7; http://dx.doi.org/10.1073/pnas.0708966105; PMID: 18234856
- Fedorov O, Müller S, Knapp S. The (un)targeted cancer kinome. Nat Chem Biol 2010; 6:166 - 9; http://dx.doi.org/10.1038/nchembio.297; PMID: 20154661
- Zhang C, Lopez MS, Dar AC, Ladow E, Finkbeiner S, Yun CH, Eck MJ, Shokat KM. Structure-guided inhibitor design expands the scope of analog-sensitive kinase technology. ACS Chem Biol 2013; 8:1931 - 8; http://dx.doi.org/10.1021/cb400376p; PMID: 23841803
- Garske AL, Peters U, Cortesi AT, Perez JL, Shokat KM. Chemical genetic strategy for targeting protein kinases based on covalent complementarity. Proc Natl Acad Sci U S A 2011; 108:15046 - 52; http://dx.doi.org/10.1073/pnas.1111239108; PMID: 21852571
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23:7198 - 209; http://dx.doi.org/10.1128/MCB.23.20.7198-7209.2003; PMID: 14517290
- Gao Y, Sartori DJ, Li C, Yu QC, Kushner JA, Simon MC, Diehl JA. PERK is required in the adult pancreas and is essential for maintenance of glucose homeostasis. Mol Cell Biol 2012; 32:5129 - 39; http://dx.doi.org/10.1128/MCB.01009-12; PMID: 23071091
- Bobrovnikova-Marjon E, Pytel D, Riese MJ, Vaites LP, Singh N, Koretzky GA, Witze ES, Diehl JA. PERK utilizes intrinsic lipid kinase activity to generate phosphatidic acid, mediate Akt activation, and promote adipocyte differentiation. Mol Cell Biol 2012; 32:2268 - 78; http://dx.doi.org/10.1128/MCB.00063-12; PMID: 22493067
- Hertz NT, Wang BT, Allen JJ, Zhang C, Dar AC, Burlingame AL, Shokat KM. Chemical genetic approach for kinase-substrate mapping by covalent capture of thiophosphopeptides and analysis by mass spectrometry. Curr Protoc Chem Biol 2010; 2:15 - 36; PMID: 23836541