
Abstract
Cyclin D1/CDK4 activity is upregulated in up to 50% of breast cancers and CDK4-mediated phosphorylation negatively regulates the TGFβ superfamily member Smad3. We sought to determine if CDK4 inhibition and doxorubicin chemotherapy could impact Smad3-mediated cell/colony growth and apoptosis in breast cancer cells. Parental and cyclin D1-overexpressing MCF7 cells were treated with CDK4 inhibitor, doxorubicin, or combination therapy and cell proliferation, apoptosis, colony formation, and expression of apoptotic proteins were evaluated using an MTS assay, TUNEL staining, 3D Matrigel assay, and apoptosis array/immunoblotting. Study cells were also transduced with WT Smad3 or a Smad3 construct resistant to CDK4 phosphorylation (5M) and colony formation and expression of apoptotic proteins were assessed. Treatment with CDK4 inhibitor/doxorubicin combination therapy, or transduction with 5M Smad3, resulted in a similar decrease in colony formation. Treating cyclin D overexpressing breast cancer cells with combination therapy also resulted in the greatest increase in apoptosis, resulted in decreased expression of anti-apoptotic proteins survivin and XIAP, and impacted subcellular localization of pro-apoptotic Smac/DIABLO. Additionally, transduction of 5M Smad3 and doxorubicin treatment resulted in the greatest change in apoptotic protein expression. Collectively, this work showed the impact of CDK4 inhibitor-mediated, Smad3-regulated tumor suppression, which was augmented in doxorubicin-treated cyclin D-overexpressing study cells.
Introduction
Breast cancer is the most common malignancy affecting women, with over 200 000 cases diagnosed and 40 000 patients dying each year from this disease.Citation1 The advent of targeted therapeutics has significantly impacted breast cancer outcomes for patients with erbB2 overexpressing cancers. Based on this bench to bedside success, the discovery of additional cancer cell targets is actively being pursued with a specific focus on cell cycle components, including mitogenic cyclins. Cyclin D1 is overexpressed at the mRNA and protein levels in up to 50% of breast cancers.Citation2-Citation4 Cyclin D1 is primarily overexpressed in estrogen receptor positive (ER+) tumors, and this overexpression is associated with poor outcomes and decreased relapse-free survival.Citation5,Citation6 As such, it is one of the most commonly overexpressed oncogenes in breast cancer and is a potentially significant therapeutic target.
Cyclins are the regulatory subunits of cyclin-dependent kinases (CDKs). Cyclin/CDK complexes permit cells to transition from the G1 to the S phase of the cell cycle. The activities of these complexes are modulated by the binding of CDK inhibitors (CDKis), including p15, p16, p21, and p27, which can sequester CDKs or bind and inhibit cyclin/CDK complexes. Cyclin D forms active complexes with either CDK4 or CDK6, which initiate the phosphorylation of the tumor suppressive retinoblastoma (Rb) family of proteins.Citation7 Hyperphosphorylation of Rb by cyclin D/CDK4 or 6 inhibits Rb from sequestering members of the E2F transcription factor family, which then drives the transcription of genes encoding the proteins required for G1/S-phase transition and S-phase progression.Citation7 Thus, cyclin D overexpression contributes to loss of cell cycle control, facilitating oncogenic progression.Citation8 Furthermore, murine studies have shown that the continued presence of active CDK4 complexes plays a key role in mammary tumor growth.Citation9,Citation10
Cyclin D/CDK4 complexes are also involved in cell cycle control through the phosphorylation and regulation of members of the transforming growth factor-β (TGFβ) superfamily.Citation11,Citation12 Several members of the TGFβ superfamily have crucial roles in mammary gland physiology, with the Smads functioning as downstream mediators of this signaling pathway.Citation13 Intact canonical TGFβ/Smad3 signaling has previously been linked to tumor suppressive cytostatic and pro-apoptotic events in early stage breast cancer.Citation14,Citation15 Simultaneously, TGFβ/Smad3 signaling has been shown to promote oncogenic progression through the induction of epithelial-to-mesenchymal transition (EMT) in advanced stage breast carcinoma. Based on these opposing actions in early and later stage disease, TGFβ/Smad3 signaling can have dichotomous actions in breast oncogenesis.Citation12 Canonical TGFβ signaling occurs through the phosphorylation of Smad3 at the C-terminus by the TGFBRI receptor. However, CDKs 4/2, in addition to other kinases, can also noncanonically phosphorylate Smad3 at multiple sites located primarily in the linker region of the protein.Citation16 This noncanonical phosphorylation of Smad3 can result in decreased tumor suppression of the Smad3 protein associated with increased c-myc activity and inhibition of CDKis.Citation17,Citation18 Conversely, transfection of the Smad3 protein mutated at five CDK phosphorylation sites (5M Smad3), was shown to restore Smad3 activity and resulted in lower c-myc mRNA levels and higher levels of the CDKi p15.Citation17,Citation18 Treatment with a CDK4i also resulted in increased Smad3 activity in cyclin D overexpressing breast cancer cells. Collectively, this data suggests that CDK4 inhibition could be a targeted treatment strategy for patients whose tumors overexpress cyclin D by promoting Smad3-regulated cell cycle arrest.
Pan-CDK inhibitors have been utilized in phase I solid tumor clinical trials, yet efficacy has thus far been modest, potentially associated with both the lack of tumor cyclin profiling and nonspecific CDK inhibition implemented in these trials.Citation19 A more thorough understanding of the role of specific cyclins and their CDK complements, in addition to the directed study of CDK inhibitors in specific cyclin-overexpressing cancers, is necessary to reveal the therapeutic potential of these agents. To date, limited study of combination CDK inhibitor/chemotherapy has demonstrated a partial response for patients in clinical trials of advanced solid tumors.Citation19 Overall, a more rational and focused implementation of CDK inhibitor therapy, combined with cytotoxic chemotherapy, could result in improved cytotoxic efficacy and improved patient outcomes.
In previous in vitro studies, CDK inhibitors and chemotherapeutics have independently induced cytostatic and apoptotic effects on cancer cells. Doxorubicin, one of the most widely implemented antitumor drugs used for breast cancer treatment, exerts cytostatic changes via increased expression of p21 in a p53-dependent manner, and apoptotic events through DNA intercalation, inhibition of DNA topoisomerase II, and induction of DNA double strand breaks.Citation20,Citation21 CDK inhibitors exert cytostatic effects through restoration of cell cycle checkpoints.Citation19 Though less well understood, it has been suggested that CDK inhibitor-associated apoptosis is related to a decrease in CDK-mediated phosphorylation of Rb and persistent E2F activation, which may induce apoptosis in cancer cells.Citation22 Elucidating the mechanisms by which CDK inhibition facilitates apoptosis would allow for more appropriate synergism with current chemotherapeutic treatments.
The apoptotic process is both positively and negatively regulated by pro- and anti- apoptotic factors, respectively. Survivin is a member of the inhibitor of apoptosis proteins (IAPs), and plays a critical role in both inhibition of apoptosis and control of cell division.Citation23,Citation24 The role of survivin in several different malignancies has been examined, including bladder, blood, liver, colon, pancreas, prostate, and breast.Citation25-Citation31 Specifically, in breast cancer, survivin expression has been linked to early recurrence and decreased survival rates.Citation32,Citation33 Survivin expression has also been associated with the promotion of cancer cell survival through the inhibition of pro-apoptotic stimuli resulting in chemoresistance.Citation34,Citation35 In a prostate cancer model, survivin downregulation was shown to occur in a TGFβ/Smad2/3-dependent manner resulting in increased apoptosis.Citation36 This mechanism involved the binding and interaction of the hypophosphorylated Rb-E2F4 complex and C-terminal phosphorylated Smad3 on the survivin promoter, inducing transcriptional repression. XIAP is a potent mammalian caspase inhibitor that binds and inhibits proteolytic activity of caspases 3, 7, and 9 in normal cells. High XIAP levels have been associated with aggressive, high grade breast cancers.Citation37 An antagonist of XIAP, second mitochondria-derived activator of caspase (Smac/DIABLO), is released from the mitochondria upon intrinsic apoptotic stimuli, and binds and inhibits XIAP, which results in the release of caspase3. Caspase 3 then cleaves PARP1, preventing DNA repair and resulting in apoptosis. To date, the impact of noncanonical phosphorylation of Smad3 has not been studied in the context of survivin, XIAP, and Smac/DIABLO expression.
