
Abstract
Chemoresistance is a major therapeutic challenge to overcome in NSCLC, in order to improve the current survival rates of <15% at 5 years. We and others have shown increased PI3K signaling in NSCLC to be associated with a more aggressive disease, and a poorer prognosis. In this study, targeted inhibition of three strategic points of the PI3K–NFκB axis was performed with the aim of exploiting vulnerabilities in cisplatin-resistant NSCLC cells. Cisplatin-resistant cell lines were previously generated through prolonged exposure to the drug. Expression of PI3K and NFκB pathway-related genes were compared between cisplatin-resistant cells and their matched parent cells using a gene expression array, qRT-PCR, DNA sequencing, western blot, and immunofluorescence. Targeted inhibition was performed using GDC-0980, a dual PI3K–mTOR inhibitor currently in Phase II clinical trials in NSCLC, and DHMEQ, an inhibitor of NFκB translocation which has been used extensively both in vitro and in vivo. Effects of the two inhibitors were assessed by BrdU proliferation assay and multiparameter viability assay. NFKBIA was shown to be 12-fold overexpressed in cisplatin-resistant cells, with no mutations present in exons 3, 4, or 5 of the gene. Corresponding overexpression of IκBα was also observed. Treatment with DHMEQ (but not GDC-0980) led to significantly enhanced effects on viability and proliferation in cisplatin-resistant cells compared with parent cells. We conclude that NFκB inhibition represents a more promising strategy than PI3K–mTOR inhibition for treatment in the chemoresistance setting in NSCLC.
Introduction
Lung cancer is the most common cause of cancer related deaths worldwide.Citation1 Non-small cell lung cancer (NSCLC) is defined as any form of epithelial lung carcinoma other than small cell lung carcinoma. This accounts for 80% of lung cancersCitation2 and includes large cell carcinoma, squamous cell carcinoma and adenocarcinoma. Despite advances in anti-cancer therapies the overall 5 y survival for lung cancer patients remains poor (<15%).
The main anti-cancer treatment for NSCLC is platinum-based chemotherapy with either cisplatin or carboplatin in combination with a secondary agent such as paclitaxel or docetaxel. Chemotherapy is used in the post-operative, adjuvant setting and concurrently with radiotherapy for the radical treatment of locally advanced unresectable disease to increase long-term survival by ~5–10% at five years.
The efficacy of cisplatin treatment is often limited by intrinsic or acquired resistance to the drug. Many possible mechanisms of cisplatin resistance have been shown in various cell lines, and recent in vivo work has further elucidated the basis of resistance to this drug. The major mechanisms of resistance are (1) increased influx/decreased influx of cisplatin, (2) elevated levels of glutathione, (3) increased DNA repair, and (4) activation of signal transduction pathways, e.g., MAPK, AKT, and NFκB.Citation3-Citation5
Phosphatidylinositol 3-kinases (PI3Ks) were discovered by Lewis Cantley and colleagues, who first published on their association with the polyoma middle T protein in 1985.Citation6 The signals that PI3K family members help to potentiate induce the cell to grow, differentiate, proliferate, and help with survival, motility, and intracellular trafficking. As such, these enzymes are strongly implicated in the development of malignant behavior as well as playing a role in resistance to chemotherapy and targeted therapies.
Upon activation by growth factor stimulation of receptor tyrosine kinases, G-protein coupled receptors or RAS phosphorylation, PI3K phosphorylates PIP2 to produce PIP3. PIP3 propagates the signal by bringing PDK1 and AKT into close proximity, allowing PDK1 to activate AKT by phosphorylation.Citation7 AKT can then help the cell to display malignant characteristics in several ways. It promotes cell survival by inhibiting BAD and BAX, two proapoptotic Bcl2 family members. AKT also phosphorylates Mdm2, resulting in antagonization of p53-mediated apoptosis, as well as negatively regulating forkhead transcription factors, resulting in reduced production of cell death-promoting proteins.Citation7 AKT can also impede negative regulation of transcription factor NFκB by IκB, leading to an increase in transcription of antiapoptotic and pro-survival genes, coding for proteins such as the antiapoptotic Bcl-2 and Bcl-XL.Citation8
One common mechanism of acquired resistance to cancer therapies involves the upregulation of pathways that promote cell survival and proliferation. It is therefore unsurprising that PI3K pathway activation, which results in the NFκB-mediated upregulation of various genes that promote this type of behavior, has been implicated in cisplatin resistance in ovarian cancer cells, where it was shown that treatment with the drug activated the PI3K pathway, and that inhibition of the pathway sensitized cells to the effects of cisplatin.Citation9 More recently, activation of AKT and ERK have been associated with resistance to cisplatin in NSCLCCitation10-Citation12 and SCLC,Citation13 though more research is needed to fully elucidate the role of these pathways in cisplatin resistance in lung cancer. Research into NFκB, PI3K, and PI3K–mTOR inhibitors in NSCLC has increased recently, with pre-clinical data showing promising results.Citation13,Citation14 Targeting the PI3K–mTOR pathway has been studied extensively in oncology, with a wide range of inhibitors investigated in cancers including NSCLC.Citation15 One such dual inhibitor is GDC-0980, which is currently in phase II trials for cancer.Citation16,Citation17
NFκB family members function as homo- or hetero-dimers to activate and repress the transcription of a large number of genes controlling immunological processes as well as cell growth and proliferation and many other cellular processes. Some of these genes include pro-survival factors such as Bcl2, vascular endothelial growth factors, and many other growth factors. In recent years the NFκB transcription factors have been studied extensively, and growing evidence suggests an important role for NFκB in various oncological processes.
