
Abstract
It has been established previously that up to 40% of mouse CD34+ hematopoietic stem cells are capable of internalizing exogenous dsDNA fragments both in vivo and ex vivo. Importantly, when mice are treated with a combination of cyclophosphamide and dsDNA, the repair of interstrand crosslinks in hematopoietic progenitors is attenuated, and their pluripotency is altered. Here we show for the first time that among various actively proliferating mammalian cell populations there are subpopulations capable of internalizing dsDNA fragments. In the context of cancer, such dsDNA-internalizing cell subpopulations display cancer stem cell-like phenotype. Furthermore, using Krebs-2 ascites cells as a model, we found that upon combined treatment with cyclophosphamide and dsDNA, engrafted material loses its tumor-initiating properties which we attribute to the elimination of tumor-initiating stem cell subpopulation or loss of its tumorigenic potential.
Introduction
There are two major models describing tumor cell heterogeneity as an organized system existing within the transformed cell population. Stochastic clonal evolution model posits that all tumor cells may have equal contribution to the induction of a new tumor.Citation1-Citation4 The second model (cancer stem cell theory) implies the existence of a hierarchical organization in a tumor, with cancer stem cells found on top of the hierarchy.Citation5,Citation6 Cancer stem cells have three main features: (1) they can self-renew indefinitely; (2) they can produce committed daughter cells with high proliferative potential, yet incapable of forming new tumors; (3) they can be re-engrafted multiple times and display the histological features of the original tumor in the grafts.
Clearly, identification of tumor-initiating stem cells (TISCs) in a pool of tumor cells is a pre-requisite for successful therapeutic targeting or direct elimination of such cells.Citation7-Citation12
Several basic approaches exist to identify TISCs. Among the most popular are limited dilution analysisCitation7,Citation8 and sphere-formation assay.Citation9,Citation10 Biochemical markers of TISCs, which can vary widely between tumor types and individual tumors are routinely used in experimental oncology applications. These markers include CD133 (prominin-1), CD44 (hyaluronan receptor), CD166, CD34, CD38 (AML markers), CD138 (marker of terminally differentiated B cells), and B5 (ABCB5, ATP-binding protein).Citation11
An alternative approach to identify TISCs takes advantage of their ability to actively efflux lipophilic dyes such as Hoechst 33342 or Rhodamine-123. Using microfluidics-based enrichment of physically deformable cells, isolation of a population of cells showing features of TISCs has been recently reported.Citation12-Citation14 Yet, no marker universally applicable for robust TISC isolation is presently available.
In the present paper, we describe a novel property characteristic of various stem cells and stem-like cells, namely, their ability to internalize extracellular dsDNA fragments. We provide experimental data showing that this property can be efficiently employed in tumor cytoreduction protocols as a tool interfering with interstrand crosslink (ICL) repair following cyclophosphamide (CP) treatment, so that tumor-initiating potential of the grafted material is completely abolished.
Results
Ascites form of Krebs-2 tumor cells internalize dsDNA fragments
Studies of exogenous dsDNA fragment internalization by mouse bone marrow cells reported an interesting fact that up to 40% of hematopoietic CD34+ progenitors can incorporate DNA.Citation15,Citation16 Subsequent analysis suggested that internalization of dsDNA fragments is a general feature of undifferentiated cells with stem cell potential. To test this idea, we first chose the ascites form of Krebs-2 mouse tumor model. Provided that the TISC concept is true, this tumor must contain a cell subpopulation that initiates tumor formation. Our experiments show that 1.5–7% of ascites cells (depending on the experiment and individual animal variation) are capable of internalizing the fragments of extracellular dsDNA. We show that ascites cells internalize both linear DNA fragments of various origin and supercoiled plasmid DNA: pUC19 (data not shown) and pEGFP-N1 ().