We have previously shown that pharmacological CDK inhibition can augment Smad3-regulated gene expression. In the current study, we implemented MCF7 cells, a prototypical ER+ breast cancer cell line. We also engineered a cyclin D overexpressing variant of this cell line to reflect the high number of ER+ breast cancers that overexpress this cyclin. We then sought to determine if CDK inhibition and chemotherapy could impact Smad3-mediated cell/colony growth and apoptosis, potentially through the regulation of survivin expression. Evidence of this relationship would provide novel insight into the mechanism behind the growth arrest and apoptosis induction mediated by both CDK inhibition and chemotherapy through a convergence on Smad3 signaling.
Results
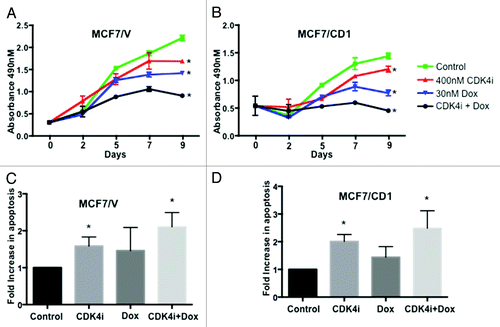
CDK4i and doxorubicin treatment decreases cell proliferation
We have previously shown that transfecting breast cancer cells with a CDK4 phosphorylation site mutant Smad3 construct or treatment with CDK4i-affected cell cycle regulation.Citation17 To further examine the impact of CDK4 inhibition and chemotherapy on cyclin D overexpressing cell proliferation, we engineered a model of cyclin D overexpression in ER+ breast cancer cells. To this end, MCF7 cells were first stably transfected with either control or cyclin D1 plasmids. An MTS assay was then used to assess the effect of treatment with CDK4i and doxorubicin on proliferation of control MCF7/V () and cyclin D1 overexpressing (CD1) MCF7 cells (). Although the two study cells lines were initially plated at equal densities, the MCF7/CD1 cells showed a higher absorbance at Day 0, indicating a greater initial degree of cellular metabolic activity, as expected, for the cyclin D-overexpressing study cells. When compared with control treatment, both MCF7/V and MCF7/CD1 breast cancer cells treated with CDK4i alone resulted in a significant decrease in cell proliferation over a period of 9 d (P < 0.05). Additionally, treatment of both cell lines with doxorubicin resulted in a greater decrease (P < 0.01), with the most significant decrease in cell proliferation occurring after combination treatment (P < 0.001). We also found that, after normalization to the non-treated cells, for the study cells treated with combination therapy, the proliferation rate was effectively reduced by 23% for the MCF7/CD1 cells, when compared with the proliferation rate for the MCF7/V cells. We observed a similar decrease in proliferation after treatment with CDK4i and doxorubicin using an alternate p53 mutated ER+ breast cancer cell line (T47D) and a cyclin D1 overexpressing cell line derivative (data not shown). As cyclin D1 overexpression is more commonly found in wild-type p53 ER+ tumors,Citation38 we pursued further work in MCF7 cells, which express wild-type p53.
Figure 1. Treatment with combination therapy results in the lowest proliferation rates and highest levels of apoptosis. (A) MCF7/V and (B) MCF7/CD1 cells were treated over the course of 9 d with control DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination therapy, and an MTS assay was used to measure cell number at the indicated time points. Absorbance for CDK4i, doxorubicin, and combination treated cells was compared with control treated cells within each cell line using a Student t test to obtain P values. Absorbance values are shown ± SE. (C) MCF7/V and (D) MCF7/CD1 cells were treated over the course of 2 d with control DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination therapy. Cells were fixed and stained using a TUNEL assay and cells undergoing apoptosis were counted and normalized to control treated cells to obtain fold changes. Experiments were repeated 3 times and representative results are shown. * indicates P < 0.05.
CDK4i and doxorubicin treatment induces apoptosis
We next assessed if combination therapy also impacted induction of apoptosis. MCF7/V and MCF7/CD1 cells were treated with control DMSO, CDK4i, doxorubicin, or combination therapy for 48 h and apoptosis was measured by TUNEL staining. When compared with control cells, we found a modest increase in apoptosis in MCF7/V cells () following individual CDK4i or doxorubicin treatment, with the highest level of apoptosis observed in response to treatment with combination therapy. For MCF7/CD1 cells, when compared with control treatment, individual CDK4i and doxorubicin treatments resulted in increased levels of apoptosis, with combination therapy resulting in the highest level of apoptosis. Overall, treatment of MCF7/CD1 cells () showed a trend toward a greater impact on apoptosis when compared with MCF7/V cells (although nonsignificant). These data further support the effect of combination therapy on induction of cancer cell death.
Transduction of Smad3 phosphorylation site mutants and CDK4i/chemotherapy treatment results in decreased colony formation in 3D culture
We implemented a more biologically relevant 3D culture model to expand upon the MTS and TUNEL 2D data and more accurately capture the impact of the study treatments in a 3D environment. While our 2D and 3D results are relatively comparable, previous studies have also characterized breast cancer cells to behave distinctly in 2D vs. 3D culture.Citation39 To this end, MCF7/V and MCF7/CD1 cell lines were transduced with control CS2 vector, WT Smad3, or 5M Smad3 constructs to assess the impact of Smad3 action and CDK4i/chemotherapy treatment on study cell colony growth. We have previously confirmed the presence of nuclear and cytoplasmic Smad3 and phosphorylated Smad3 in the study cell lines. We have also previously shown the relative increase in CDK4 kinase activity found in MCF7/CD1 cells when compared with MCF7/V cells.Citation17 Currently, we confirmed expression of Smad3 and cyclin D1 in both study cell lines. As expected, Smad3 expression increased in the study cells transduced with WT or 5M Smad3 (). Expression of cyclin D1 also increased in MCF7/CD1 cells when compared with MCF7/V (). Following transduction, tumor colony formation was assessed in Matrigel at days 4 and 12. When examining the control treated cells, MCF7/V cells () formed smaller colonies when compared with MCF7/CD1 cells () after 12 d, demonstrating the effect of cyclin D overexpression on increased cellular proliferation in this setting. For MCF7/V cells on day 12, transduction of WT Smad3 and 5M Smad3 constructs resulted in a similar decrease in colony area over time (P < 0.05), when compared with transduction of the CS2 vector. While WT Smad3 transduction resulted in a significant reduction in colony growth in MCF7/V cells, in the context of cyclin D overexpression (MCF7/CD1 cells), this growth suppression was not observed. Notably, when compared with transduction with the CS2 vector, MCF7/CD1 cells transduced with the 5M Smad3 construct showed a significant decrease in colony area on day 12 (P < 0.0001) vs. MCF7/CD1 cells transduced with the WT Smad3 construct. These findings support the role of intact Smad3 signaling, resistant to CDK phosphorylation, in the suppression of tumor cell proliferation.