The IκB family plays a key role in regulating NFκB. IκB proteins consist of an N regulatory domain, a series of six or more ankyrin repeats, and a PEST domain near the C terminus. Interactions with NFκB are mediated by the ankyrin repeat domains.Citation18 IκB proteins inhibit NFκB function by masking NFκB nuclear translocation signals, and sequestering the transcription factor in the cytosplasm ().
Figure 1. PI3K signaling and inhibition PI3K-AKT-mTOR activation leads to regulation of proliferation, apoptosis, angiogenesis and other key cellular processes, through the transcription of hundreds of target genes by NFκB. Inactive NFκB is bound by its inhibitor IκBα in the cytoplasm, until activation leads to dissociation of the two proteins and proteosomal degradation of IκBα. NFκB is then free to translocate to the nucleus where it acts as a transcription factor. One of NFκB’s many target genes is IκBα, which removes NFκB from the nucleus, returning it to its original inactive form in the cytoplasm. GDC-0980 is a dual PI3K–mTOR inhibitor. DHMEQ is an inhibitor of NFκB translocation to the nucleus. AKT, Protein Kinase B; IκBa, Nuclear factor of kappa light polypeptide gene enhancer in B-cellsinhibitor, α; mTOR, mammalian target of rapamycin; NFκB, Nuclear factor kappa-light-chain-enhancer ofactivated B cells; PI3K, Phosphatidylinositide 3-kinase.
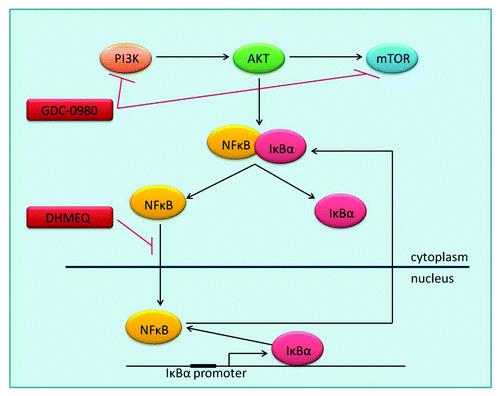
The most widely studied IκB family member is IκBα, which is encoded by NFKBIA. In order to allow NFκB to function, signaling molecules such as AKT activate IκB kinase 1 (IKK1), which phosphorylates IκBα on serines 32 and 36. This targets the protein for degradation in the ubiquitin proteasome, thus allowing the dissociation and translocation of NFκB to the nucleus.Citation19 In the nucleus, NFκB activates the transcription of κB-dependent genes, one of which is IκBα. Newly synthesized IκBα is responsible for post-induction repression of NFκB, through the removal of NFκB from DNA, and transportation of the transcription factor back to the cytoplasm.Citation20 Mutations in the IκBα gene have been reported in Hodgkin/Reed-Sternberg cells resulting in overexpression of a C-terminally truncated protein and constitutive activation of NFκB.Citation21,Citation22
In cancer cells, NFκB has been shown to act in an anti-apoptotic, pro-proliferative manner. As such, inhibition of NFκB has been investigated in various types of cancer as a potential therapeutic intervention strategy. One such inhibitor is Dehydroxymethylepoxyquinomicin (DHMEQ), which prevents translocation of NFκB to the nucleus. Derived from the antibiotic epoxyquinomycin C, DHMEQ is a specific inhibitor of NFκB nuclear translocation,Citation23 which has shown both anti-inflammatory effects and anti-tumor effects in multiple cancer types both in vitro and in vivo.Citation24-Citation31
Here we investigate the effect of targeting key signaling molecules within the PI3K/NFκB pathway in two in vitro models of cisplatin-resistant NSCLC.
Results
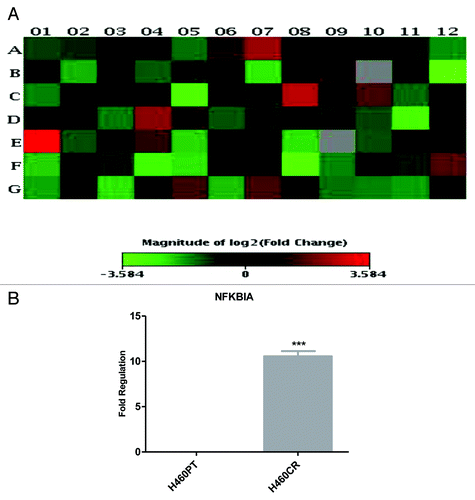
An RT2 Profiler array (SABiosciences) was used to quantify the expression of 84 genes associated with the PI3K-Akt pathway and to identify any differences in mRNA expression between the H460PT (parent) and H460CR (cisplatin-resistant) cell lines. NFKBIA displayed an 11.99-fold upregulation in H460CR cells (). Four genes were upregulated greater than 2-fold and 11 genes were downregulated greater than 2-fold in cisplatin-resistant cells compared with parent cells. Of particular interest was NFKBIA as it displayed the highest differential expression between parent and resistant cell lines. This result was validated by QPCR using primers specific to NFKBIA, which identified 10.56-fold upregulation of NFKBIA in H460CR cells ().