Figure 1. Internalization of exogenous dsDNA by ascites cells of mouse Krebs-2 tumor. (A) Fluorescence analysis of TAMRA-labeled Alu DNA and TAMRA-dUTP precursor by ascites form of Krebs-2 tumor cells. Notably, ascites cells fail to incorporate TAMRA-dUTP. Bar corresponds to 50 µm. (B) Same as above, zoom-in, bars correspond to 10 µm. Top and side views of a TAMRA-positive cell are shown on the bottom right panel. (C) Fluorescence analysis of total human TAMRA-labeled DNA fragments (0.2–6 kb) internalized by ascites Krebs-2 tumor cells. Bars correspond to 10 µm. (D) Molecular analysis of dsDNA internalization by Krebs-2 ascites cells. Upper panel: αP32-labeled PCR-amplified GFP fragment was used to directly monitor DNA internalization by ascites cells (probe size is shown on the *GFP lane), M, DNA weight marker; CP+DNA, Krebs-2 ascites cells collected 18 h post CP injection and incubated with labeled DNA (for 1, 2, 4 and 8 h); DNA, Krebs-2 ascites cells from CP-untreated mice and incubated with labeled DNA (for 1, 2, 4 and 8 h); ВMC, bone marrow cells from an intact mouse incubated with labeled DNA (for 1, 2, 4 and 8 h). Lower panel: Non-labeled linearized pEGFP-N1/HindIII DNA was used to detect internalization of exogenous DNA by cells. Internalization was visualized by Southern blot using 32P-labeled GFP DNA as a probe. Treatments and collection timepoints are the same as on the upper panel. Southern blot and gel images are shown. Bone marrow cells were used as a positive control, where DNA internalization is well-documented.Citation15 (E) FACS profiles showing dsDNA internalization upon increasing concentration of the labeled substrate in the medium. Graph summarizing FACS data are shown below. (F) Flow cytometry analysis of dynamics of Alu-TAMRA dsDNA internalization in course of incubation with Krebs-2 tumor cells. Data shown on the FACS plots are converted into a graph format (bottom right).
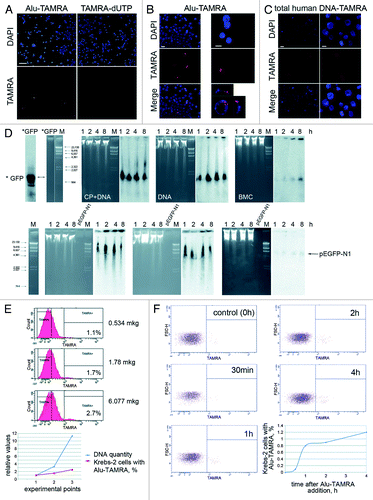
Next, we progressively increased (12-fold) the amount of the labeled probe in the culture medium, but observed only 2.5-fold increase in the percentage of DNA-internalizing ascites cells (). Similarly, increasing the incubation time from 30 min to 4 h resulted in only subtle increase in dsDNA-internalizing cells, from 0.8% to 1.2% (% of TAMRA-dsDNA internalizing cells was calculated relatively to the total number of undamaged fixed cells in the sample, with degraded cells and cell clumps omitted from the analysis) ().
Analysis of the cell cycle in ascites cells showed that 51.1%, 17.8%, and 28.3% of cells were in G1, S, and G2-M phases, respectively (, 1). Cell cycle profiling of TAMRA positive cells indicated that their cell cycle distribution was very similar to that of the total cell population, hence the property of DNA internalization is S phase-independent (data not shown).
