
Abstract
DNA damage response (DDR) that includes cell cycle check points, DNA repair, apoptosis, and senescence is intimately linked with cancer. It shields an organism against cancer development when genomic integrity fails. DNA repair pathways protect the cells from tumor progression caused as a result of DNA damage induced by irradiation or due to chemotherapeutic treatment. Many promising anticancer agents have been identified that target specific DNA repair pathways in response to DNA damage thereby leading to apoptosis. Here we identified a novel bisindole-PBD conjugate that possess potent anticancer activity in breast cancer cells. Further studies aimed at understanding the mechanism of action of the molecule showed its role in DNA damage induced apoptosis via inhibition of DNA repair pathway. Trypan blue and BrdU assay exhibited a dose-dependent effect. Single-stranded DNA damage was observed by COMET assay. In addition DNA damage induced ROS generation with simultaneous activation of ATM and ATR upon compound treatment was observed. Further downregulation of Bcl-XL and activation of Bax showed DNA damage induced apoptosis in MCF-7 and MDAMB-231 cells. In conclusion, it can be summarized that bisindole-PBD conjugate induces DNA damage in a dose dependent (2, 4, and 8 μM) manner by inhibiting the DNA repair genes.
Keywords: :
Introduction
Cellular homeostasis and organism viability are maintained by intact genome condition. Continuous exposure to endogenous and environmental DNA damaging agents disturbs the genomic integrity. Higher the level of DNA damage, higher is the possibility for the cells to undergo arrest or death.
Of all the DNA damage, DNA double-strand breaks (DSBs) are most harmful. They can induce chromosomal rearrangements which in turn activate different oncogenes and suppress tumor suppressor genes thereby causing malignant transformation.Citation1 Cytotoxic lesions like DSBs are produced by radiomimetic drugs other than ionizing radiation. It is one of the most destructive forms of DNA damage. Unrepaired DSBs causes genomic instability via breakage in chromosome, translocations and hence help in tumorigenesis.Citation2 To defend themselves against DSBs, eukaryotes employ DNA damage response (DDR) pathway to detect DNA damage, arrest cell cycle until the damaged DNA is repaired, and to induce cell death if the damage is unrepairable. DDR initiates two major stages: identifying DNA breaks followed by downstream events leading to cell cycle arrest and DNA damage repair and subsequent cell cycle resumption. DDR pathway can activate cell cycle checkpoints to arrest the cell or they can activate the DNA repair pathways. Cells depend on two major pathways to repair DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ). Both the pathways are complementary and function under different conditions. Whereas HR requires homologous template, NHEJ can function throughout the cell cycle without any template.Citation3 DDR pathway involves sensor proteins such as DNA-dependent protein kinase catalytic subunit (DNA-PKcs), ATR, and ATM. These proteins have the ability to recognize damaged DNA and convey the damage signal to the effector proteins that may be regulator of cell cycle progression, or chromatin modifier and result in DNA repair.Citation4-Citation6 Several factors influence DSB processing, signaling, and repair that accumulate at damaged sites in focal structures termed IR-induced foci (IRIF). Detection of DSBs performed by Mre11–Rad50–Nbs1 (MRN) and Ku70–Ku80 complexes, which activates ataxia telangiectasia mutated (ATM).Citation7 This MRN complex plays a pivotal role in double stranded DNA break repair and DNA recombination.Citation8-Citation10 The activated ATM connects DDR to cell cycle via p53. The activation and stabilization of p53 is performed by ATM as a result of phosphorylation of p53 at serine 15.Citation11 In response to DNA damage, there is increase in p53 in cells through posttranscriptional mechanism. As a result transactivation is enhanced and thereby activation of downstream genes occurs.Citation12 Apart from this, ATM phosphorylates Chk2 and Chk1 as response to DNA damageCitation13 and recruits phosphorylated form of H2AX (gamma H2AX) at the site of DSB that in turn decondenses the chromatin to assemble repair and apoptotic cascade factors..
Another factor maintaining genomic integrity and cellular response to DNA damage is the level of intracellular reactive oxygen species (ROS). DNA damage results in the increase of ROS levels which is crucial in the regulation of cell death and survival. This ROS is partly known to regulate p53 activity. Various stimuli like chemotherapeutic drugs, TNF-α, etc. generate ROS.Citation14 Intracellular ROS is generated through some enzymes called NAD(P)H oxidases (Noxes) and Nox induced ROS has been implicated in oncogenic signaling.Citation15 However, exact mechanism by which DNA damage induces ROS and its involvement of DDR protein is still not clear.Citation16,Citation17
Mechanism behind the cause of cancer is mainly established as genetic instability. Though the exact mechanism of cancer cell initiation and progression and the pathways involved remain unclear, many hypotheses, research, and resources are being dedicated toward it. Breast cancer is one of the most common types of cancer that occur in women and it is the main cause of cancer-associated mortality. Targeting the DNA molecule has shown to be most common for almost all the clinically active anticancer drugs.Citation18 The naturally occurring pyrrolo[2,1-c][1,4]benzodiazepines (PBD) isolated from Streptomyces species, exhibit efficient anticancer activity. They bind to DNA in a sequence specific manner (via covalent binding). Likewise bis-(indolyl) methane/bisindole and its derivativesCitation19 are also known to have antiproliferative and apoptotic effect both in vitro and in vivo.Citation20-Citation25 The apoptosis-inducing capability as well as HDAC inhibitory role of the bisindole-PBD conjugate (5b) has been well established in breast cancer cellsCitation26 but its in-depth mechanism on its role in DNA damage repair pathway has not been investigated. Here in this study the role of bisindole-PBD conjugate 5b on DNA damage and DNA repair pathway has been discussed.
Results
Bisindole-linked pyrrolo[2,1-c][1,4]benzodiazepine induce cell death in MCF-7 and MDAMB-231 cells
Bisindole linked prrrolo[2,1-c][1,4]benzodiazepine (5b) () was synthesized by coupling active moiety of bisindolyl methane derivative through 3- and 4-OH groups to DC-81, a well-known PBD via alkyl spacers. To examine the role of the compound (5b) Trypan Blue cell exclusion assay was performed. The number of cells stained with trypan blue, indicated the percentage of cell death. In both the cell lines MCF-7 and MDMB-231, the compound 5b produced increased antiproliferative activity as compared with SAHA to induce death at 4 and 8 μM concentrations at 24 h (). In MCF-7 cells, there was marked increase in percentage cell death i.e.; approximately 35%, 60%, and 72% at 2, 4, and 8 μM respectively, in comparison to untreated control that showed 10% cell death and well known inhibitor SAHA having 25% cell death at 1 μM concentration (). In MDAMB-231 cells, after incubation with the compound 5b for 24 h produced a 5- to 7-fold increase in cell death at 4 and 8 μM concentrations as compared with 2-fold cell death in cells treated with SAHA at 5 μM dose for the same time period ().