Figure 2. Treatment with CDK4i and doxorubicin combination therapy or transduction with 5M Smad3 suppresses colony growth. (A) MCF7/V and MCF7/CD1 cells were transduced with CS2 vector, WT Smad3, or 5M Smad3. Protein was extracted and immunoblotted with Smad3, cyclin D, and GAPDH. (B) MCF7/V and (C) MCF7/CD1 cells were transduced with CS2 vector control, WT Smad3, or 5M Smad3 and treated over the course of 12 d with control DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination therapy. Colony area was measured at the indicated time points using Metamorph software. Experiments were repeated 3 times and representative results are shown. Error bars indicate ± SE. * indicates P < 0.05 for comparisons between vector transduced cells and WT and 5M transduced cells treated with control. ^ indicates P < 0.05 for comparisons between control treatment and study treatments for each transduced construct within each cell line.
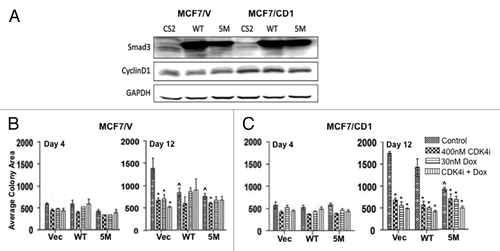
We next compared the impact of the study treatments to control within the CS2 vector, WT and 5M Smad3-transduced cells. For the MCF7/CD1 CS2 vector transduced cells, all treatment conditions resulted in a more significant decrease in colony size (P < 0.0001), when compared with vector-transduced MCF7/V cells (P < 0.05). For the WT and 5M-transduced MCF7/CD1 cells, combination treatment resulted in a significant decrease in colony size (P < 0.01), while no significant change was seen after combination treatment for the WT and 5M-transduced MCF7/V cells. Lastly, for both study cell lines, transduction of the 5M Smad3 construct resulted in a similar decrease in colony size when compared with treatment with the CDK4 inhibitor, suggesting that restoration of Smad3-mediated tumor suppressive activity could be achieved through the pharmacological inhibition of CDK-mediated phosphorylation of Smad3.
Expression of apoptotic proteins is affected by CDK4i/doxorubicin combination therapy
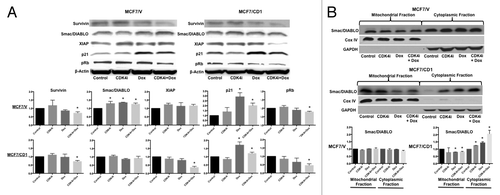
To further examine the potential relationship between CDK4i, doxorubicin, and the induction of apoptosis, we explored multiple candidate factors contributing to this mechanism. The Proteome Profiler Human Apoptosis Antibody Array was employed to characterize expression of critical apoptosis-related proteins after CDK4i and doxorubicin treatment, as well as combination therapy. The impact of treatment on expression levels of five selected proteins is shown for MCF7/V () and MCF7/CD1 () cells. Levels of Fas, a key protein in the extrinsic apoptotic pathway, were not significantly altered upon CDK4i treatment in either study cell line, but were increased with doxorubicin and combination therapy. Levels of the CDKi p21 increased in a similar manner after CDK4i or doxorubicin treatment in MCF7/V cells, with the greatest increase observed in response to combination therapy. While CDK4i treatment led to a minimal increase in expression of p21 in MCF7/CD1 cells, doxorubicin alone and combination treatment resulted in a greater (and similar) increase in p21 expression. Of particular interest, survivin and XIAP levels significantly decreased and Smac/DIABLO levels generally increased after treatment with the study drugs alone and in combination. Collectively, these findings point toward the contribution of CDK4i/doxorubicin to the repression of survivin and the potential to facilitate apoptosis in the study cells. Acknowledging the limitations of high-throughput assays, we then used standard immunoblotting to further delineate the changes found in the array studies. We confirmed a significant decrease in survivin protein levels after combination treatment, when compared with control, for the MCF7/V and MCF7/CD1 cells (). Additionally, expression of XIAP significantly decreased in MCF7/CD1 cells upon combination CDK4i/doxorubicin treatment (). While MCF7/V cells responded to individual and combination treatment with a trend toward an increase in Smac/DIABLO expression, no significant change in Smac/DIABLO expression was detected in either the MCF7/V or CD1 cells using whole cell lysates. Once activated, Smac/DIABLO is released from the mitochondria into the cytoplasm to bind XIAP. Thus, we next examined if the sub-cellular localization of Smac/DIABLO was altered upon treatment. We found that treatment with CDK4i/doxorubicin resulted in significantly decreased Smac/DIABLO expression in the mitochondrial fraction, and increased expression in the cytoplasmic fraction for MCF7/CD1 cells (). p21 expression increased in both doxorubicin and combination-treated MCF7/V and MCF7/CD1 cells, while pRb levels decreased in both MCF7/V and MCF7/CD1 combination-treated cells ().
Figure 3. The array shows that CDK4i and doxorubicin therapy affect expression of proteins associated with apoptosis. (A) MCF7/V and (B) MCF7/CD1 were treated with control DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination therapy. Protein was collected and applied to the array. Protein expression was quantified using Multigauge software. The array was repeated 3 times and representative results are shown. Error bars indicate ± SE.
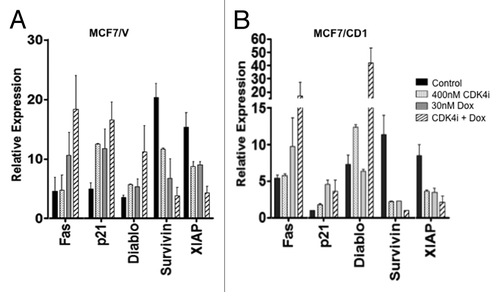
Figure 4. Survivin expression decreases upon treatment with CDK4 inhibitor and doxorubicin alone or in combination. (A) MCF7/V and MCF7/CD1 cells were treated with DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination treatment for 48 h. Protein was extracted, followed by immunoblotting with survivin, XIAP, Smac/DIABLO, p21, pRb, and β-Actin antibodies. (C) MCF7/V and MCF7/CD1 cells were treated with DMSO, 400 nM CDK4i, 30 nM doxorubicin, or combination treatment for 48 h. Mitochondrial and cytoplasmic fractions were isolated, followed by immunoblotting with Smac/DIABLO, Cox IV, and GAPDH antibodies. Experiments were repeated 3 times and representative results are shown. * indicates P < 0.05.
Intact Smad3 signaling and doxorubicin treatment alter apoptotic protein expression
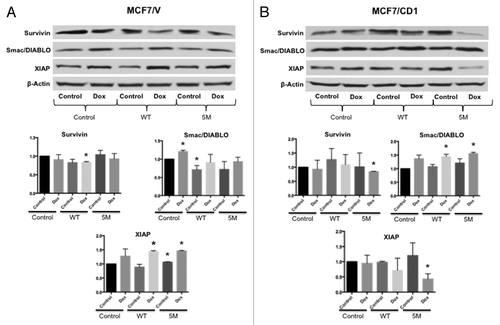
To determine if survivin expression was regulated by Smad3 action, we transduced MCF7/V and MCF7/CD1 cells with vector control, WT Smad3, or 5M Smad3 constructs. We then assessed the expression of survivin, XIAP, and SMAC/Diablo in the transduced cells in the presence or absence of doxorubicin. For both MCF7/V () and MCF7/CD1 () cells, WT or 5M Smad3 transduction did not result in significant changes in survivin expression. XIAP expression increased upon 5M Smad3 transduction, in MCF7/V, but not MCF7/CD1, cells. Smac/DIABLO expression decreased upon WT Smad3 transduction in the MCF7/V cells, attributed to the negative feedback mediated by TGFβ signaling after Smad3 overexpression ().Citation40 We then tested for potential synergy between doxorubicin treatment and transduction with the 5M Smad3 construct. To this end, we found that doxorubicin treatment and expression of the Smad3 5M protein resulted in a significant decrease in survivin and XIAP expression in MCF7/CD1 cells (). Correspondingly, a significant increase in Smac/DIABLO expression was seen when 5M Smad3-transduced MCF7/CD1 cells were treated with doxorubicin.