Figure 2. Analysis of tumorigenic properties of TAMRA-positive cells. (A) Overlap between CD34+ and TAMRA+ ascites cell populations: 1, cell cycle profile for Krebs-2 ascites cells; 2, FACS analysis confirming the presence of CD34+ and TAMRA+ cells in the same sample of ascites; 3 and 4, FACS analysis of TAMRA and/or FITC positive cells. Vast majority of CD34+ cells are also TAMRA+ (3), whereas about half of TAMRA+ cells are also CD34+ (4); 5, cytofluorescence analysis showing co-staining of ascites Krebs-2 cells in a sample used for FACS profiling. Bars correspond to 10 µm. (B) Analysis of engraftment potential of TAMRA+, TAMRA− cells. 1, The plots illustrate the dynamics of tumor formation (percentage of mice with detectable tumors); 2, FACS profile of Krebs-2 ascites cells following incubation with TAMRA-DNA; 3, FACS profile of an enriched sample of TAMRA positive cells to be used for engraftment. (C) Analysis of engraftment potential of CD34+ and CD34− cells. 1, The plots illustrate the dynamics of tumor formation (percentage of mice with detectable tumors); 2, FACS analysis of Krebs-2 ascites cell samples after incubation with isotype control (left) and CD34 specific antibodies (right). (D) 1–6 spatial organization of TAMRA-positive cells in solid form of Krebs-2 tumor. Bars correspond to 20 µm (1–4) and 10 µm (5). Well-defined foci of cells internalizing TAMRA-DNA. Each focus includes tens to hundreds of cells. 7, Histology analysis of a solid form of Krebs-2 tumor. Tumor tissue displays pronounced cellular atypia and aggressive growth pattern. Stretches of tumor cells (arrows) are interlaced with the fragments of muscle fibers. 8 and 9, FACS quantification of TAMRA positive cells in a solid form of Krebs-2 tumor, bar corresponds to 20 µm. (E) Spatial organization of TAMRA positive cells in spleen and liver. 1 and 2, upon co-incubation of tissue samples with TAMRA-labeled DNA, followed by cryosectioning and confocal imaging; 3 and 4, upon physical homogenization of the tumor down to individual cells, co-incubation with TAMRA-labeled DNA and FACS analysis. Spleen samples lack any cells showing TAMRA-DNA internalization. Single TAMRA positive cells were detected in the liver. Bars correspond to 50 µm. (F) 3D reconstruction of a solid form of Krebs-2 tumor showing localization of TAMRA-DNA internalizing cell clusters.
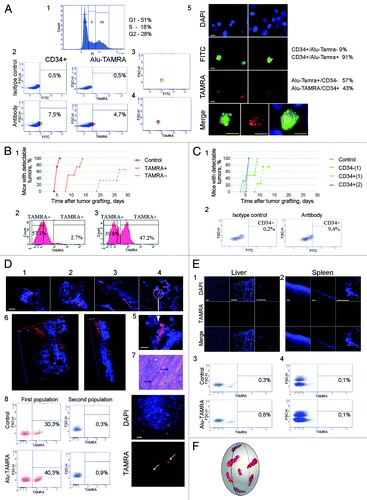
Surface glycoprotein CD34 is one of the markers of stemness in mouse hematopoietic progenitors. We hypothesized that among ascites cells there is a subpopulation of stem-like CD34+ cells. FACS analysis showed that this was indeed the case and that up to 7.0% of ascites tumor cells expressed CD34 marker (, 2). Follow-up analysis demonstrated that 40–90% of CD34+ cells were TAMRA-positive, and that ~50% of TAMRA-positive cell population was CD34+. We observed a substantial proportion of cells showing non-specific signal across all detection channels, as our double-labeling experiments involved lengthy incubations and washes (see Materials and Methods). This obstacle prevented us from accurately quantifying the overlap between TAMRA+ and CD34+ cell populations via FACS analysis. So, we resorted to the fluorescence microscopy analysis of cell samples incubated with TAMRA-labeled DNA and stained with CD34-specific conjugates (, 3–5).
Extensive overlap observed for the cell populations that were positive for markers showing distinct cellular localization allowed us to proceed to comparative experiments on cell engraftment of flow-sorted cell populations.
Specifically, we tested the tumorigenic potential of TAMRA+ and CD34+ ascites cells (altogether, 7 experimental series were performed). Suspension of ascites cells was incubated with either TAMRA-labeled DNA, or with CD34-specific antibodies. Next, the cells were sorted into “positive” and “negative” populations. We observed that TAMRA+ cells induced graft proliferation 11 d sooner than TAMRA- cells. Surprisingly, the transplant growth began in TAMRA+ samples later than in the control, where both the cells capable of internalizing DNA and more differentiated as well as stromal cells were simultaneously present (; Fig. S1).
Our analysis of engraftment potential indicates that CD34– cells display delayed onset of growth as compared with CD34+ cells. Yet, the dynamics of tumor growth induction is very similar for CD34+ cells and the control ascites (; Fig. S1).