Figure 1. Induction of cell death by bisindole-PBD hybrid. (A) Chemical structure of SAHA and bisindole-PBD hybrid conjugates. Bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine induces cell death in MCF-7 and MDAMB-231 cells. Trypan blue exclusion assay in MCF-7 (B) and in MDAMB-231 (C) showing cell death at 2, 4, and 8 µM. Each experiment was performed thrice. Error bar represents standard deviation from three different experiments. BrdU cell proliferation assay in MCF-7 (D) and MDAMB-231 (E). MCF-7 cells were seeded at a density of 10 000 cells and were grown for 24 h, BrdU was incubated for 5 h at RT followed by treatment 1 µM SAHA (in MCF-7), 5 µM SAHA (in MDAMB-231), 2, 4, and 8 µM 5b for 24 h. The intensity of blue color was measured at 450 nm which is proportional to the amount of BrdU incorporated in proliferating cells. Data presented from three independent experiments. Error bar represents standard deviation from three different experiments
![Figure 1. Induction of cell death by bisindole-PBD hybrid. (A) Chemical structure of SAHA and bisindole-PBD hybrid conjugates. Bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine induces cell death in MCF-7 and MDAMB-231 cells. Trypan blue exclusion assay in MCF-7 (B) and in MDAMB-231 (C) showing cell death at 2, 4, and 8 µM. Each experiment was performed thrice. Error bar represents standard deviation from three different experiments. BrdU cell proliferation assay in MCF-7 (D) and MDAMB-231 (E). MCF-7 cells were seeded at a density of 10 000 cells and were grown for 24 h, BrdU was incubated for 5 h at RT followed by treatment 1 µM SAHA (in MCF-7), 5 µM SAHA (in MDAMB-231), 2, 4, and 8 µM 5b for 24 h. The intensity of blue color was measured at 450 nm which is proportional to the amount of BrdU incorporated in proliferating cells. Data presented from three independent experiments. Error bar represents standard deviation from three different experiments](/cms/asset/7cae0907-f6f4-4a80-9757-1c0cde2195c2/kcbt_a_10929705_f0001.gif)
The viability of the cells after compound 5b exposure was again confirmed by BrdU cell proliferation assay. The results further confirmed the cytotoxicity of the compound at different concentrations, such as 2, 4, and 8 µM. There was slight decrease in fold change of DNA synthesis at 4 and 8 µM concentrations as detected by level of less incorporated bromodeoxyuridine () in both the cell lines. A decrease in percentage up to 46% and 42% at 4 and 8 µM concentrations in MCF-7 cells () and 65% and 48% at 4 and 8 µM concentrations in MDAMB-231 cells percentage was observed (). SAHA at 1 µM concentration in MCF-7 and 5 µM in MDAMB-231 showed no significant effect.
Bisindole-linked pyrrolo[2,1-c][1,4]benzodiazepine-mediated induction of reactive oxygen species
Reactive oxygen species (ROS) and mitochondria play important role in apoptosis. ROS generation is seen during HDACi-induced cell death.Citation27-Citation29 However, ROS also plays an indirect role by modulating the signaling pathways. In previous studies, we have shown the HDAC inhibitory role of the compound 5b.Citation26 We have checked the production of hydrogen peroxide by carboxy-H2DCFDA staining method, after incubation of both the cell lines with the compound 5b at 4 µM concentration for 30 min. Pronounced ROS generation was observed under fluorescent microscope upon compound after the treatment ().
Figure 2. Exposure to DNA damaging agents leads to increased level of ROS. Induction of ROS in MCF-7 and MDAMB-231 cells after treatment with SAHA (1 and 5 µM) and 5b at 4 µM for 24 h. Treated cells were incubated with carboxy-H2DCFDA for 30 min at 37 °C and ROS were observed by fluorescence microscopy. In both the cell lines there was significant increase of ROS as compared with controls and SAHA treated samples.
Administration of bisindole-linked pyrrolo[2,1-c][1,4]benzodiazepine leads to enhanced mitochondrial membrane disruption and DNA damage leading to apoptosis
Mitochondria play a key role in the apoptotic process. It controls apoptosis at several levels; mainly in maintenance of ATP production and mitochondrial membrane potential for the release of several apoptotic factors from the inter membrane space to the cytosol. Therefore mitochondrial membrane potential serves as an important marker for measuring the status of the cells. Loss of mitochondrial membrane potential directly correlates with percentage of apoptosis. Upon treatment of the compound there was a clear decrease in the mitochondrial membrane potential. SAHA treated cells also exhibited a similar pattern ().
Figure 3. Assessment of mitochondrial membrane potential and induction of apoptosis after compound treatment. (A and B) Mitochondrial membrane potential was measured by JC-1 staining and analyzed by flowcytometry. Cells were grown in 6-well plate at a density of 2 × 105 per well. Treatment was given at 5 µM (in MDAMB-231), 1 µM (MCF-7) concentration of SAHA, 2, 4, and 8 µM 5b for 24 h. After trypsinization, cells were incubated with 10 μg/mL of JC-1 for 20 min at 37 °C. Red/green fluorescence intensity ratio was measured by flow cytometry. (C and D) Western blot analysis of proteins associated with apoptosis. MCF-7 AND MDAMB-231 cell were treated with SAHA (1 µM in MCF-7 and 5 µM in MDAMB-231) and 5b (2, 4, 8 µM) and were incubated for 24 h. Proteins were extracted for western blots analysis with different antibodies. β-actin was used as loading control.
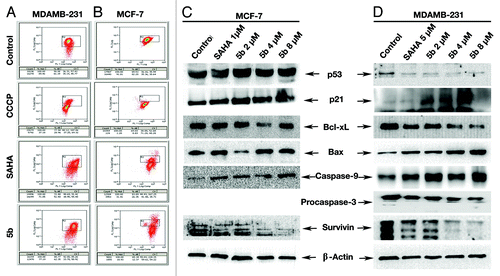
The intracellular signaling pathway leading to the onset of apoptosis is very diverse. One of the possible explanations to the activation of Caspases via mitochondrial pathway involves apoptotic signaling through Bcl-2 super family that includes Bax, Bak, and Bid. Instead of directly activating Caspases, these members induce caspase signaling through mitochondrial involvement in release of pro apoptotic proteins such as cytochrome c.Citation30 We have performed western blot analysis with a series of anti-apoptotic proteins after isolating total protein from the cells upon treatment with the compound. We have also isolated total protein from untreated cells and SAHA-treated cells. Upon hybridization using anti-Bax antibody, we found a significant increase in BAX production in compound treated samples (). Similarly, there was a significant increase in Caspase-9 in response to compound treatment (). The anti-apoptotic protein Bcl-xL was downregulated along with Survivin. Induction of p21 was produced in MCF-7 cells. The level of p53 was also highly elevated showing a dependency of p21 induction with elevation of p53. MDAMB-231 cells are deficient in p53. p21 levels were also increased in these cell showing that the induction is independent of p53 concluding that the induction of apoptosis is due to induced DNA damage which involves both p53 dependent and independent pathway.