Figure 5. Intact Smad3 signaling and doxorubicin treatment result in the greatest reduction in survivin expression. (A) MCF7/V and (B) MCF7CD1 cells were transduced with CS2 vector control, WT Smad3, or 5M Smad3 and then treated with either DMSO control or 30 nM doxorubicin for 48 h. Protein was extracted, followed by immunoblotting with survivin, XIAP, Smac/DIABLO, and β-actin antibodies. Experiments were repeated 3 times and representative results are shown. * indicates P < 0.05.
Discussion
We sought to determine the effect of CDK4 inhibition, alone or in combination with doxorubicin, on cell proliferation, colony growth, and apoptosis in an in vitro model of cyclin D overexpressing ER+ breast cancer. While cell proliferation was effectively reduced in response to treatment with CDK4i or doxorubicin, a greater effect was found when the therapies were combined. Additionally, cyclin D-overexpressing breast cancer cells showed the highest level of apoptosis induction following combination CDK4 inhibitor and doxorubicin treatment. 3D Matrigel culture studies expanded upon the proliferation/apoptosis findings in an environment that allowed the breast cancer cells to grow into colonies, more closely reflecting in vivo cancer growth. Colony growth was decreased after treatment with CDK4i. Cells transduced with 5M Smad3 construct showed a similar decrease in colony size, demonstrating that pharmacologically or genetically inhibiting CDK-mediated Smad3 phosphorylation resulted in breast cancer cell growth inhibition. Consistent with the proliferation/apoptosis assays, combination CDK4i/doxorubicin treatment also resulted in the smallest colony size. Lastly, we identified pro- and anti-apoptotic factors including survivin, XIAP and Smac/DIABLO, to be critical proteins associated with CDK4i/doxorubicin treatment-induced cellular changes.
We have previously shown that cyclin D-overexpressing ER+ breast cancer cells transfected a CDK4 phosphorylation-resistant Smad3 construct or treated with CDK4i resulted in increased cdki p15 and decreased c-myc mRNA expression.Citation17 Additionally, we had found that treatment with CDK4i, as well as transfection with 5M Smad3, resulted in higher Smad3 reporter activity, and CDK4 inhibition-induced cell cycle arrest.Citation17 These results prompted us to further investigate the impact of CDK4i on cancer cell proliferation and apoptosis. Previous studies with PD0332991, a CDK4/6 inhibitor, showed the greatest inhibition of proliferation occurring in ER+ breast cancer cells.Citation41 PD0332991 also inhibited tumor growth in the setting of hormone resistant ER+ breast cancer cells in vivo.Citation42 Our current work adds to this prior data supporting the anti-proliferative impact of specific CDK4 inhibition in ER+ breast cancer. Although results from previous clinical trials using single-agent pan-CDKis have been inconsistent, the outcomes associated with combination CDKi therapy with cytotoxic drugs have been more encouraging.Citation19,Citation43 Our in vitro proliferation and 3D tumor colony formation data provide further pre-clinical evidence of the potential of these agents. Furthermore, clinical trials implementing CDK inhibitors have indiscriminately included patients irrespective of their tumor cyclin expression profile, and, to date, one clinical trial (NCT00721409) examining the efficacy of letrozole with and without PD0332991, has specifically targeted ER+ breast tumors.Citation43 The recently published results of the randomized phase II PALOMA-1 trial are very promising with combination letrozole and PD0332991 resulting in significantly improved progression free survival (20.2 mo) when compared with treatment with letrozole alone (10.2 mo).Citation44 Our work suggests that future clinical studies using CDK inhibitors may also benefit from stratifying patients with greater discrimination directed toward cyclin expression status.
Smad3 is a transcription factor that regulates the expression of genes critical to several phenotypic endpoints, including cell cycle control and apoptosis.Citation45 The 3D tumor colony data in this study showed that the effect of WT Smad3 vs. 5M Smad3 transduction on colony growth was nearly equivalent for the control MCF7/V cells. It follows that for MCF7/V cells, basal cyclin D/CDK4 activity was not likely sufficient to repress WT Smad3, and thus transduction of both plasmids resulted in a similar significant decrease in colony area. However, in the setting of control-treated, cyclin D-overexpressing MCF7/CD1 cells, 5M Smad3 transduction was more effective in inhibiting colony growth when compared with transduction with the WT construct. These findings suggest that WT Smad3 is phosphorylated and inhibited by the highly active CDK4 in these cells. Conversely, cells transduced with the 5M Smad3 construct were resistant to inhibitory CDK4 phosphorylation, and likely retained tumor suppressive actions including repression of c-myc and promotion of cdki p15 transcription. Additionally, the impact of individual and combination treatment was mitigated in WT Smad3- or 5M Smad3-transduced MCF7/V cells, but remained intact particularly for the WT Smad3-transduced MCF7/CD1 cells. These data provide further evidence for the potential of CDK4 inhibitors to reduce tumor cell growth through the restoration of Smad3-mediated cell cycle control in cyclin D overexpressing breast cancer cells. Lastly, prior work has shown that cyclin D overexpressing cancer cells were more sensitive to doxorubicin.Citation46 Consequently, irrespective of Smad3 expression status, overexpression of cyclin D1 in the MCF7/CD1 cells should result in increased cell division, and thus greater sensitivity to the cytotoxic effects of doxorubicin chemotherapy, found in .
The induction of cellular apoptosis concomitant with cell cycle arrest is a key mechanism by which optimal treatments induce breast cancer regression. Doxorubicin is a widely used anthracycline that has strong antineoplastic activity.Citation21 We observed a greater increase in apoptosis in response to CDK4i/doxorubicin combination therapy, with MCF7/CD1 cells showing the highest levels of apoptosis after treatment. Additionally, it has previously been shown that doxorubicin treatment activates TGFβ signaling in breast cancer cells through the induction of C-terminal phospho-Smad3 expression.Citation47 As C-terminal phosphorylation of Smad3 enables subsequent nuclear translocation, doxorubicin treatment may augment the expression of apoptotic proteins after WT or 5M Smad3 transduction, in part, by increasing the amount of nuclear Smad3. To further examine potential apoptotic factors linked to these findings, we used a Proteome Profiler Human Antibody Array to perform a high-throughput analysis of 35 apoptosis-related proteins. Among these, p21, Fas, survivin, XIAP, and Smac/DIABLO exhibited differential expression patterns as a result of cyclin overexpression and the study treatments. It has been reported that high survivin expression is an independent predictor of poor prognosis for breast cancer patients, and survivin is not typically detectable in healthy adult tissues with low cell turnover.Citation24,Citation48 Increased survivin levels are also associated with repression of apoptosis and chemoresistance, while inhibition of survivin has resulted in enhanced chemotherapy-mediated apoptotic induction.Citation49 As such, survivin may be an important therapeutic target.Citation24,Citation48
Furthermore, high expression of other factors noted in our array study, including Smac/DIABLO, have been previously shown to enhance the apoptotic effects associated with chemotherapy, including doxorubicin, in multiple breast cancer cell lines.Citation50 Expression levels of survivin and Smac/DIABLO mRNA were also found to be reciprocal in primary breast cancer tissues and benign human tissues, respectively.Citation51 Smac/DIABLO inhibits members of the IAP family, and overexpression of this protein was shown to potentiate the effectiveness of chemotherapy in breast cancer cells previously resistant to caspase-mediated apoptosis.Citation51 Specifically, Smac/DIABLO directly interacts with and inhibits XIAP. Conversely, overexpression of XIAP can repress the release of Smac/DIABLO from the mitochondria.Citation52 In our studies, the cytoplasmic expression of Smac/DIABLO significantly increased in CD1 overexpressing breast cancer cells treated with combination CDK4i/doxorubicin (), which correlated with significantly lower levels of XIAP (). Thus in MCF7/CD1 cells, high XIAP expression may be preventing the release of Smac/DIABLO from the mitochondria until a treatment-induced reduction of XIAP occurs. In MCF7/V cells, treatment with CDK4i/doxorubicin did not result in significant changes in XIAP expression, and correspondingly, no significant changes in Smac/DIABLO localization occurred. Recent reports have linked XIAP to expression of CDK4 and cyclin D,Citation53,Citation54 supporting the observed discrepancies in Smac/DIABLO expression/localization and study treatment impact found for the MCF7/V and MCF7/CD1 cells. In addition, the reciprocal expression of survivin and Smac/DIABLO has been described where after induction of apoptotic stimuli, survivin was released from the mitochondria to then bind and inhibit Smac/DIABLO.Citation55 This resulted in decreased Smac/DIABLO available to bind and inhibit other IAPs, such as XIAP, allowing XIAP to directly interact with caspases and block cell death (). Thus, combination CDK4i/doxorubicin treatment may also limit therapeutic resistance through the inhibition of survivin and the promotion of Smac/DIABLO expression.