One of the intriguing questions in cancer biology is how TISCs are organized in a tumor mass. Using the propensity of TISCs to internalize dsDNA, one can visualize them on tumor sections, and so analyze the spatial organization of cells with high tumorigenic potential. It was shown that in the solid form of Krebs-2 tumor, the cells with tumorigenic potential are found in groups that are randomly scattered throughout the tumor, which appears as a composite of cell masses, trabecules, and cavities. Labeled cell foci are buried inside the tumor sac. FACS analysis of dissociated tumor samples demonstrated solid Krebs-2 grafts encompass up to 10% dsDNA-internalizing cells. As a control, cryosections from liver and spleen of the same animal were used. No evidence of DNA-internalizing cells was found in spleen cryosections, whereas liver samples showed the presence of internalized TAMRA-labeled DNA probe in 0.5% of cells ().
Ability of human glioma neurosphere cells to internalize dsDNA
Here we analyze the ability of human glioma cells grown in cell culture to internalize the fragments of extracellular dsDNA. The primary culture of glioblastoma cells was obtained from a patient undergoing surgical resection of histology-verified grade IV glioma. After two more passages, in addition to adherent cells, we observed spontaneous formation of neurosphere-like detached clusters of cells. These neurosphere-forming cells displayed pronounced proliferative activity and were capable of forming secondary neurospheres. Both adherent cells and neurosphere-forming cells were used in our analyses ().
Figure 3. Internalization of Alu fragment DNA by human glioma cells. (A) Phase-contrast image of adherent cell fraction and neurospheres from human glioblastoma cell culture. (B) Fluorescence microscopy analysis of TAMRA-labeled DNA internalization by glioblastoma cells. Background images show the magnitude of non-specific autofluorescence. Notably, labeled DNA probe displays specific nuclear localization in the cells from neurosphere fraction, and only a single specific positive signal was observed across all adherent cells analyzed. (C) Cytofluorescence analysis of TAMRA-labeled DNA in freely-floating neurosphere cell cultures. Several neurospheres are shown. Bottom image represents cytospinned neurospheres stained with DAPI. (D) 3D reconstituted image of a neurosphere with cells internalizing TAMRA-labeled dsDNA (arrowheads). (E) Visualization of GFP expression in neurospheres that have internalized pEGFP-N1 plasmid. As a control, we provide the fluorescence image of a neurosphere (1) and dying glioma cells (2) neither of which were incubated with plasmid DNA. Unless specified otherwise, bars correspond to 50 µm.
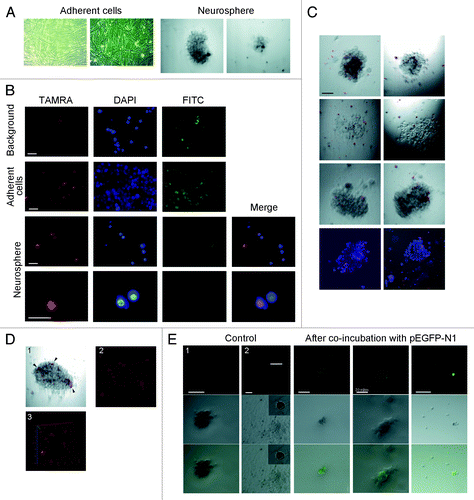
Initially we tested the ability of cultured human glioma cells to internalize extracellular dsDNA fragments using dissociated adherent cells and neurospheres. Specifically, we directly counted the numbers of TAMRA-positive cells using fluorescence microscopy. The cells from adherent fraction failed to internalize TAMRA-labeled dsDNA fragments. In contrast, 7.8% of cells from dissociated neurospheres displayed TAMRA labeling (). Next, we proceeded to quantify DNA internalization by whole neurospheres and to quantify engraftment efficiency of adherent cell and neurosphere subpopulations in NOD/SKID mice. Our time-lapse imaging experiments showed that neurosphere cells actively internalized TAMRA-labeled dsDNA and became saturated within 1 h. shows a panel of neurospheres and a 3D reconstruction, with TAMRA-positive cells clearly present within the neurospheres. We performed direct quantification of TAMRA-dsDNA internalization in squashed preparations of DAPI-stained neurospheres (7 neurospheres altogether), which indicated that about 20% of neurosphere cells were capable of internalizing dsDNA.