Figure 2. NFKBIA is more highly expressed in H460CR cells than H460 cells An RT2 profiler PI3K-Akt pathway array was performed to compare gene expression in H460 (cisplatin-sensitive) and H460CR (cisplatin-resistant) cell lines. (A) A heat map was constructed using SABiosciences online software, corresponding to . Four genes were upregulated greater than 2-fold and 11 genes were downregulated greater than 2-fold in cisplatin-resistant cells compared with parent cells. Position E01 corresponds to an increase in NFKBIA. (B) Upregulation of NFKBIA in H460CR cells was validated by QRTPCR, using NFKBIA and ACTB specific primers, in three independent experiments. Fold regulation is shown for NFKBIA in H460CR cells compared with H460PT cells, calculated via the ΔΔCt method. ***P < 0.001. H460PT, H460 parent cells; H460CR, H460 cisplatin-resistant cells.
The gene product of NFKBIA, IκBα, was then compared in H460 parent () and H460 cisplatin-resistant cells (), IκBα was shown to be more highly expressed in H460CR cells than H460PT cells by analysis of immunofluorescence () and western blotting (). IκBα was also shown to be more highly expressed in A549CR cells than A549PT cells by densitometry analysis of western blots ().
Figure 3. IkBa is more highly expressed in H460CR cells than H460parent cells. (A) Immunofluorescence image: H460 parent untreated cells were stained with IκBα primary antibody and Alexa Fluor488 conjugated secondary antibody (green). Cells were also stained with Höechst nuclear stain (blue). Alexa Fluor conjugated secondary antibody (green) indicates IκBα expression in the cytoplasm and peri-nuclear space. (B) Immunofluorescence image: H460CR untreated cells were stained and imaged as in (A). The increased intensity of green staining here indicates the presence of more IκBα here than in H460 cells. (C) Immunofluorescence analysis: Cells were either left untreated (as in [A and B]) or pretreated with TNAα (25 ng/mL) for 20 min. Cells were imaged using High Content Analysis. Each treatment was performed in triplicate and three independent experiments were performed. Cell intensity (amount of IκBα fluorescence) was measured in the nucleus and the cytoplasm using HCA software. (D) H460 and A549 parent and cisplatin-resistant cell protein samples were run on an SDS-PAGE gel and analyzed by western blot three independent times for expression of IκBα and αβTubulin. (E) Densitometry analysis was performed using ImageJ software to compare expression of IκBα between H460 and A549 parent and cisplatin-resistant cells. *P < 0.05 ***P < 0.001. H460PT, H460 parent cells; H460CR, H460 cisplatin-resistant cells.
![Figure 3. IkBa is more highly expressed in H460CR cells than H460parent cells. (A) Immunofluorescence image: H460 parent untreated cells were stained with IκBα primary antibody and Alexa Fluor488 conjugated secondary antibody (green). Cells were also stained with Höechst nuclear stain (blue). Alexa Fluor conjugated secondary antibody (green) indicates IκBα expression in the cytoplasm and peri-nuclear space. (B) Immunofluorescence image: H460CR untreated cells were stained and imaged as in (A). The increased intensity of green staining here indicates the presence of more IκBα here than in H460 cells. (C) Immunofluorescence analysis: Cells were either left untreated (as in [A and B]) or pretreated with TNAα (25 ng/mL) for 20 min. Cells were imaged using High Content Analysis. Each treatment was performed in triplicate and three independent experiments were performed. Cell intensity (amount of IκBα fluorescence) was measured in the nucleus and the cytoplasm using HCA software. (D) H460 and A549 parent and cisplatin-resistant cell protein samples were run on an SDS-PAGE gel and analyzed by western blot three independent times for expression of IκBα and αβTubulin. (E) Densitometry analysis was performed using ImageJ software to compare expression of IκBα between H460 and A549 parent and cisplatin-resistant cells. *P < 0.05 ***P < 0.001. H460PT, H460 parent cells; H460CR, H460 cisplatin-resistant cells.](/cms/asset/a9058b05-33dc-458c-8175-fa667111c6d1/kcbt_a_10929841_f0003.gif)
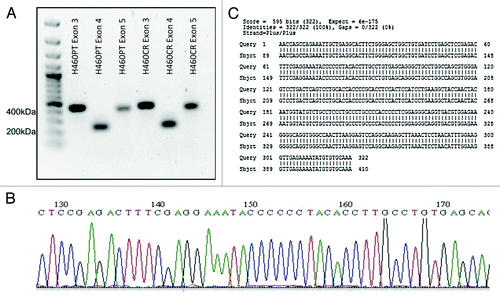
A point mutation in exon 5 of NFKBIA has been previously identified which results in a truncated form of the protein IκBα, which cannot bind and inhibit NFκB.Citation21,Citation22 Exons 3, 4, and 5 of NFKBIA were amplified via nested PCR and PCR products were visualized on an agarose gel (). These amplicons were then sequenced using the ABI3130 sequencer (). Sequences were compared with wild-type sequences using BLASTN, with no mutations being found ().
Figure 4. NFKBIA Sequence Trace. (A) Exons 3, 4, and 5 of IκBα were amplified via nested PCR from H460PT and H460CR RNA. PCR products were visualized on a 1% agarose gel, and compared with a 100 bp DNA ladder (Fermentas). (B) Amplicons were sequenced using the AB3130 sequencer. Sequence trace files were obtained for each exon in each cell line, an example of which is shown here. No double peaks were observed in any trace file. (C) Forward and reverse sequences were compared against wild-type sequences for each NFKBIA exon in both cell lines using the basic local alignment search tool for nucleotides (BLASTN). No discrepancies were identified between our sequences and wild-type sequences. A sample sequence of H460 NFKBIA exon 3 is shown here, aligned with wild type IκBα exon 3. H460PT, H460 parent cells; H460CR, H460 cisplatin-resistant cells.