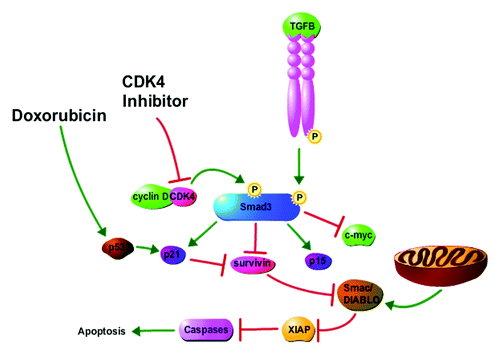
Figure 6. Proposed model for impact of CDK4i and doxorubicin on cancer cell cycle arrest and apoptosis. Cyclin D overexpression mediates CDK4 phosphorylation of Smad3, resulting in lower expression of the Smad3-regulated cdkis p15 and p21 and higher expression of c-myc and survivin. Increased survivin levels inhibit apoptosis through the regulation of Smac/DIABLO and XIAP. CDK4i treatment results in cell cycle arrest and induction of apoptosis through inhibition of CDK4-mediated phosphorylation of Smad3 and the consequent increase of p15 and p21 expression and decrease of c-myc and survivin expression. Doxorubicin treatment results in cell cycle arrest and apoptosis, in part, through the induction of p21 and the direct action of p21 on inhibition of survivin. The figure was made with tools from http://www.proteinlounge.com.
Yang et al. have also previously shown that Smad3- and Rb/E2F4-mediated induction of apoptosis occurred through the transcriptional repression of survivin, and that decreased levels of Rb/E2F4 complex formation correlated with higher levels of survivin.Citation36 It is possible that these increased survivin levels resulted from Rb inactivation, which occurs primarily through CDK mediated phosphorylation. Accordingly, we found that in MCF7/V and MCF7/CD1 cells, combination CDK4i/doxorubicin treatment resulted in decreased pRb expression. The role of doxorubicin in regulation of survivin has also been studied, in which a doxorubicin-induced and p53-mediated increase of p21 expression were shown to inhibit survivin in HepG2 human hepatocellular carcinoma cells.Citation56 The reduction in survivin and XIAP and increase in Smac/DIABLO expression, found after the addition of doxorubicin treatment to 5M Smad3-transduced cells, further demonstrate how combination therapy can amplify the efficacy of either agent alone through a potential Smad3, Rb/E2F4, and doxorubicin-induced p53/p21 mediated repression of survivin and subsequent increase in apoptosis.
Combination CDK inhibitor and doxorubicin therapy has not been specifically studied in the context of Smad3 signaling in breast cancer cells. Previous work found that this combination therapy may be suboptimal in the context of hormone insensitive breast cancer cells, where CDK inhibition may protect the RB-proficient cancer cells from the impact of doxorubicin.Citation57 However, in our studies implementing a hormone sensitive MCF7 breast cancer model, combination therapy was found to be effective. Simultaneously, synergism of Seliciclib, a pan-CDK inhibitor, and doxorubicin has also been observed in a breast cancer MCF7 xenograft model.Citation58 In that study, synergism was found to occur through the induction of cell cycle arrest associated with the upregulation of p21. Our immunoblotting studies also showed significant induction of p21 for both the MCF7/V and CD1 cells treated with doxorubicin. As, stated, the impact of doxorubicin was expected as this agent induces expression of p21 in a p53-dependent manner.Citation20 Furthermore, both MCF7/V and MCF7/CD1 cells treated with combination CDK4i/doxorubicin showed significantly increased p21 expression. Expression of p21 has also been linked to Smad3 in association with Sp1 transcription factor activation.Citation59 Thus, as we have previously shown, CDK4i-mediated restoration of canonical Smad3 action may also contribute to the induction of p21, particularly for the MCF7/V cells.Citation17 These data highlight the benefit of selective treatment strategies that employ combination therapy, rationally impacting several signaling pathways, to collectively contribute to cancer cell cytostatic and cytotoxic events. To this end, Fas, which also increased after combination therapy in our array studies, is a potentially important protein for future work associated with the mediation of extrinsic apoptosis.
Collectively, our work demonstrates the potential therapeutic benefit of combining CDK4i with chemotherapy in an ER+ cyclin D-overexpressing breast cancer model. We showed that decreased cell proliferation, smaller colony formation, and higher apoptosis levels occurred in breast cancer cells treated with combination CDK4i/doxorubicin. We also found that these tumor suppressive events are, in part, regulated by Smad3, and that pharmacological and genetic inhibition of CDK4-mediated Smad3 phosphorylation result in decreased cell proliferation and increased apoptosis. We have previously shown that noncanonical phosphorylation of Smad3 by CDKs affects the expression of p15 and c-myc in breast cancer cells. The current study points toward expression of survivin as an additional key protein regulated by Smad3 signaling, contributing to important work focusing on cancer cell specificity, co-activators, and co-repressors, and also allowing for greater comprehension of the broad-spectrum of influences mediated by the TGFβ superfamily. Further study of CDK-mediated regulation of survivin may have implications for cyclin D-overexpressing ER+ breast cancers in which chemoresistance may be associated with high survivin levels. Overall, the incorporation of CDK inhibition into chemotherapeutic regimens may result in decreased survivin levels, increased apoptosis, and tumor suppression, and contribute to improved patient outcomes.
Methods
Cell culture and generation of stable cell lines
MCF7 cells were obtained from the American Type Culture Collection and maintained in DMEM-F12 according to supplier recommendations. Culture medium was supplemented with 1% Pen Step/Glutamine, 1% nonessential amino acids, 1% sodium pyruvate, and 10% fetal bovine serum. Human cyclin D1 cDNA was inserted into the cytomegalovirus-driven pcDNA-DEST40 expression vector (Invitrogen) containing a neomycin resistance gene to obtain the pEXPR-cyclin D1-expressing plasmid. To create the vector control and cyclin D1-overexpressing (CD1) cell lines, cells were stably transfected with empty vector (pcDNA-DEST40) or pEXPR-cyclin D1 plasmids. Two days after transfection, stable clones were selected in medium containing 500 mg/mL G418 (Invitrogen). The resistant clones were maintained in G418 for 21 d. Cyclin D1 overexpression was verified by immunoblot analysis. Study cells stably expressing empty vector or cyclin D1 were maintained in the same manner as the parental lines.