Our experiments have an obvious translational application, as exogenous dsDNA internalization by human glioma cells may be considered as a promising therapeutic target. So, we explored the ability of neurosphere cells to internalize extracellular dsDNA in a form of supercoiled plasmid DNA. We used pEGFP-N1 plasmid for this purpose, as it was shown to transiently transfect eukaryotic cells and produce a fluorescent protein GFP. The results of this analysis are summarized in . Some isolated cells as well cells within neurospheres were GFP-positive. When total DNA from glioma cells incubated with pEGFP-N1 was transformed in competent E. coli, colonies with intact pEGFP-N1 were observed, so we could argue that the plasmid was present in glioma cells in its native form (data not shown). Our experiments thus show that functional GFP protein is produced from the plasmid DNA internalized into glioma cell compartments.
Both subpopulations of cells from human glioma (adherent cells and neurospheres) were tested for their tumorigenicity. Cell suspensions were injected into the left hemispheres of NOD/SKID mice. Graft proliferation was visualized 26 d after injections (). All animals injected with neurosphere cells had clearly discernible transplants. In contrast, no engraftment or transplant growth was observed in mice injected with adherent fraction of human glioma cells, even at a day 49 post-injection.
Figure 4. Pathomorphology, histofluorescence and PCR analysis of glioblastoma xenotransplants from brains of NOD/SCID mice. (A) Brain MRI of experimental animals. Upper panel: Serial mouse brain MRI sections on day 28 after glioma adherent cell fraction engraftment. Middle panel: Serial mouse brain MRI sections on day 28 after glioma neurosphere cell injection. Lower panel: Serial mouse brain manganese-enhanced MRI sections (same animal as above). Arrows indicate localization of the growing transplant. (B) Histology analysis of paraffin sections of glioma transplants and original glioblastoma biopsy. Black arrows on mouse brain sections indicate graft cells, and white arrows show normal brain tissue (mouse 1). Tumor displays surface localization and proliferation in interlobar gyri (central sulcus in this case). HE staining, 200× magnification. Mouse 2: large tumor lining the brain ventricle wall. Central part of the tumor is infiltrated with inflammatory cells (mostly granulocytes). Black arrows point to the cell infiltrate; white arrows show normal brain tissue. HE staining, 400× magnification. Original human tumor: 1, endothelial hyperplasia, vessel proliferation, 200× magnification; 2, cell and nuclear polymorphisms, 400× magnification; 3, endothelial hyperplasy, focal necrosis in the tumor, 200× magnification. AE staining. (C) PCR analysis of DNA isolated from the brain tissue carrying glioma graft and from the liver of the same animal (negative control). Human Alu-specific primers were used in amplification reaction. (D) Histofluorescence analysis of cryosections of mouse brains with human glioma graft. 1 and 2, overview of a tumor focus cross-section. Arrow indicates a demarcation line between the brain and the tumor. Zoom-in insets are shown in 3 and 4. Red signals correspond to graft cells that internalized TAMRA-Alu DNA. Bars correspond to 10 µm (1 and 2) and 50 µm (3–5); 6, 3D reconstruction of the tumor fragment. Red dots correspond to individual cells or clusters of cells.
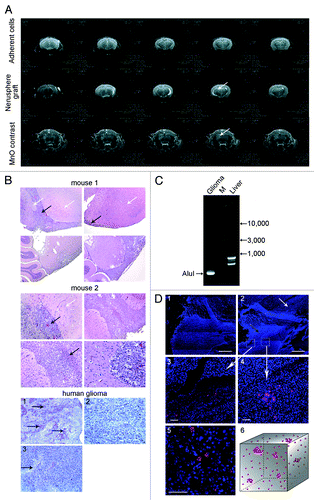
To characterize the neurosphere-derived grafts in more detail, we performed histology analysis of brain sections (). Light microscopy analysis of paraffin sections of mouse brains with developed glioma detected pathological changes restricted to large hemispheres. We observed large groups of tumor cells preferentially located in pia, outgrowing toward the surface of large hemispheres and via brain ventricles. Tumors had large and small caliber vessels and hyperemic foci. The entire tumor mass was profusely infiltrated with inflammatory cells, predominantly neutrophil granulocytes. Curiously, the surrounding brain tissues remained intact and showed no evidence of dystrophy. We noted a pronounced “surface” localization of the tumor with very weak invasive growth. When progressing beyond the injection site, the tumor spread along the brain gyri and was clearly separable from the brain cells. Only weak invasive growth was observed deeper in the brain tissue, which is characteristic of glioblastoma. Pathology description of tumors developed in mice matched the original biopsy description: presence of polymorphic nuclei, rare mitoses, extensive vascularization with pronounced endothelial proliferation, local necrosis and interstitial lymphoid infiltrations (astrocytic glioma) (). Our histological comparison thus confirms the identity of tumor material obtained from mice to the original human tissue ().