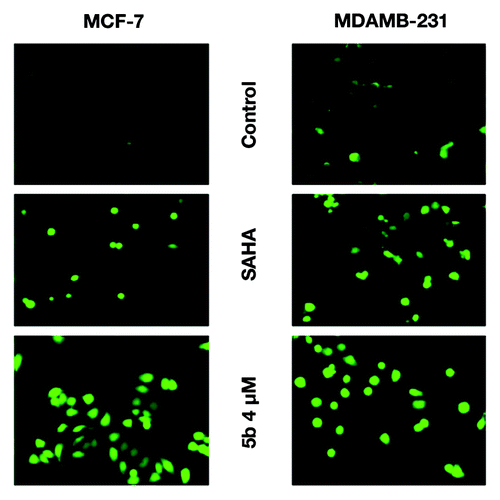
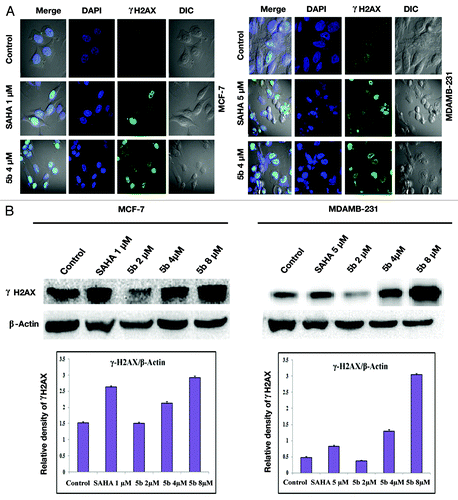
Cells exposed to DNA damaging agents generate DSBs and result in the phosphorylation of histone H2A variant H2AX.Citation31 γ-H2AX or phosphorylated H2AX is generated by phosphorylation of histone H2AX on serine residue at 139 (γ-H2AX). This foci generation is an early marker for DNA double strand break. Bisindole-PBD conjugate induce formation of γ-H2AX foci as a result of DNA damage. After treatment with the compound at 4 µM, concentration, there was an increase in γ-H2AX foci clearly depicting DSB breaks as observed under confocal laser microscope (). To further confirm the induction of gamma H2AX, we performed western blot with anti- γ-H2AX antibody. There was an elevated level of γ-H2AX in cells treated with compound at 4 and 8 μM concentrations ().
Figure 4. Identification of γ-H2AX foci and γ-H2AX protein expression induced from DSBs. (A) Immunofluorescence studies of γ-H2AX after treatment with compounds. Cells were incubated with 5 µM (in MDAMB-231), 1 µM (MCF-7) concentration of SAHA, or 4 µM of 5b for 24 h. Cells were fixed and processed for immunofluorescence using and γH2AX antibody. Nuclei were counterstained with DAPI. Images were acquired in Olympus confocal laser microscope. (B) Study of phosphorylation of H2AX was performed by western blot analysis. Proteins were extracted from compound treated cells at different concentrations—1 µM SAHA (in MCF-7), 5 µM SAHA (in MDAMB-231), 2, 4, and 8 µM 5b for 24 h—and immunoblot analysis was performed to verify γ-H2AX protein expression after treatment. Error bar represents standard deviation from three different experiments.
To investigate the possibility of other types of DNA damage, both the cell lines were treated with SAHA (1 µM in MCF-7, 5 µM in MDAMB-231) and 5b at 4 µM for 24 h. The cells were then subjected to single cell gel electrophoresis/COMET assay performed under alkaline conditions. This assay helps to identify damage in DNA including DSBs and single strand break. There was a significant increase in COMET tail length (). There was 65% increase in comet length after incubation with 5b for 24 h in comparison to control MCF-7 cells. SAHA treated cells produced a 22% increase in comet length in the same cell line. MDAMB-231 cells also produced a significant increase in comet tail length upon 5b treatment. There was almost 55% increase in comet length after 5b administration whereas 32% increase was seen in SAHA treated cells.
Figure 5. Bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine induces single strand DNA break in both MCF-7 and MDAMB-231 cells. Cells were treated with 1 µM SAHA (in MCF-7), 5 µM SAHA (in MDAMB-231), and 4 µM of 5b for 24 h. DNA damage was determined by the percentage of DNA tail area. Each experiment was performed thrice. Error bar represents standard deviation obtained from three different independent experiments. (A) MCF-7 and (B) MDAMB-231.
![Figure 5. Bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine induces single strand DNA break in both MCF-7 and MDAMB-231 cells. Cells were treated with 1 µM SAHA (in MCF-7), 5 µM SAHA (in MDAMB-231), and 4 µM of 5b for 24 h. DNA damage was determined by the percentage of DNA tail area. Each experiment was performed thrice. Error bar represents standard deviation obtained from three different independent experiments. (A) MCF-7 and (B) MDAMB-231.](/cms/asset/f84c2a89-d552-4a78-a3a3-33ac9dabf9e9/kcbt_a_10929705_f0005.gif)
Bisindole-linked pyrrolo[2,1-c][1,4]benzodiazepine induced DNA damage activates ATM and ATR
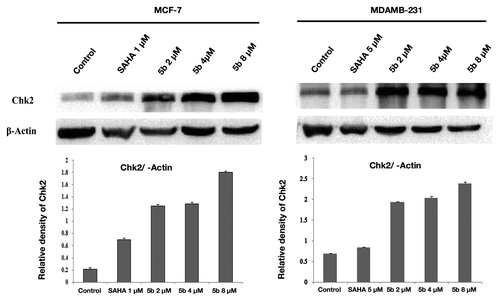
Cell cycle check points are activated upon production of DNA damage. These check point activation are controlled by two major kinases, ATM and ATR. ATM is a serine/threonine protein kinase that is recruited and activated by DSBs.Citation32 It plays an important role in cell cycle machinery and activates checkpoint signaling in response to DSBs. ATR has specific activation that is mainly activated when there is a stop in replication fork. ATM and ATR kinases are placed near DNA breaks. Recruitment of ATM at the DNA damage sites occurs via human MRN complex (Mre11–Rad50–NBS1).Citation33 ATM upon DNA damage autophosphorylates and that in turn activates DNA damage checkpoints including Chk1 and Chk2.Citation34 Treatment with SAHA (1 µM in MCF-7, 5 µM in MDAMB-231) and 5b (2, 4, and 8 µM) for 24 h upregulated the expression of DNA damage specific genes ATM, ATR () and check point protein Chk2 level in both the cell lines (), confirming further that upon treatment with the compound produced DNA damage in both the breast cancer cell lines that led to the upregulation of the check point proteins.