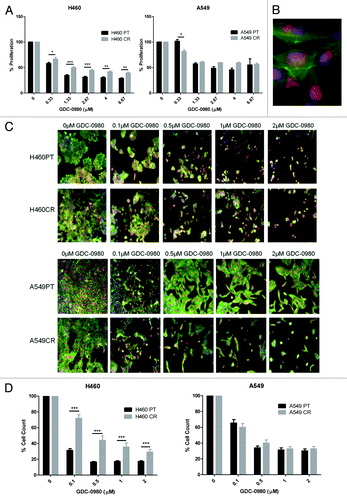
The effects of PI3K–mTOR inhibitor GDC-0980 on cell proliferation were analyzed by BrdU assay. GDC-0980 was found to induce a reduction in proliferation in H460PT, H460CR, A549PT, and A549CR cell lines. GDC-0980 treatment was significantly more effective at all doses in H460PT cells when compared with the H460CR cells. At the lowest dose (0.33 μM), GDC-0980 treatment was significantly more effective in A549CR cells than A549PT cells; however at all other doses, there was a trend toward GDC-0980 treatment being more effective in A549PT cells than A549CR cells (). The effect of GDC-0980 on these four cell lines was further investigated by a multiparameter cell viability assay using HCA. Imaging of cells treated with GDC-0980 compared with untreated cells allowed for visualization of cellular damage induced by the drug () which was then quantified by cell count using HCA software (). Cell count was significantly reduced in H460PT cells compared with H460CR cells in response to GDC-0980 treatment at all doses. There was no significant difference in cell count between A549PT and A549CR cells ().
Figure 5. The effects of GDC-0980 treatment on proliferation and cell viability in H460 parent and cisplatin-resistant cells. (A) H460 and parent and cisplatin-resistant cells were treated in triplicate at noted concentrations of GDC-0980 for 72 h. Cells were analyzed via BrdU assay (n = 3). Percentage proliferation is shown here. (B) Sample image: Cells were stained with Höechst blue nuclear stain, mitotracker red mitochondrial stain and phalloidin green F-actin stain, and imaged using the In Cell Analyzer 1000. (C) H460 and parent and cisplatin-resistant cells were treated in triplicate at noted concentrations of GDC-0980 for 48 h. Eight fields were imaged per well at 10× magnification using the In Cell Analyzer 1000. (D) Images from (C) were analyzed using In Cell software which identified cell count automatically from nuclear staining. *P < 0.05 **P < 0.005 ***P < 0.001. H460PT, H460 parent cells; H460CR, H460 cisplatin-resistant cells
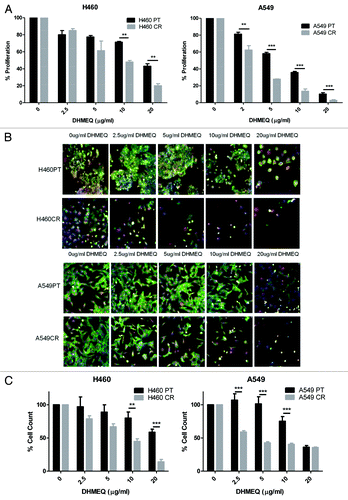
The effects of NFκB translocation inhibitor DHMEQ on cell proliferation were analyzed by BrdU assay. DHMEQ was found to induce a reduction in proliferation in H460PT, H460CR, A549PT, and A549CR cell lines. In contrast to the response to GDC-0980 treatment, DHMEQ treatment led to a significantly increased reduction of proliferation in cisplatin-resistant cells compared with parent cells at all doses for A549 cells, and at higher doses for H460 cells (). This increased efficacy of DHMEQ treatment in cisplatin-resistant cells was also observed by multiparameter cell viability assay using HCA. Imaging of cells treated with DHMEQ compared with untreated cells allowed for qualitative observation of the significant difference in response between parent and cisplatin-resistant cells (). This difference in response was quantified by cell count using HCA software. Cell count was significantly reduced in cisplatin-resistant H460 and A549 cells compared with matched parent cells at noted concentrations ().
Figure 6. The effects of DHMEQ treatment on proliferation and cell viability in H460 parent and cisplatin-resistant cells. (A) H460 and parent and cisplatin-resistant cells were treated in triplicate at noted concentrations of DHMEQ for 72 h. Cells were analyzed via BrdU assay (n = 3). Percentage proliferation is shown here. (B) H460 and parent and cisplatin-resistant cells were treated in triplicate at noted concentrations of DHMEQ for 48 h. Cells were stained with Höechst blue nuclear stain, mitotracker red mitochondrial stain and phalloidin green F-actin stain, and imaged using the In Cell Analyzer 1000. Eight fields were imaged per well at 10x magnification. (C) Images from (B) were analyzed using In Cell software which identified cell count automatically from nuclear staining. *P < 0.05 **P < 0.005 ***P < 0.001 H460PT: H460 parent cells H460CR: H460 cisplatin-resistant cells.
Discussion
Activation of the PI3K/NFκB pathway has been shown previously to play a role in cisplatin resistance in ovarian cancer.Citation3 As such, in-depth gene expression analysis of matched cisplatin-sensitive (H460P) and cisplatin-resistant (H460CR) cell lines was performed using the human PI3K-AKT Signaling Pathway RT2 profiler array. Of the 84 genes quantified only 4 upregulated genes, including an 11.99-fold increase in NFKBIA (gene encoding IκBα) expression, were identified in H460CR cells in comparison to H460 cells. Significant upregulation of NFKBIA in the H460CR cell line was validated by a gene-specific qPCRassay. Investigation of IκBα expression at the protein level was performed via immunofluorescence and western blot. Immunofluorescence imaging indicated increased levels of IκBα in H460 cisplatin-resistant cells compared with H460 parent cells. Western blot analysis indicated higher levels of IκBα in both H460 and A549 cisplatin-resistant cells compared with their respective parent cell lines. Such activated levels of this negative regulator of NFκB in both chemoresistant models implies that IκBα could play a role in cisplatin resistance in NSCLC.