Antibodies and reagents
Antibodies used were as follows: anti-cyclin D1, anti-Smad3, anti-pSmad3 (Ser423/425), anti-XIAP, anti-Smac/DIABLO, anti-COXIV, anti pRb (Cell Signaling), anti-CDK4, anti-GAPDH, anti-survivin, anti-p21 (Santa Cruz), and anti-β-actin (Sigma). CDK4 inhibitor II (IC50 value of 200 nmol/L) was from EMD Biosciences and doxorubicin (IC50 value of 0.1 μmol/L) was from Sigma. Each drug was resuspended in dimethyl sulfoxide (DMSO), and DMSO was used as the control treatment. Where indicated, cells were treated with the following doses: 0.05% DMSO, 400 nM CDK4i (based on prior dose response studies),Citation17 and 30 nM doxorubicin.
Cell proliferation
The effect of CDK4 inhibitor, doxorubicin, or combination treatment on MCF7 cell proliferation was examined using the MTS (3-[4,5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium, inner salt) assay in the CellTiter 96 aqueous non-radioactive cell proliferation assay kit (Promega), performed according to the manufacturer's instructions. Briefly, study cells were seeded at a density of 2500 cells/100 μL media in triplicate wells of a 96-well plate and incubated for 24 h in 5% CO2/air at 37 °C. The medium was aspirated and replaced with 100 μL fresh medium containing treatment regimen or control DMSO. All drugs were reconstituted in MCF7 media. Cells were incubated with a media change and cell proliferation assessed at days 0, 2, 5, 7, and 9. After a 2-h incubation, the plates were read at 490 nM in a Biorad Microplate Reader. Experiments were repeated 3 times and representative results are shown. Drug concentrations were based on initial dosage studies.Citation17
Apoptosis
Apoptosis was measured for each of the treatment groups using the Terminal deoxynucleotidyl transferase mediated dUTP Nick End Labeling (TUNEL) assay. Cells were treated for 48 h as described above with control DMSO, CDK4 inhibitor, doxorubicin, or combination therapy. The supernatant was collected to isolate any floating apoptotic cells. Following trypsinization, all cells were spun down, rinsed with cold phosphate buffered saline (PBS), and fixed in 1 mL 10% neutral buffered formalin. Fixed cells were embedded into paraffin blocks and TUNEL analysis was performed at the Pathology Core Facility of Northwestern University. Impact of treatments was normalized to control treatment within each cell line. Experiments were repeated 3 times and representative results are shown.
3D cell culture
MCF7/V and MCF7/CD1 cells were infected with GFP expressing lentiviral constructs containing CS2 vector control, WT Smad3, or 5M Smad3. The 5M Smad3 expression plasmid has been described previously, and contains multiple CDK phosphorylation site mutations (T8/T179/S204/S208/S213) that result in the inhibition of CDK4/2 phosphorylation.Citation11 This Smad3 mutant was a generous gift from Dr Fang Liu (Rutgers, State University of New Jersey). GFP-positive cells were isolated on the Beckman Coulter Elite at the Northwestern Flow Cytometry core facility. After multiple passages of stable cell lines, sorted Smad3 expressing cells were trypsinized and resuspended in growth medium containing 2% Matrigel (BD Bioscience). Cells were seeded at a density of 1000–1500 cells/well in a BD Matrigel Matrix 96-well plate (BD Bioscience) containing an underlying 1-mm thick bed of Matrigel. Cells were treated as described above. Cells received fresh medium every 48 h. Pictures of growing colonies were taken on days 4 and 12. Average colony size was measured using MetaMorph software (Molecular Devices). Experiments were conducted three times, and representative results are shown.
Apoptosis antibody array
The Proteome Profiler Human Apoptosis Antibody Array Kit (R&D Systems, Inc.) was used to determine the relative levels of apoptosis-related proteins in the treated cells. According to manufacturer instructions, 400 µg of protein lysate from treated cells was incubated with array membranes for 4 d. Membrane images were taken using an LAS 3000 imaging system and quantitative analysis was performed with MultiGauge software (FujiFilm). Numerical values corresponding to the signal intensity of each spot on the array were exported into a spreadsheet file. The background signal was subtracted from total signal values of each spot and each pair of duplicate spots was averaged for each protein in each treatment group. Experiments were repeated 3 times and representative results are shown.
Immunoblotting
Where indicated, cells were either treated as described above for 48 h or transduced with lentivirus expressing CS2+ control vector, WT Smad3, or 5M Smad3 mutant construct (5M Smad3). Thereafter, cells were scraped, pelleted, and rinsed with ice-cold PBS, then lysed in ice-cold M-PER (Thermo Scientific) containing protease inhibitors. Cellular lysates were spun, supernatants were recovered, and the protein concentration was determined using the Bio-Rad protein assay kit. For the lysates subjected to mitochondrial fractionation, cells were scraped and homogenized in sucrose buffer (250 mM sucrose, 10 mM TRIS-HCl, 0.1 mM EGTA) and spun at 100 × g for 10 min. The supernatant was collected in a new tube and spun at 10 000 × g for 10 min. The supernatant (cytoplasmic fraction) was transferred to another tube and the pellet (mitochondrial fraction) was suspended in 50 uL of sucrose buffer. Cox IV, a mitochondrial protein, and GAPDH, a cytoplasmic protein, served as the control for the mitochondrial fractionation. Fifty micrograms of lysate was diluted in 2× SDS-PAGE sample buffer (1:1,v/v), electrophoresed, and transferred to a PVDF membrane. Membranes were blocked with 5% milk or 5% BSA in TBS-T at room temperature for 1 h and incubated with appropriate primary antibody at 4 °C overnight. After rinsing with TBS-T, the membrane was incubated with secondary antibody in 5% milk or 5% BSA for 1 h at room temperature. Protein bands were visualized by an ECL detection system (Supersignal, Thermo Scientific). For reblotting, membranes were agitated with Restore PLUS Stripping Buffer (Thermo Scientific) for 15 min at room temperature. Experiments were repeated 3 times and representative results are shown.
Statistical analysis
Results were presented as mean values ± SE and analyzed by the unpaired, two tailed Student t test. P < 0.05 was considered statistically significant.
| Abbreviations: | ||
| ER | = | estrogen receptor |
| CDK | = | cyclin-dependent kinase |
| CDKi | = | cyclin-dependent kinase inhibitor |
| Rb | = | retinoblastoma |
| TGFβ | = | transforming growth factor beta |
| IAP | = | inhibitor of apoptosis |
| EMT | = | epithelial-to-mesenchymal transition |
| DMSO | = | dimethyl sulfoxide |
| Smac | = | second mitochondria-derived activator of caspase |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Stacey Tobin and Megan Novak for their editorial assistance and Dr Vamsi Parini for assessment of TUNEL staining.
Financial Support
J.S.J. is a Lynn Sage Scholar supported by the NIH K22 CA138776 and R01GM097220 research grant, the Central Surgical Association Foundation, the Society of Surgical Oncology, the Saslow family, and A Sister’s Hope. E.T. is a Dr John N. Nicholson Fellow, a Chicago Biomedical Consortium Scholar supported by the Searle Funds at the Chicago Community Trust, and is also supported by the NIH/NCI training grant T32CA09560 and the National Cancer Institute Kirchstein-NRSA Fellowship F31 CA168106-02.