Figure 6. DNA damage activates ATM and ATR gene expressions. Treatment was given at 5 µM (in MDAMB-231), 1 µM (MCF-7) concentration of SAHA, 2, 4, and 8 µM 5b for 24 h. Total RNA was extracted from treated cells. RT-PCR was conducted using specific primers. PCR products were separated on 1.2% agarose gel electrophoresis and visualized under UV light. GAPDH was used as loading control. Experiment was repeated three times. Error bar represents standard deviation from three different experiments.
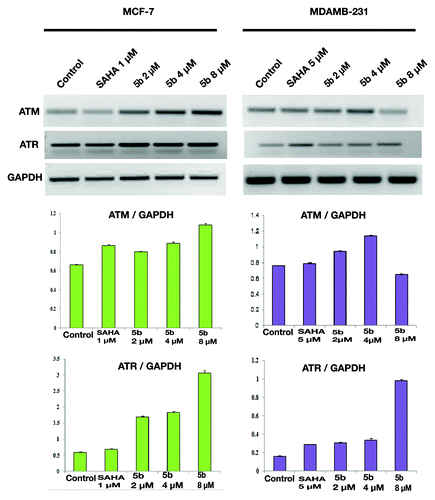
Figure 7. Effect of bisindole-PBD hybrid on DNA damage associated protein. MCF-7 and MDAMB-231 cells were incubated with SAHA and 5b for 24 h. SAHA was administered at 1 µM concentration in MCF-7 and 5 µM concentration in MDAMB-231 cells. 5b was used at 2, 4, and 8 µM concentration. Cell lysates were collected, and protein expression level of Chk2 was determined by Immunoblot analysis. β-actin was used as loading control.
Bisindole-linked pyrrolo[2,1-c][1,4]benzodiazepine suppresses mRNA expression of the genes involved in DNA repair pathway
There are two major types of DSBs: homologous recombination (HR) and non-homologous end joining (NHEJ).Citation35 Breast and ovarian tumor suppressor BRCA1 is a hereditary gene that activates p53 dependent pathway gene expression. It is a multifunctional protein that is involved in various cellular processes like DNA repair, transcriptional regulation, and cell cycle checkpoint control.Citation36 Its role in maintaining genome integrity comes during protecting cells from DSBs via homologous recombination and non-homologous end-joining, respectively.Citation37,Citation38 BRCA1 is also a target for several kinases such as ATM and ATR.Citation39 We therefore tested our compound for its effect in BRCA1 gene expression after treating both the cell lines with 5b at 4 µM concentration for 24 h. Both the cell lines were also treated with SAHA in the above mentioned concentration. BRCA1 was downregulated significantly in both the cell lines upon treatment with the compound. The downregulation was much significant even in comparison to SAHA ().
Figure 8. mRNA levels of different DNA repair genes were suppressed by bisindole-PBD hybrid which was independent of ATR expression. (A and B) Expression of DNA repair associated genes was studied by RT-PCR. Treatment was given at 5 µM (in MDAMB-231), 1 µM (MCF-7) concentration of SAHA, and 2, 4, and 8 µM 5b for 24 h. in MCF-7 and MDAMB-231 cells and were incubated for 24 h. Total RNA extraction was done by Trizol method. PCR amplification of DNA repair associated gene (BRCA1, Rad50, Rad51, Ku70, and Ku80) product was observed by agarose gel. (C and D) RT-PCR amplification of DNA repair genes under reduced level of ATR. ATR-specific siRNA transfection was performed to see the expression pattern of DNA repair genes.
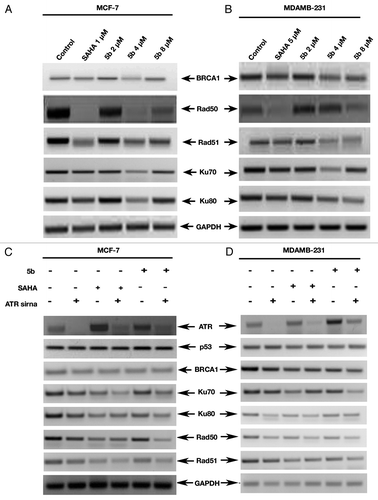
It was originally suggested that BRCA1 protects genome by controlling cell cycle indirectly.Citation40 Recently, involvement of Rad50 as seen from colocalization studies with BRCA1 establishes the fact that Rad50 is directly involved in HR. Rad50 forms a tight complex with Mre11 and Nijmegen breakage syndrome gene 1 (MRN1). This complex is implicated both in HR and NHEJ. Protein foci of both Rad50 and Mre11 are observed constantly at the region of DSBs. We therefore studied the expression level of Rad. When MCF-7 cells were treated with 5b at 4 µM concentration, RAD50 expression was lowered whereas in MDAMB-231, RAD51 was downregulated at 4 µM ().
In case of non-homologous end joining complex (NHEJ) DNA DSBs are initially stabilized. It has been shown that BRCA1 promotes NHEJ via Ku DNA-PK-dependent pathway.Citation41,Citation42 In its first step it identifies and binds Ku heterodimer at the point of double strand break and regulate error prone end joining. This heterodimer comprises of two subunits Ku70 and Ku80. Similarly to understand the complete pathway for the involvement of BRCA1 in both kinds of DSBs, the cell lines were treated with compound 5b at 2, 4, and 8 µM concentrations. Total RNA was isolated and gene expression analysis performed by RT-PCR. There was a drastic in the expression of both Ku70 and Ku80 in both the cell lines treated with the compound at 4 µM concentration for 24 h. There was no significant change at 2 µM concentration. This led us to understand that compound 5b interrupts the DNA repair NHEJ process allowing the cells to proceed toward death.