It has been shown previously that truncated forms of the IκBα protein resulted in reduced IκBα–NFκB binding and prevention of post-induction repression of NFκB. Deregulated IκBα results in constitutive NFκB activity and the emergence of a chemoresistant phenotype due to upregulation of survival and anti-apoptotic genes transcriptionally regulated by NFκB. NFKBIA exons 3, 4, and 5 are the most common sites for deletions and mutations which result in truncated forms of the IκBα protein.Citation21,Citation22,Citation32 Here we screened these exons, in H460 parent and resistant cells, for the presence of mutations by Sanger sequencing. These sequences were compared with wild-type sequences using BLASTN. No mutations or deletions were detected.
In order to assess the viability of targeting the PI3K pathway as a second line treatment in NSCLC, we investigated the effects of targeted inhibition on two strategic points in the PI3K–NFκB pathway in parent and cisplatin-resistant cells. First, treatment with GDC-0980, a dual PI3K–mTOR inhibitor, led to significantly reduced proliferation and viability in parent H460 cells compared with their matched cisplatin-resistant cells. This is unsurprising due to the 11 upstream PI3K pathway genes which were seen to be more highly expressed in parent cells than cisplatin-resistant cells on the gene expression array. This data demonstrates that while the PI3K pathway maybe a potential therapeutic target in H460P cells the H460CR cell lines are less reliant on this pathway. GDC-0980 was not as effective in the A549PT cells and this may be due in part to the fact that these cells are PIK3CA mutation negative whereas the H460PT cells harbor a PIK3CA mutation. In both A549PT and A549CR cells, a similar trend in proliferation and cell viability was observed at higher concentrations of the drug, while at the lowest concentrations a significantly increased effect on proliferation in cisplatin-resistant cells was observed. As such, we believe that direct inhibition of PI3K–mTOR may not be a useful strategy in this setting.
We hypothesize based on our initial protein expression data that a deregulated IκBα/NFκB interaction may play a significant role in cisplatin resistance in our NSCLC models therefore we investigated the effect of targeting this aberrant IκBα/NFκB pathway. To this end we examined the effect of a specific inhibitor of NFκB translocation and binding, DHMEQ, in our cisplatin-resistant models. This inhibitor, a derivative of the antibiotic epoxyquinomicin C, has shown promise as a treatment for rheumatoid arthritis as well as being effective in suppressing hormone-refractory prostate cancer in an in vivo model without any toxicity and is currently being progressed toward clinical trials.Citation27-Citation29 A dose-response proliferation assay and a multiparameter HCA viability assay were performed using H460 and A549 parent and cisplatin-resistant cell lines. In both models of resistance DHMEQ induced a reduction in proliferation to a significantly greater extent in the cisplatin-resistant cell lines than in parent cell lines. These data further highlight that a deregulated IκBα/NFκB interaction may play a key role in cisplatin resistance in NSCLC cell lines. We hypothesize that inhibition of NFκB or its downstream targets may be an ideal strategy to resensitize cisplatin-resistant cells to cisplatin, and this should be investigated thoroughly with a view to designing new targeted therapeutic protocols for cisplatin-resistant NSCLC patients.
Further investigations to elucidate the exact mechanism involved in these IκBα activated resistant cells as well as cisplatin and DHMEQ combination treatment are warranted both in vitro and in vivo. Based on these data, we believe that a non-toxic specific inhibitor of NFκB such as DHMEQ may play a key role in future treatment of NSCLC patients with either intrinsic or acquired cisplatin resistance.
This study was performed on the basis of previous published evidence supporting a role for the PI3K–NFκB axis in cisplatin resistance,Citation3,Citation9-Citation13 with the aim of identifying strategic points within this pathway to target in order to overcome this resistance in NSCLC. With this promising data implying a major role for IκBα/NFκB interaction in NSCLC cisplatin resistance, inhibition of NFκB by DHMEQ or other targeted inhibitors could provide a beneficial treatment strategy for NSCLC patients who progress on cisplatin. We believe this data underpins the importance of determining which point in a signaling cascade is critical to therapeutic targeting, in order to ensure maximal benefit in specific clinical settings such as chemoresistance.
Materials and Methods
Cell culture
H460 cells were grown in RPMI1640 media (Lonza) supplemented with 10% FBS and 1% penicillin/streptomycin at 37 °C and 5% CO2. A549 cells were grown in Ham’s F-12 media (Lonza) supplemented with 10% FBS, 1% penicillin/streptomycin and 1% L-glutamine at 37 °C and 5% CO2. Cisplatin-resistant cell lines had previously been developed in this laboratory via continuous exposure of H460 and A549 cells to cisplatin.Citation33 H460 parent cells (H460PT) could then be compared with H460 cisplatin-resistant cells (H460CR), and A549 parent cells (A549PT) could be compared with A549 cisplatin-resistant cells (A549CR).