References
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin 2010; 60:277 - 300; http://dx.doi.org/10.3322/caac.20073; PMID: 20610543
- Buckley MF, Sweeney KJ, Hamilton JA, Sini RL, Manning DL, Nicholson RI, deFazio A, Watts CK, Musgrove EA, Sutherland RL. Expression and amplification of cyclin genes in human breast cancer. Oncogene 1993; 8:2127 - 33; PMID: 8336939
- Keyomarsi K, Pardee AB. Redundant cyclin overexpression and gene amplification in breast cancer cells. Proc Natl Acad Sci U S A 1993; 90:1112 - 6; http://dx.doi.org/10.1073/pnas.90.3.1112; PMID: 8430082
- Gillett C, Fantl V, Smith R, Fisher C, Bartek J, Dickson C, Barnes D, Peters G. Amplification and overexpression of cyclin D1 in breast cancer detected by immunohistochemical staining. Cancer Res 1994; 54:1812 - 7; PMID: 8137296
- Aaltonen K, Amini RM, Landberg G, Eerola H, Aittomäki K, Heikkilä P, Nevanlinna H, Blomqvist C. Cyclin D1 expression is associated with poor prognostic features in estrogen receptor positive breast cancer. Breast Cancer Res Treat 2009; 113:75 - 82; http://dx.doi.org/10.1007/s10549-008-9908-5; PMID: 18240019
- Hui R, Cornish AL, McClelland RA, Robertson JF, Blamey RW, Musgrove EA, Nicholson RI, Sutherland RL. Cyclin D1 and estrogen receptor messenger RNA levels are positively correlated in primary breast cancer. Clin Cancer Res 1996; 2:923 - 8; PMID: 9816251
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene 2009; 28:2925 - 39; http://dx.doi.org/10.1038/onc.2009.170; PMID: 19561645
- Kim JK, Diehl JA. Nuclear cyclin D1: an oncogenic driver in human cancer. J Cell Physiol 2009; 220:292 - 6; http://dx.doi.org/10.1002/jcp.21791; PMID: 19415697
- Yu Q, Sicinska E, Geng Y, Ahnström M, Zagozdzon A, Kong Y, Gardner H, Kiyokawa H, Harris LN, Stål O, et al. Requirement for CDK4 kinase function in breast cancer. Cancer Cell 2006; 9:23 - 32; http://dx.doi.org/10.1016/j.ccr.2005.12.012; PMID: 16413469
- Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 2006; 9:13 - 22; http://dx.doi.org/10.1016/j.ccr.2005.12.019; PMID: 16413468
- Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004; 430:226 - 31; http://dx.doi.org/10.1038/nature02650; PMID: 15241418
- Tarasewicz E, Jeruss JS. Phospho-specific Smad3 signaling: impact on breast oncogenesis. Cell Cycle 2012; 11:2443 - 51; http://dx.doi.org/10.4161/cc.20546; PMID: 22659843
- Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol Ther 2001; 91:1 - 34; http://dx.doi.org/10.1016/S0163-7258(01)00143-7; PMID: 11707292
- Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res 2005; 65:7968 - 75; PMID: 16140969
- Jeruss JS, Sturgis CD, Rademaker AW, Woodruff TK. Down-regulation of activin, activin receptors, and Smads in high-grade breast cancer. Cancer Res 2003; 63:3783 - 90; PMID: 12839974
- Sundqvist A, Ten Dijke P, van Dam H. Key signaling nodes in mammary gland development and cancer: Smad signal integration in epithelial cell plasticity. Breast Cancer Res 2012; 14:204; http://dx.doi.org/10.1186/bcr3066; PMID: 22315972
- Zelivianski S, Cooley A, Kall R, Jeruss JS. Cyclin-dependent kinase 4-mediated phosphorylation inhibits Smad3 activity in cyclin D-overexpressing breast cancer cells. Mol Cancer Res 2010; 8:1375 - 87; http://dx.doi.org/10.1158/1541-7786.MCR-09-0537; PMID: 20736297
- Cooley A, Zelivianski S, Jeruss JS. Impact of cyclin E overexpression on Smad3 activity in breast cancer cell lines. Cell Cycle 2010; 9:4900 - 7; http://dx.doi.org/10.4161/cc.9.24.14158; PMID: 21150326
- Dickson MA, Schwartz GK. Development of cell-cycle inhibitors for cancer therapy. Curr Oncol 2009; 16:36 - 43; PMID: 19370178
- Bar-On O, Shapira M, Hershko DD. Differential effects of doxorubicin treatment on cell cycle arrest and Skp2 expression in breast cancer cells. Anticancer Drugs 2007; 18:1113 - 21; http://dx.doi.org/10.1097/CAD.0b013e3282ef4571; PMID: 17893511
- Hortobágyi GN. Anthracyclines in the treatment of cancer. An overview. Drugs 1997; 54:Suppl 4 1 - 7; PMID: 9361955
- Shapiro GI. Cyclin-dependent kinase pathways as targets for cancer treatment. J Clin Oncol 2006; 24:1770 - 83; http://dx.doi.org/10.1200/JCO.2005.03.7689; PMID: 16603719
- Reed JC, Bischoff JR. BIRinging chromosomes through cell division--and survivin’ the experience. Cell 2000; 102:545 - 8; http://dx.doi.org/10.1016/S0092-8674(00)00076-3; PMID: 11007472
- Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997; 3:917 - 21; http://dx.doi.org/10.1038/nm0897-917; PMID: 9256286
- Swana HS, Grossman D, Anthony JN, Weiss RM, Altieri DC. Tumor content of the antiapoptosis molecule survivin and recurrence of bladder cancer. N Engl J Med 1999; 341:452 - 3; http://dx.doi.org/10.1056/NEJM199908053410614; PMID: 10438269
- Adida C, Haioun C, Gaulard P, Lepage E, Morel P, Briere J, Dombret H, Reyes F, Diebold J, Gisselbrecht C, et al. Prognostic significance of survivin expression in diffuse large B-cell lymphomas. Blood 2000; 96:1921 - 5; PMID: 10961895
- Ito T, Shiraki K, Sugimoto K, Yamanaka T, Fujikawa K, Ito M, Takase K, Moriyama M, Kawano H, Hayashida M, et al. Survivin promotes cell proliferation in human hepatocellular carcinoma. Hepatology 2000; 31:1080 - 5; http://dx.doi.org/10.1053/he.2000.6496; PMID: 10796883
- Kawasaki H, Altieri DC, Lu CD, Toyoda M, Tenjo T, Tanigawa N. Inhibition of apoptosis by survivin predicts shorter survival rates in colorectal cancer. Cancer Res 1998; 58:5071 - 4; PMID: 9823313
- Satoh K, Kaneko K, Hirota M, Masamune A, Satoh A, Shimosegawa T. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer 2001; 92:271 - 8; http://dx.doi.org/10.1002/1097-0142(20010715)92:2<271::AID-CNCR1319>3.0.CO;2-0; PMID: 11466679
- Xing N, Qian J, Bostwick D, Bergstralh E, Young CY. Neuroendocrine cells in human prostate over-express the anti-apoptosis protein survivin. Prostate 2001; 48:7 - 15; http://dx.doi.org/10.1002/pros.1076; PMID: 11391682
- Ryan B, O’Donovan N, Browne B, O’Shea C, Crown J, Hill AD, McDermott E, O’Higgins N, Duffy MJ. Expression of survivin and its splice variants survivin-2B and survivin-DeltaEx3 in breast cancer. Br J Cancer 2005; 92:120 - 4; http://dx.doi.org/10.1038/sj.bjc.6602314; PMID: 15611790
- Yamashita S, Masuda Y, Kurizaki T, Haga Y, Murayama T, Ikei S, Kamei M, Takeno S, Kawahara K. Survivin expression predicts early recurrence in early-stage breast cancer. Anticancer Res 2007; 27:4C 2803 - 8; PMID: 17695451
- Hinnis AR, Luckett JC, Walker RA. Survivin is an independent predictor of short-term survival in poor prognostic breast cancer patients. Br J Cancer 2007; 96:639 - 45; http://dx.doi.org/10.1038/sj.bjc.6603616; PMID: 17285125