To further investigate the role of ATR upon these repair genes, we knocked down ATR with siRNA and performed RT-PCR analysis of the genes involved in the DNA repair pathway. But we did not observe any significant change in the expression pattern in the mRNA levels of DNA repair genes (BRCA1, Rad50, Rad51, Ku70, and Ku80) in both MCF-7 and MDAMB-231 cell lines ().
Discussions
Bisindoles exhibit potent anticancer activities in different cancer cell lines.Citation20-Citation24 The antitumor pyrrolo[2,1-c][1,4]-benzodiazepines (PBDs) conjugates are well known anticancer compounds that covalently bind to C2–NH2 group of guanine base through the electrophilic C11-position of PBDs in the minor groove of DNA.Citation43 The hybrid molecule bisindole-PBD conjugate exhibited very high anticancer activity against breast cancer cell MCF-7. Breast cancer is the most prevalent type of cancer diagnosed in women.Citation44 In this present study we tried to explore the in depth role of the conjugate 5b in two different breast cancer cell lines MCF-7 and MDAMB-231 that induce DNA damage via activation of ATM and ATR pathway.
Multicellular organisms are made up of highly organized and tightly packed cells. It is maintained by a unique balance of cell division and cell death. In normal condition, induction of DNA damage leads to apoptosis that leads to a blockage in DNA replication. Abrogation of apoptosis leads to carcinogenesis.Citation45,Citation46 Membrane blebbing, cell shrinkage, and DNA fragmentation are the characteristics feature of apoptosis. In this study, the induction of cell death after treatment with the conjugate compound 5b was confirmed by several assays Trypan blue exclusion assay and BrdU assay. This induced level of cell death was a result of DNA damage which in turn leads to the increase of intracellular ROS production via p53 mediated or independent mechanism.Citation47,Citation48 Simultaneously there was increase of oxidative stress in association with disruption of mitochondrial membrane resulting in decreased in mitochondrial membrane potential and oxidative repair upon treatment with the compound 5b.
Change in the MMP initiates the release of apoptogeneic factors like cytochrome c from outer membrane to cytosol.Citation49,Citation50 Cytochrome c forms apoptosomes with procaspase-9, Apaf-1, and ATP, which in turn activate the downstream apoptosis signals like Caspase-3 and Caspase-7. Caspases are a family of cysteine proteases that help in the execution of apoptotic cascade that may be effectors and initiators. Initiator caspases are associated with pro-apoptotic cascades/signals. Upon activation, these caspases cleave and activates different downstream effector caspases like Caspase-3, 6, and 7. Disruption of outer mitochondrial membrane releases cytochrome c, which is responsible for activation of procaspase-9. This cytochrome c released from intermembrane space binds to apoptosis protease activating factor-1 and helps in recruiting Caspase-9 and proteolytically induces the expression of Caspase-3. Incubation with the compound 5b in a dose-dependent manner activates the expression of Caspase-9 and induces Caspase-3 resulting in mitochondrial mediated apoptosis. MCF-7 is deficient in Caspase-3. In this cell line, DNA fragmentation leads to activation of Caspase-7 and cleavage of PARP.Citation51,Citation52
Bcl-2 family proteins regulate cell death or survival. Both Bcl-2 and Bcl-xL are anti-apoptotic proteins and hence inhibits apoptosis. Our present data showed that cells treated with 5b resulted in reduced expression of Bcl-xL. Bcl-xL interacts with Apaf1 and prevents apoptosis by inhibiting Apaf1-dependent overexpression of Caspase-9. Our results show a downregulation of Bcl-xL after treatment of 5b creating a loss of MMP with release of cytochrome c and activation of caspase cascade. Moreover incubation with 5b decreased the protein expression of Survivin (inhibitor of apoptosis family) that lead to cell death by activating the Caspase-3 level.
The tumor suppressor p53 mRNA and protein levels were upregulated after incubation with 5b in case of MCF-7 cells, thereby inducing p21 expression and pushing the cells toward apoptosis. Interestingly in case of in MDAMB-231 cells that carry p53 mutation produced reduced expression of p53 both at mRNA and protein level along with, elevated level of p21 explaining a mechanism independent of p53 status.
Induction of DSB break was marked by increase in γ-H2AX foci formation after treatment at 4 µM for 24 h in both the cell lines. Formation of phosphorylated form of H2AX is the first event involved in DNA damage response. Immunoblot analysis with γ-H2AX antibody also confirmed the increase in DNA damage in response to 5b exposure. Single strand DNA break was observed through COMET assay. Histone modification was initiated by the well-known phosphatidylinositol-3 kinase-related kinase (PIKK) members.Citation53 The PIKK family members ataxia telangiectasia mutated (ATM), AT-related (ATR), and DNA-dependent protein kinase (DNA-PK) are implicated in DNA repair process in mammals. ATM is the major kinase responsible for histone H2AX phosphorylationCitation54 as a result of DNA double strand break following blockage in replication fork. Our results showed the compound 5b activated DNA damage checkpoint genes ATM and ATR at different doses. ATM senses double stranded break and ATR senses single strand break. Once activated ATM phosphorylates CHK2 and ATR phosphorylates CHK1. We have observed the elevated levels of p-chk1 and p-Chk2 protein in both the cell lines.
DNA repair genes helps to maintain the genomic stability. Unrepaired DNA leads to cell death. There was a significant downregulation of DNA repair genes BRCA1, Ku70, Ku80, Rad50, and Rad51 after incubation with the compound 5b. In another set of experiment we examined the ATR activating role of 5b by transfecting cells with ATR specific siRNA. We observed significant knockdown of ATR and the efficacy of the compound in activating ATR expression. Moreover there was no significant change in DNA repair gene expression and p53 expression after transfection with ATR siRNA.
In conclusion, the profound activity of bisindole-PBD conjugates 5b in MCF-7 and MDAMB-231 cells have been shown. The present study put an insight to DNA damage induced apoptosis by the bisindole-PBD conjugate. This conjugate induces apoptosis via ROS generation and downregulation of anti-apoptotic Bcl-xL. Activation of ATM and ATR with an increase in γ-H2AX foci indicated marked DNA damage. These observations put immense importance and or potential value of the conjugate in understanding and developing new therapeutics in breast cancer. Further in vivo experiment is needed to validate its efficiency.
Materials and Methods
Cell lines and reagents
Human breast cancer cell lines MDAMB-231 and MCF-7 were purchased from American Type Culture collection. Both the cell lines were cultured in RPMI-1640 and Dulbecco’s modified Eagle’s medium (DMEM) (Sigma) respectively, supplemented with 10% fetal bovine serum and 100 U/mL Pencillin and 100 mg/ml streptomycin sulfate (Sigma) at 37 °C with 5% CO2 in a 95% humidified atmosphere. Vorinostat (SAHA) and DMSO were obtained from Sigma.