Gene expression array
RNA was isolated from parent and resistant cell lines using TriReagent. Two RT2 Profiler PCR arrays were used (SABiosciences PI3K–AKT pathway array: PAHS-058). One 96 well array was performed for H460PT RNA and the other for H460CR RNA. cDNA was added to RT2 qPCR Master Mix, which contains SYBR Green and reference dye. The experimental cocktail of cDNA, Master Mix, and H2O was added to the 96 well array (25 μL per well). Real-time PCR thermal cycling was performed using the ABI 7500 thermal cycler. Changes in gene expression between H460PT and H460CR cell lines were analyzed using SABiosciences online software which incorporates the ΔΔCT method.
qRT-PCR
qRT-PCR validation of array results was performed for NFKBIA. Roche FastStart Universal SYBR green master (Rox) was used with cDNA prepared from H460PT and H460CR cells. NFKBIA and β-actin-specific primers were used (SABiosceinces).
NFKBIA nested PCR
Nested PCR was performed for exons 3, 4, and 5 of the NFKBIA gene. In the first PCR reaction, forward primers were used. In the second PCR reaction, inner forward primers were used. For both reactions, the same reverse primers were used. Primer sequences and annealing temperatures are shown in as adapted from.Citation31 The nested PCR Products were run on a 1% agarose gel with 1× TBE buffer. A 100 bp DNA ladder was used to determine the size of the amplicons. PCR product purification was performed using a QIAquick PCR Purification Kit (Qiagen). The DNA was purified according to the manufacturer’s protocol, using the buffers and spin columns provided. The purified DNA was eluted in 30 µL Buffer EB. Cycle sequencing was then performed using BigDye Terminator v3.1. Each reaction contained 1 µL primer, 3 µL BigDye terminator mix v3.1, 50 ng template DNA and dH2O to a total volume of 20 µL. A control tube contained 1 µL pGem, 2 µL M13 primer, 3 µL BigDye terminator mix v.3.1, and 14 µL dH2O. The tubes were then placed in the GeneAmp 2400 thermal cycler using the following program: Step 1: 96 °C for 1 min, step 2: 96 °C for 10 min, step 3: 50 °C for 5 s, step 4: 60 °C for 4 min, step 5: repeat steps 2–4, 25 times, step 6: go to 4 °C. The sequencing products were then cleaned using DyeEx spin columns (Qiagen). The clean-up was performed as per the manufacturer’s protocol, and the recovered reaction was dried using a speedy vac at medium heat until dry. The pellet was then resuspended in 10 µL HiDiformamide, and loaded onto an ABI 96-well plate. The plate was then placed on the AB3130 sequencing platform, which read the sequences.
Table 1. NFKBIAa primers used in this study
Western blot
Protein was isolated from H460PT, H460CR, A549PT, and A549CR cells using RIPA buffer supplemented with sodium orthovandate (50 mM), PMSF (100 mM), protease inhibitor cocktail, β-glycerophosphate (500 mM) and sodium fluoride (500 mM). Thirty micrograms of total protein from each cell line was loaded onto a 12% acrylamide-SDS gel. Proteins were resolved by electrophoresis and transferred onto a PVDF membrane. IκBα was detected using a rabbit IκBα (N-terminal) antibody (Cell Signaling). Membranes were probed as per manufacturer’s protocol. Bound antibody complexes were detected using the Supersignal West Pico Chemiluminescent substrate kit (Pierce). The membrane was then exposed to scientific imaging X-ray film and developed.
Immunofluorescence
Expression levels of IκBα in H460 and A549 parent and cisplatin-resistant cell lines were also investigated at the protein level by Immunofluorescence using High Content Analysis (HCA) imaging. Cells were seeded in 96-well plates and either pretreated with TNFα (25 ng/mL for 20 min) or left untreated, then fixed with 4% PFA for 15 min at RT. Cells were subsequently blocked in blocking buffer (100 μL per well, 1× PBS containing 5% normal goat serum and 0.3% Triton X-100, Sigma) at room temperature for 60 min.
The blocking solution was aspirated, and diluted primary antibody was added to each well (100 μL per well, 1× PBS containing 1% BSA, 0.3% Triton X-100, and 0.8% IκBα antibody). The plate was incubated at 4 °C overnight.
Cells were washed 3 times for 5 min each in PBS (100 μL), and then incubated with Alexa Fluor 488 conjugated goat-anti-mouse secondary antibody (Invitrogen) for 1 h at room temperature in the dark. Cells were washed 3 times for 5 min each in PBS (100 μL), and then stained with Höechst nuclear stain (bisbenzimide H33348, Fluka Biochemika, 4 µM, 20 min). One hundred microliters of PBS was finally added to all 96 wells of the plate for imaging.
The cells were imaged using the InCell Analyzer1000 (GE Lifesciences) and images were analyzed using the High Content Analyzer software, which quantified the intensity of green fluorescence in the cytosol and nucleus for untreated and TNFα treated cells.
Proliferation
Cells were treated with either GDC-0980 (Genentech) or DHMEQ (Umezawa Lab) at noted concentrations for 72 h in 96 well plates at 37 °C and 5% CO2. The Cell Proliferation ELISA, BrdU (colorimetric) kit (Roche) was used to assess percentage proliferation in relation to untreated cells. BrdU reagent was added directly to each well for the last 4 h of treatment, allowing it to incorporate into the genome of proliferating cells. The cells were then fixed and exposed to an anti-BrdU antibody for 90 min before finally adding substrate and stop solutions as per manufacturer’s protocol. Absorbance was measured @ 595 nm by a spectrophotometer to quantify corresponding alterations in proliferation.