- Yonesaka K, Tamura K, Kurata T, Satoh T, Ikeda M, Fukuoka M, Nakagawa K. Small interfering RNA targeting survivin sensitizes lung cancer cell with mutant p53 to adriamycin. Int J Cancer 2006; 118:812 - 20; http://dx.doi.org/10.1002/ijc.21350; PMID: 16108013
- Olie RA, Simões-Wüst AP, Baumann B, Leech SH, Fabbro D, Stahel RA, Zangemeister-Wittke U. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res 2000; 60:2805 - 9; PMID: 10850418
- Yang J, Song K, Krebs TL, Jackson MW, Danielpour D. Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced apoptosis and tumor progression. Oncogene 2008; 27:5326 - 38; http://dx.doi.org/10.1038/onc.2008.165; PMID: 18504435
- Jaffer S, Orta L, Sunkara S, Sabo E, Burstein DE. Immunohistochemical detection of antiapoptotic protein X-linked inhibitor of apoptosis in mammary carcinoma. Hum Pathol 2007; 38:864 - 70; http://dx.doi.org/10.1016/j.humpath.2006.11.016; PMID: 17350670
- Bukholm IK, Berner JM, Nesland JM, Børresen-Dale AL. Expression of cyclin Ds in relation to p53 status in human breast carcinomas. Virchows Arch 1998; 433:223 - 8; http://dx.doi.org/10.1007/s004280050240; PMID: 9769125
- Jeruss JS, Liu NX, Chung Y, Magrane G, Waldman F, Edgerton S, Yang X, Thor AD. Characterization and chromosomal instability of novel derived cell lines from a wt-erbB-2 transgenic mouse model. Carcinogenesis 2003; 24:659 - 64; http://dx.doi.org/10.1093/carcin/bgg001; PMID: 12727793
- Li Y, Wang M, Carra C, Cucinotta FA. Modularized Smad-regulated TGFβ signaling pathway. Math Biosci 2012; 240:187 - 200; http://dx.doi.org/10.1016/j.mbs.2012.07.005; PMID: 22892478
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ, Ginther C, Atefi M, Chen I, Fowst C, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 2009; 11:R77; http://dx.doi.org/10.1186/bcr2419; PMID: 19874578
- Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H, Dowsett M, Jiang A, Smith RA, Maira SM, et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 2011; 1:338 - 51; http://dx.doi.org/10.1158/2159-8290.CD-11-0101; PMID: 22049316
- Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011; 11:558 - 72; http://dx.doi.org/10.1038/nrc3090; PMID: 21734724
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO, Ettl J, Patel R, Pinter T, Schmidt M, et al. Final results of a randomized Phase II study of PD 0332991, a cyclin-dependent kinase (CDK)-4/6 inhibitor, in combination with letrozole vs letrozole alone for first-line treatment of ER+/HER2- advanced breast cancer (PALOMA-1; TRIO-18). AACR Meeting; 2014; San Diego Convention Center, San Diego, CA; 2014.
- Heldin CH, Moustakas A. Role of Smads in TGFβ signaling. Cell Tissue Res 2012; 347:21 - 36; http://dx.doi.org/10.1007/s00441-011-1190-x; PMID: 21643690
- Shao J, Teraishi F, Katsuda K, Tanaka N, Fujiwara T. p53 inhibits adriamycin-induced down-regulation of cyclin D1 expression in human cancer cells. Biochem Biophys Res Commun 2002; 290:1101 - 7; http://dx.doi.org/10.1006/bbrc.2001.6314; PMID: 11798189
- Bandyopadhyay A, Wang L, Agyin J, Tang Y, Lin S, Yeh IT, De K, Sun LZ. Doxorubicin in combination with a small TGFbeta inhibitor: a potential novel therapy for metastatic breast cancer in mouse models. PLoS One 2010; 5:e10365; http://dx.doi.org/10.1371/journal.pone.0010365; PMID: 20442777
- Ryan BM, Konecny GE, Kahlert S, Wang HJ, Untch M, Meng G, Pegram MD, Podratz KC, Crown J, Slamon DJ, et al. Survivin expression in breast cancer predicts clinical outcome and is associated with HER2, VEGF, urokinase plasminogen activator and PAI-1. Ann Oncol 2006; 17:597 - 604; http://dx.doi.org/10.1093/annonc/mdj121; PMID: 16403812
- Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003; 22:8581 - 9; http://dx.doi.org/10.1038/sj.onc.1207113; PMID: 14634620
- Fandy TE, Shankar S, Srivastava RK. Smac/DIABLO enhances the therapeutic potential of chemotherapeutic drugs and irradiation, and sensitizes TRAIL-resistant breast cancer cells. Mol Cancer 2008; 7:60; http://dx.doi.org/10.1186/1476-4598-7-60; PMID: 18590557
- Mansour A, Nabil M, Ali-Labib R, Said H, Annos F. Reciprocal expression of survivin and SMAC/DIABLO in primary breast cancer. Med Oncol 2012; 29:2535 - 42; http://dx.doi.org/10.1007/s12032-011-0129-0; PMID: 22161156
- Flanagan L, Sebastià J, Tuffy LP, Spring A, Lichawska A, Devocelle M, Prehn JH, Rehm M. XIAP impairs Smac release from the mitochondria during apoptosis. Cell Death Dis 2010; 1:e49; http://dx.doi.org/10.1038/cddis.2010.26; PMID: 21364655
- Cao Z, Zhang R, Li J, Huang H, Zhang D, Zhang J, Gao J, Chen J, Huang C. X-linked inhibitor of apoptosis protein (XIAP) regulation of cyclin D1 protein expression and cancer cell anchorage-independent growth via its E3 ligase-mediated protein phosphatase 2A/c-Jun axis. J Biol Chem 2013; 288:20238 - 47; http://dx.doi.org/10.1074/jbc.M112.448365; PMID: 23720779
- Che Y, Ye F, Xu R, Qing H, Wang X, Yin F, Cui M, Burstein D, Jiang B, Zhang DY. Co-expression of XIAP and cyclin D1 complex correlates with a poor prognosis in patients with hepatocellular carcinoma. Am J Pathol 2012; 180:1798 - 807; http://dx.doi.org/10.1016/j.ajpath.2012.01.016; PMID: 22429965
- Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem 2003; 278:23130 - 40; http://dx.doi.org/10.1074/jbc.M300957200; PMID: 12660240
- Xiong J, Hu L, Li Y, Dou L, Cai P, Tang Z, Wang L. Effect of survivin regulation of transcription level by p21waf1 overexpression in HepG2 hepatocellular carcinoma cells. J Huazhong Univ Sci Technolog Med Sci 2008; 28:308 - 13; http://dx.doi.org/10.1007/s11596-008-0318-z; PMID: 18563330
- Sun B, Wingate H, Swisher SG, Keyomarsi K, Hunt KK. Absence of pRb facilitates E2F1-induced apoptosis in breast cancer cells. Cell Cycle 2010; 9:1122 - 30; http://dx.doi.org/10.4161/cc.9.6.10990; PMID: 20237430
- Appleyard MV, O’Neill MA, Murray KE, Paulin FE, Bray SE, Kernohan NM, Levison DA, Lane DP, Thompson AM. Seliciclib (CYC202, R-roscovitine) enhances the antitumor effect of doxorubicin in vivo in a breast cancer xenograft model. Int J Cancer 2009; 124:465 - 72; http://dx.doi.org/10.1002/ijc.23938; PMID: 19003963
- Pardali K, Kurisaki A, Morén A, ten Dijke P, Kardassis D, Moustakas A. Role of Smad proteins and transcription factor Sp1 in p21(Waf1/Cip1) regulation by transforming growth factor-beta. J Biol Chem 2000; 275:29244 - 56; http://dx.doi.org/10.1074/jbc.M909467199; PMID: 10878024