Trypan blue exclusion assay
Trypan blue exclusion assay is used to estimate cell viability in a cell suspension. It relies on the property of the cell membrane, i.e., viable cells due to their permeable membrane are unable to up take trypan blue whereas dead cells are stained (0.4% w/v in saline). Both the cell lines were treated with the compound 5b at 2, 4, and 8 μM concentrations for 24 h. SAHA was treated at 1 μM in case of MCF-7 and at 5 μM for MDAMB-231. Thereafter, the cells were trypsinized and resuspended in the media. For every 100 μL of cell suspension, 10 μL of 0.4% Trypan blue was added. After thorough mixing, a small volume was loaded on to the hemocytometer and percentage of dead cells calculated. Each sample was assayed in triplicates.
BrdU cell proliferation assay
This assay is based on the principle of incorporation of bromodeoxyuridine in newly synthesized DNA. Assay was performed as per company protocol (BrdU Cell Proliferation Kit, Millipore, 2750).
ROS detection
MCF-7 and MDAMB-231 cells were treated with the compound 5b at a concentration of 4 μM and incubated for 24 h. Simultaneously, both the cell lines were separately treated with SAHA that serves as positive control at a concentration of 1 μM for MCF-7 and 5 μM for MDAMB-231 cells. After 24 h the cells were further incubated with carboxy-H2DCFDA for 30 min at 37 °C followed by washes in PBS. The cells were then viewed under fluorescence microscope at 20× objective (Olympus microscope).
Detection of Δψm by flow cytometry analysis with JC-1 staining
JC-1, a cationic dye, is used as an indicator of mitochondrial potential. It exhibits mitochondrial potential-dependent accumulation which is detected by a fluorescence emission shift from green to red. After the incubation of the cell lines at the above mentioned concentration for 24 h, cells were trypsinized. They were thereafter resuspended in media followed by 10 min of incubation with 10 μg/mL of JC-1 (Invitrogen) at 37 °C. Red and green fluorescence emissions were analyzed by flow cytometry.
Immunofluorescence of γ-H2AX
Cells were seeded at a density of 2.5 × 105 on coverslips. After 24 h, cells were treated at the concentration mentioned. After the desired treatment time, the media was removed and cells were washed with PBS. Cells were fixed in 4% paraformaldehyde for 20 min followed by incubation with 0.2% Triton X-100 in PBS for 5 min. Cells were washed twice with PBS and blocked with 1% BSA in PBS for 1 h. Cells were incubated with anti-γH2AX antibody (Origene, TA301078, dilution 1:100) for 2 h in PBST at room temperature, followed by three washes (10 min each) in PBS. For detection the cells were incubated in dark with FITC-conjugated anti-rabbit secondary antibody (1:50 dilution) (Jackson Immuno Research Laboratories Inc.) at room temperature for 1 h. After washes for three times (10 min each in PBS), the coverslips were mounted with DAPI and observed under confocal microscope (Olympus FV1000). Images were processed with flow view version 1.7c software program.
COMET assay
The assay was performed according to neutral/alkaline conditions as described earlier.Citation55 Cells after treatment were put in low melting agarose and spread over glass slide. It was further incubated in lysis buffer (2.5 M NaCl 146.1 g, 100 mM EDTA 37.2 g, 10 mM Trizma base 1.2 g, then added fresh 1% Triton X-100, and 10% DMSO, and then refrigerated for at least 30 min prior to slide addition) for overnight at 4 °C. The slides were thereafter washed, and kept for incubation in alkaline lysis buffer for 30 min. Then the slides were subjected to electrophoresis at 30 V and 240 mA for 30 min and subjected to washes with distilled water, fixed in 70% ethanol, and stained with ethidium bromide (1 mg/mL). Images were taken in fluorescent microscope (Olympus). Images acquired were analyzed with Comet score software.
Gene expression analysis by semi-quantitative reverse transcription PCR (RT-PCR)
Total RNA from compound (5b) treated, SAHA treated and untreated cells was extracted using Trizol reagent obtained from Invitrogen and reverse transcribed into cDNA using RNA to cDNA EcoDry™ Premix kit (Double Primed, Clontech). PCR was performed using specific primers in Eppendorf Mastercycler Gradient PCR machine. PCR products were electrophoresed on agarose gel (1.2%) and visualized under U.V. light. Respective band signal was measured by Quantity one version 4.1.1 software. Primer sets used for PCR amplification are listed in Table S1.
ATR siRNA transfection
ATR siRNA (Dharmacon) transfection was performed by the Lipofectamine® RNAiMAX Transfection Reagent following the manufacturer’s method. Briefly MCF-7 and MDAMB-231 cells were plated in 6-well plates at density of 100 000/well and transfected with ATR siRNA at a final concentration of 25 nM in serum/antibiotic free media. After 6 h of transfection, the media was replaced with complete growth media following which the cells were allowed to recover for 24 h. Cells were incubated with the compound and proper controls for 24 h. mRNA obtained from cells were amplified and analyzed for ATR and other DNA repair gene specific primers (Table S1).
Protein extraction and immunoblot analysis
Total cell lysates were obtained by lysing the cells with ice cold RIPA buffer (Sigma R 0278) containing protease inhibitor (Roche). Lysates were centrifuged at 12 000 rpm for 15 min at 4 °C. The protein obtained from supernatant was quantified by Bradford method (BIO-RAD) using Multimode Varioskan instrument (Thermo-Fisher Scientifics). Proteins were separated by SDS-PAGE and transferred onto PVDF membrane. Primary antibodies used for immunoblot analysis were anti-γ-H2AX (1:1000; Origene), anti-Chk2 (1:1000; Cell Signaling Technology), anti-β-actin (1:1000; Imgenex), anti- p21 (1:500; Millipore), anti-p53 (1:500; Abbiotec), anti- Bax (1:300; Santa Cruz Biotechnology), anti-Bcl-xL (1:1000; Santa Cruz Biotechnology), anti-Caspase-9 (1;1000; Abbiotec), anti-Caspase-3 (1:1000; Imgenex), anti-survivin (1:500; Imgenex). Western blot was performed on lysate to detect protein expressions taking β-actin as loading control. Rabbit and mouse polyclonal secondary antibodies were purchased from Santa Cruz Biotechnology, Inc..