Multiparameter high content analysis assay
Cells were treated with either GDC-0980 or DHMEQ at noted concentrations for 24 h in 96-well plates at 37 °C and 5% CO2. Cells were stained simultaneously with Höechst nuclear stain (bisbenzimide H33348, FlukaBiochemika, 1 µM, 30 min) and mitotracker red mitochondrial stain (Invitrogen, 0.5 µM, 30 min), then washed in PBS and fixed in PFA (4%, 10 min), before permeablizing (0.5% Triton-X-100 in PBS) and finally staining with Alexa Fluor 488 Phalloidin (Invitrogen, 10 µM, 30 min). The cells were imaged using the InCell Analyzer1000 (GE Lifesciences) and images were analyzed using High Content Analysis (HCA) software, which quantified cell count.
| Abbreviations: | ||
| AKT | = | protein kinase B |
| IκBa | = | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha |
| mTOR | = | mammalian target of rapamycin |
| NFκB | = | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSCLC | = | non-small cell lung cancer |
| PI3K | = | phosphatidylinositide 3-kinase |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Connla Edwards and Anthony Davies for their assistance related to the High Content Analysis aspects of this study, as well as Genentech for gifting GDC-0980 to us for use in this study. The authors wish to acknowledge the Irish Cancer Society for funding this research (CRS11HEA).
References
- Parkin DM. Global cancer statistics in the year 2000. Lancet Oncol 2001; 2:533 - 43; http://dx.doi.org/10.1016/S1470-2045(01)00486-7; PMID: 11905707
- Pfannschmidt J, Muley T, Bülzebruck H, Hoffmann H, Dienemann H. Prognostic assessment after surgical resection for non-small cell lung cancer: experiences in 2083 patients. Lung Cancer 2007; 55:371 - 7; http://dx.doi.org/10.1016/j.lungcan.2006.10.017; PMID: 17123661
- Godwin P, Baird AM, Heavey S, Barr MP, O’Byrne KJ, Gately K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front Oncol 2013; 3:120; http://dx.doi.org/10.3389/fonc.2013.00120; PMID: 23720710
- Godwin AK, Meister A, O’Dwyer PJ, Huang CS, Hamilton TC, Anderson ME. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc Natl Acad Sci U S A 1992; 89:3070 - 4; http://dx.doi.org/10.1073/pnas.89.7.3070; PMID: 1348364
- Yang H, Kong W, He L, Zhao JJ, O’Donnell JD, Wang J, Wenham RM, Coppola D, Kruk PA, Nicosia SV, et al. MicroRNA expression profiling in human ovarian cancer: miR-214 induces cell survival and cisplatin resistance by targeting PTEN. Cancer Res 2008; 68:425 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-07-2488; PMID: 18199536
- Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature 1985; 315:239 - 42; http://dx.doi.org/10.1038/315239a0; PMID: 2987699
- Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 1999; 337:575 - 83; http://dx.doi.org/10.1042/0264-6021:3370575; PMID: 9895304
- Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochem J 2008; 415:333 - 44; http://dx.doi.org/10.1042/BJ20081056; PMID: 18842113
- Peng DJ, Wang J, Zhou JY, Wu GS. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem Biophys Res Commun 2010; 394:600 - 5; http://dx.doi.org/10.1016/j.bbrc.2010.03.029; PMID: 20214883
- Zhang B, Zhang K, Liu Z, Hao F, Wang M, Li X, Yin Z, Liang H. Secreted clusterin gene silencing enhances chemosensitivity of a549 cells to cisplatin through AKT and ERK1/2 pathways in vitro. Cell Physiol Biochem 2014; 33:1162 - 75; http://dx.doi.org/10.1159/000358685; PMID: 24751980
- Wang M, Liu ZM, Li XC, Yao YT, Yin ZX. Activation of ERK1/2 and Akt is associated with cisplatin resistance in human lung cancer cells. J Chemother 2013; 25:162 - 9; http://dx.doi.org/10.1179/1973947812Y.0000000056; PMID: 23783141
- Lee MW, Kim DS, Min NY, Kim HT. Akt1 inhibition by RNA interference sensitizes human non-small cell lung cancer cells to cisplatin. Int J Cancer 2008; 122:2380 - 4; http://dx.doi.org/10.1002/ijc.23371; PMID: 18224693
- Belyanskaya LL, Hopkins-Donaldson S, Kurtz S, Simões-Wüst AP, Yousefi S, Simon HU, Stahel R, Zangemeister-Wittke U. Cisplatin activates Akt in small cell lung cancer cells and attenuates apoptosis by survivin upregulation. Int J Cancer 2005; 117:755 - 63; http://dx.doi.org/10.1002/ijc.21242; PMID: 15981204
- Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol 2010; 28:1075 - 83; http://dx.doi.org/10.1200/JCO.2009.25.3641; PMID: 20085938
- Heavey S, O'Byrne KJ, Gately K. Strategies for co-targeting the PI3K/AKT/mTOR pathway in NSCLC. Cancer Treat Rev 2014; 40:445 - 56; http://dx.doi.org/10.1016/j.ctrv.2013.08.006; PMID: 24055012
- Genentech. Study of GDC-0941 or GDC-0980 With Fulvestrant Versus Fulvestrant in Advanced or Metastatic Breast Cancer in Patients Resistant to Aromatase Inhibitor Therapy. ClinicalTrialsgov [Internet]. Available from: http://clinicaltrials.gov/ct2/show/NCT01437566?term=GDC-0980&rank=6: Accessed 28/04/13, Identifier: NCT01437566; 2011.
- Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, Lesnick JD, Lewis C, Nonomiya J, Pang J, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther 2011; 10:2426 - 36; http://dx.doi.org/10.1158/1535-7163.MCT-11-0446; PMID: 21998291
- Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998; 95:749 - 58; http://dx.doi.org/10.1016/S0092-8674(00)81698-0; PMID: 9865693
- Régnier CH, Song HY, Gao X, Goeddel DV, Cao Z, Rothe M. Identification and characterization of an IkappaB kinase. Cell 1997; 90:373 - 83; http://dx.doi.org/10.1016/S0092-8674(00)80344-X; PMID: 9244310
- Arenzana-Seisdedos F, Turpin P, Rodriguez M, Thomas D, Hay RT, Virelizier JL, Dargemont C. Nuclear localization of I kappa B alpha promotes active transport of NF-kappa B from the nucleus to the cytoplasm. J Cell Sci 1997; 110:369 - 78; PMID: 9057089
- Emmerich F, Meiser M, Hummel M, Demel G, Foss HD, Jundt F, Mathas S, Krappmann D, Scheidereit C, Stein H, et al. Overexpression of I kappa B alpha without inhibition of NF-kappaB activity and mutations in the I kappa B alpha gene in Reed-Sternberg cells. Blood 1999; 94:3129 - 34; PMID: 10556199
- Cabannes E, Khan G, Aillet F, Jarrett RF, Hay RT. Mutations in the IkBa gene in Hodgkin’s disease suggest a tumour suppressor role for IkappaBalpha. Oncogene 1999; 18:3063 - 70; http://dx.doi.org/10.1038/sj.onc.1202893; PMID: 10340377
- Ariga A, Namekawa J, Matsumoto N, Inoue J, Umezawa K. Inhibition of tumor necrosis factor-alpha -induced nuclear translocation and activation of NF-kappa B by dehydroxymethylepoxyquinomicin. J Biol Chem 2002; 277:24625 - 30; http://dx.doi.org/10.1074/jbc.M112063200; PMID: 11983688
- Kikuchi E, Horiguchi Y, Nakashima J, Kuroda K, Oya M, Ohigashi T, Takahashi N, Shima Y, Umezawa K, Murai M. Suppression of hormone-refractory prostate cancer by a novel nuclear factor kappaB inhibitor in nude mice. Cancer Res 2003; 63:107 - 10; PMID: 12517785
- Starenki DV, Namba H, Saenko VA, Ohtsuru A, Maeda S, Umezawa K, Yamashita S. Induction of thyroid cancer cell apoptosis by a novel nuclear factor kappaB inhibitor, dehydroxymethylepoxyquinomicin. Clin Cancer Res 2004; 10:6821 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-04-0463; PMID: 15501958
- Matsumoto G, Namekawa J, Muta M, Nakamura T, Bando H, Tohyama K, Toi M, Umezawa K. Targeting of nuclear factor kappaB Pathways by dehydroxymethylepoxyquinomicin, a novel inhibitor of breast carcinomas: antitumor and antiangiogenic potential in vivo. Clin Cancer Res 2005; 11:1287 - 93; PMID: 15709200
- Watanabe M, Dewan MZ, Okamura T, Sasaki M, Itoh K, Higashihara M, Mizoguchi H, Honda M, Sata T, Watanabe T, et al. A novel NF-kappaB inhibitor DHMEQ selectively targets constitutive NF-kappaB activity and induces apoptosis of multiple myeloma cells in vitro and in vivo. Int J Cancer 2005; 114:32 - 8; http://dx.doi.org/10.1002/ijc.20688; PMID: 15523684
- Katsman A, Umezawa K, Bonavida B. Reversal of resistance to cytotoxic cancer therapies: DHMEQ as a chemo-sensitizing and immuno-sensitizing agent. Drug Resist Updat 2007; 10:1 - 12; http://dx.doi.org/10.1016/j.drup.2007.01.002; PMID: 17306602
- Katsman A, Umezawa K, Bonavida B. Chemosensitization and immunosensitization of resistant cancer cells to apoptosis and inhibition of metastasis by the specific NF-kappaB inhibitor DHMEQ. Curr Pharm Des 2009; 15:792 - 808; http://dx.doi.org/10.2174/138161209787582156; PMID: 19275643
- Castro-Gamero AM, Borges KS, da Silva Silveira V, Lira RC, de Paula Gomes Queiroz R, Valera FC, Scrideli CA, Umezawa K, Tone LG. Inhibition of nuclear factor-κB by dehydroxymethylepoxyquinomicin induces schedule-dependent chemosensitivity to anticancer drugs and enhances chemoinduced apoptosis in osteosarcoma cells. Anticancer Drugs 2012; 23:638 - 50; http://dx.doi.org/10.1097/CAD.0b013e328350e835; PMID: 22382389
- Umezawa K. Possible role of peritoneal NF-κB in peripheral inflammation and cancer: lessons from the inhibitor DHMEQ. Biomed Pharmacother 2011; 65:252 - 9; http://dx.doi.org/10.1016/j.biopha.2011.02.003; PMID: 21723080
- Lake A, Shield LA, Cordano P, Chui DT, Osborne J, Crae S, Wilson KS, Tosi S, Knight SJ, Gesk S, et al. Mutations of NFKBIA, encoding IkappaB alpha, are a recurrent finding in classical Hodgkin lymphoma but are not a unifying feature of non-EBV-associated cases. Int J Cancer 2009; 125:1334 - 42; http://dx.doi.org/10.1002/ijc.24502; PMID: 19507254
- Barr MP, Gray SG, Hoffmann AC, Hilger RA, Thomale J, O’Flaherty JD, Fennell DA, Richard D, O’Leary JJ, O’Byrne KJ. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS One 2013; 8:e54193; http://dx.doi.org/10.1371/journal.pone.0054193; PMID: 23349823