Densitometry analysis
Densitometry analysis was done by Image J software. The region (area) of interest of the gel picture was selected and the densitometry value was estimated by Image J. Relative densitometry values were calculated considering β-actin (in case western blot) and GAPDH (in PCR) as an internal control and were plotted in Microsoft Office Excel. Standard deviation and standard error were calculated in excel from the values obtained from experiments. Variables presented in histogram were obtained from three different independent experiments.
| Abbreviations: | ||
| DDR | = | DNA damage response |
| DSBs | = | DNA double-strand breaks |
| HR | = | homologous recombination |
| NHEJ | = | non-homologous end joining |
| ROS | = | reactive oxygen species |
| IRIF | = | IR-induced foci |
| ATM | = | ataxia telangiectasia mutated |
| BrdU | = | 5-bromo-2'-deoxyuridine |
| RT-PCR | = | Reverse Transcriptase-Polymerase chain reaction |
Additional material
Download Zip (32.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors acknowledge CSIR 12th FYP (CSC0111) for the financial support. P.S. thanks CSIR for providing the SRF (ID 8169). Special thanks go to CCMB Cell Culture Facility and Y. Suresh for conducting all the flow cytometry experiments and P. Devender for maintaining the mammalian cell culture.
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 8; http://dx.doi.org/10.1038/nature08467; PMID: 19847258
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 9; http://dx.doi.org/10.1038/35044005; PMID: 11100718
- Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res 2004; 32:3683 - 8; http://dx.doi.org/10.1093/nar/gkh703; PMID: 15252152
- Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science 2002; 297:547 - 51; http://dx.doi.org/10.1126/science.1074740; PMID: 12142523
- Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 2002; 14:237 - 45; http://dx.doi.org/10.1016/S0955-0674(02)00312-5; PMID: 11891124
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605 - 11; http://dx.doi.org/10.1038/nature03442; PMID: 15758953
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR 3rd, Hays L, Morgan WF, Petrini JH. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 1998; 93:477 - 86; http://dx.doi.org/10.1016/S0092-8674(00)81175-7; PMID: 9590181
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462 - 7; http://dx.doi.org/10.1074/jbc.C100466200; PMID: 11571274
- Hopfner KP, Karcher A, Craig L, Woo TT, Carney JP, Tainer JA. Structural biochemistry and interaction architecture of the DNA double-strand break repair Mre11 nuclease and Rad50-ATPase. Cell 2001; 105:473 - 85; http://dx.doi.org/10.1016/S0092-8674(01)00335-X; PMID: 11371344
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998; 281:1674 - 7; http://dx.doi.org/10.1126/science.281.5383.1674; PMID: 9733514
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323 - 31; http://dx.doi.org/10.1016/S0092-8674(00)81871-1; PMID: 9039259
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A 2000; 97:10389 - 94; http://dx.doi.org/10.1073/pnas.190030497; PMID: 10973490
- Simizu S, Takada M, Umezawa K, Imoto M. Requirement of caspase-3(-like) protease-mediated hydrogen peroxide production for apoptosis induced by various anticancer drugs. J Biol Chem 1998; 273:26900 - 7; http://dx.doi.org/10.1074/jbc.273.41.26900; PMID: 9756937
- Rowe LA, Degtyareva N, Doetsch PW. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae.. Free Radic Biol Med 2008; 45:1167 - 77; http://dx.doi.org/10.1016/j.freeradbiomed.2008.07.018; PMID: 18708137
- Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 2010; 35:505 - 13; http://dx.doi.org/10.1016/j.tibs.2010.04.002; PMID: 20430626
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000; 5:415 - 8; http://dx.doi.org/10.1023/A:1009616228304; PMID: 11256882
- Bragado P, Armesilla A, Silva A, Porras A. Apoptosis by cisplatin requires p53 mediated p38alpha MAPK activation through ROS generation. Apoptosis 2007; 12:1733 - 42; http://dx.doi.org/10.1007/s10495-007-0082-8; PMID: 17505786
- Neidle S, Thurston DE. Chemical approaches to the discovery and development of cancer therapies. Nat Rev Cancer 2005; 5:285 - 96; http://dx.doi.org/10.1038/nrc1587; PMID: 15803155
- Su Y, Vanderlaag K, Ireland C, Ortiz J, Grage H, Safe S, Frankel AE. 1,1-Bis(3′-indolyl)-1-(p-biphenyl)methane inhibits basal-like breast cancer growth in athymic nude mice. Breast Cancer Res 2007; 9:R56; http://dx.doi.org/10.1186/bcr1761; PMID: 17764562
- Shiri M, Zolfigol MA, Kruger HG, Tanbakouchian Z. Bis- and trisindolylmethanes (BIMs and TIMs). Chem Rev 2010; 110:2250 - 93; http://dx.doi.org/10.1021/cr900195a; PMID: 20041637
- Andreani A, Burnelli S, Granaiola M, Leoni A, Locatelli A, Morigi R, Rambaldi M, Varoli L, Landi L, Prata C, et al. Antitumor activity of bis-indole derivatives. J Med Chem 2008; 51:4563 - 70; http://dx.doi.org/10.1021/jm800194k; PMID: 18598018
- Gaisina IN, Gallier F, Ougolkov AV, Kim KH, Kurome T, Guo S, Holzle D, Luchini DN, Blond SY, Billadeau DD, et al. From a natural product lead to the identification of potent and selective benzofuran-3-yl-(indol-3-yl)maleimides as glycogen synthase kinase 3beta inhibitors that suppress proliferation and survival of pancreatic cancer cells. J Med Chem 2009; 52:1853 - 63; http://dx.doi.org/10.1021/jm801317h; PMID: 19338355
- Queiroz MJ, Abreu AS, Carvalho MSD, Ferreira PMT, Nazareth N, São-José Nascimento M. Synthesis of new heteroaryl and heteroannulated indoles from dehydrophenylalanines: Antitumor evaluation. Bioorg Med Chem 2008; 16:5584 - 9; http://dx.doi.org/10.1016/j.bmc.2008.04.004; PMID: 18439831
- Vine KL, Matesic L, Locke JM, Ranson M, Skropeta D. Cytotoxic and anticancer activities of isatin and its derivatives: a comprehensive review from 2000-2008. Anticancer Agents Med Chem 2009; 9:397 - 414; http://dx.doi.org/10.2174/1871520610909040397; PMID: 19442041
- Lee SO, Abdelrahim M, Yoon K, Chintharlapalli S, Papineni S, Kim K, Wang H, Safe S. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res 2010; 70:6824 - 36; http://dx.doi.org/10.1158/0008-5472.CAN-10-1992; PMID: 20660371
- Kamal A, Srikanth YV, Ramaiah MJ, Khan MN, Kashi Reddy M, Ashraf M, Lavanya A, Pushpavalli SN, Pal-Bhadra M. Synthesis, anticancer activity and apoptosis inducing ability of bisindole linked pyrrolo[2,1-c][1,4]benzodiazepine conjugates. Bioorg Med Chem Lett 2012; 22:571 - 8; http://dx.doi.org/10.1016/j.bmcl.2011.10.080; PMID: 22104151
- Rowe LA, Degtyareva N, Doetsch PW. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae.. Free Radic Biol Med 2008; 45:1167 - 77; http://dx.doi.org/10.1016/j.freeradbiomed.2008.07.018; PMID: 18708137
- Salmon TB, Evert BA, Song B, Doetsch PW. Biological consequences of oxidative stress-induced DNA damage in Saccharomyces cerevisiae.. Nucleic Acids Res 2004; 32:3712 - 23; http://dx.doi.org/10.1093/nar/gkh696; PMID: 15254273
- Evert BA, Salmon TB, Song B, Jingjing L, Siede W, Doetsch PW. Spontaneous DNA damage in Saccharomyces cerevisiae elicits phenotypic properties similar to cancer cells. J Biol Chem 2004; 279:22585 - 94; http://dx.doi.org/10.1074/jbc.M400468200; PMID: 15020594
- Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes Dev 1999; 13:1899 - 911; http://dx.doi.org/10.1101/gad.13.15.1899; PMID: 10444588
- Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 2012; 920:613 - 26; http://dx.doi.org/10.1007/978-1-61779-998-3_40; PMID: 22941631
- Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741 - 8; http://dx.doi.org/10.1038/sj.onc.1210872; PMID: 18066086
- You Z, Chahwan C, Bailis J, Hunter T, Russell P. ATM activation and its recruitment to damaged DNA require binding to the C terminus of Nbs1. Mol Cell Biol 2005; 25:5363 - 79; http://dx.doi.org/10.1128/MCB.25.13.5363-5379.2005; PMID: 15964794
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73 - 112; http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0; PMID: 21034966
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002; 23:687 - 96; http://dx.doi.org/10.1093/carcin/23.5.687; PMID: 12016139
- Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP. The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst 2004; 96:1659 - 68; http://dx.doi.org/10.1093/jnci/djh312; PMID: 15547178
- Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 1997; 88:265 - 75; http://dx.doi.org/10.1016/S0092-8674(00)81847-4; PMID: 9008167
- Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999; 285:747 - 50; http://dx.doi.org/10.1126/science.285.5428.747; PMID: 10426999
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177 - 96; http://dx.doi.org/10.1101/gad.914401; PMID: 11544175
- Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res 2005; 3:531 - 9; http://dx.doi.org/10.1158/1541-7786.MCR-05-0192; PMID: 16254187
- Mari PO, Florea BI, Persengiev SP, Verkaik NS, Brüggenwirth HT, Modesti M, Giglia-Mari G, Bezstarosti K, Demmers JAA, Luider TM, et al. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc Natl Acad Sci U S A 2006; 103:18597 - 602; http://dx.doi.org/10.1073/pnas.0609061103; PMID: 17124166
- Uematsu N, Weterings E, Yano K, Morotomi-Yano K, Jakob B, Taucher-Scholz G, Mari PO, van Gent DC, Chen BP, Chen DJ. Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol 2007; 177:219 - 29; http://dx.doi.org/10.1083/jcb.200608077; PMID: 17438073
- Hurley LH, Petrusek R. Proposed structure of the anthramycin-DNA adduct. Nature 1979; 282:529 - 31; http://dx.doi.org/10.1038/282529a0; PMID: 503235
- Coughlin SS, Ekwueme DU. Breast cancer as a global health concern. Cancer Epidemiol 2009; 33:315 - 8; http://dx.doi.org/10.1016/j.canep.2009.10.003; PMID: 19896917
- Blagosklonny MV. Targeting cancer cells by exploiting their resistance. Trends Mol Med 2003; 9:307 - 12; http://dx.doi.org/10.1016/S1471-4914(03)00111-4; PMID: 12900218
- Rodriguez-Nieto S, Zhivotovsky B. Role of alterations in the apoptotic machinery in sensitivity of cancer cells to treatment. Curr Pharm Des 2006; 12:4411 - 25; http://dx.doi.org/10.2174/138161206779010495; PMID: 17168751
- Rowe LA, Degtyareva N, Doetsch PW. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic Biol Med 2008; 45:1167 - 77; http://dx.doi.org/10.1016/j.freeradbiomed.2008.07.018; PMID: 18708137
- Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000; 5:415 - 8; http://dx.doi.org/10.1023/A:1009616228304; PMID: 11256882
- Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta 2011; 1807:735 - 45; http://dx.doi.org/10.1016/j.bbabio.2011.03.010; PMID: 21453675
- Ballot C, Kluza J, Martoriati A, Nyman U, Formstecher P, Joseph B, Bailly C, Marchetti P. Essential role of mitochondria in apoptosis of cancer cells induced by the marine alkaloid Lamellarin D. Mol Cancer Ther 2009; 8:3307 - 17; http://dx.doi.org/10.1158/1535-7163.MCT-09-0639; PMID: 19952118
- Kottke TJ, Blajeski AL, Meng XW, Svingen PA, Ruchaud S, Mesner PW Jr., Boerner SA, Samejima K, Henriquez NV, Chilcote TJ, et al. Lack of correlation between caspase activation and caspase activity assays in paclitaxel-treated MCF-7 breast cancer cells. J Biol Chem 2002; 277:804 - 15; http://dx.doi.org/10.1074/jbc.M108419200; PMID: 11677238
- Mc Gee MM, Hyland E, Campiani G, Ramunno A, Nacci V, Zisterer DM. Caspase-3 is not essential for DNA fragmentation in MCF-7 cells during apoptosis induced by the pyrrolo-1,5-benzoxazepine, PBOX-6. FEBS Lett 2002; 515:66 - 70; http://dx.doi.org/10.1016/S0014-5793(02)02440-7; PMID: 11943196
- Smith GC, Jackson SP. The DNA-dependent protein kinase. Genes Dev 1999; 13:916 - 34; http://dx.doi.org/10.1101/gad.13.8.916; PMID: 10215620
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 2001; 276:42462 - 7; http://dx.doi.org/10.1074/jbc.C100466200; PMID: 11571274
- Olive PL, Banáth JP. The comet assay: a method to measure DNA damage in individual cells. Nat Protoc 2006; 1:23 - 9; http://dx.doi.org/10.1038/nprot.2006.5; PMID: 17406208