To reconfirm the human origin of gliomas that had formed in experimental mice, we performed PCR analysis of DNA isolated from the injected brain areas and from the liver (). We used human Alu repeat as a marker of human DNA, which we extensively used previously.Citation17 Genomic DNA isolated from glioma-engrafted mouse brain tissue specifically produces Alu fragment of the expected size and sequence (97% homology with human Alu repeat). This is unlike the PCR product obtained from liver mass DNA, where sequencing of one of the major PCR bands confirmed its mouse origin (Fig. S2).
Similarly to the solid form of Krebs-2 tumor, we characterized spatial organization of proliferating foci within the glioma transplant cell mass. The general features of glioma structure suggested the following organization of TISCs within the tumor. A broad region of glioma encompassing up to 40% cross-section surface harbors many dispersed TAMRA-labeled cells. Besides these cells, we identified clusters of TAMRA-positive cells, typically 5–6 clusters per section (~50 cells in total) ().
Direct synergistic cytotoxic activity of cyclophosphamide and dsDNA preparation on TISCs in ascites form of Krebs-2 tumor
Analysis of activation of adaptive immune response by dsDNA preparations revealed that immune system contribution accounted for only partial Krebs-2 transplant regression.Citation18,Citation19 At the same time, we showed that synergistic action of CP and dsDNA may alter hematopoietic stem cells so that some of their pluripotency properties are lost.Citation15,Citation16
By testing the effects of dsDNA preparation with or without CP treatment on Krebs-2 ascites tumor cells, we found that over 80% of tumor cells underwent apoptosis and that mice developed multiple pathologies, which led to the rapid and synchronized death of treated animals. Given that ascites were shown to encompass two cell populations—internalizing DNA (1–3%) and ignoring DNA (97–99%)—it was important to know, which cells were targeted by these treatments. These numbers clearly indicated that the treatments predominantly targeted non-internalizing Krebs-2 ascites cells (likely via bystander effect).Citation20,Citation21 Notably, if combined DNA and CP injections were performed to target fresh ascites cells (<4 d), massive apoptosis was likewise observed, yet it was not accompanied with serious pathologies: mice tended to survive and showed long-term remission (manuscript in preparation). It was demonstrated above that dsDNA-internalizing Krebs-2 cells display features of TISCs. So, we hypothesized that CP+dsDNA treatment may also affect TISC population, and this may inhibit their function or destroy these cells, which can be manifested as inhibition of tumor graft growth. Hence we asked, what happens to the DNA-internalizing cells? We could not address this question directly, because treatments were performed using excessive amount of DNA, and this saturated the cells with exogenous DNA molecules, leaving no chance for internalization of TAMRA-labeled DNA probe (as is specified in ). Use of the second tentative marker of TISCs, CD34+, to trace the tumorigenic population of ascites cells was also somewhat problematic. We showed that CD34 was found in ~50% of DNA-internalizing cells, and that 40–90% of CD34+ cells were capable of internalizing TAMRA-DNA. Clearly, overlap between TAMRA+ and CD34+ cell populations is incomplete and unequal. So, if elimination of TAMRA+ CD34+ cells has occurred, or if such cells for some reasons have lost CD34 expression, we measured CD34+ cells that failed to internalize the DNA. In this scenario, it is impossible to determine the real pattern of how TAMRA+ cells are affected by the treatments. To overcome these obstacles, we developed an approach allowing us to estimate the biological impact of CP+dsDNA treatment on TISCs. This approach provides a qualitative assessment of TISCs presence in the grafted material. If the transplant grows, then viable TISCs survived the treatments. If no transplant growth is observed, the TISCs are eliminated or lost their tumorigenic potential.
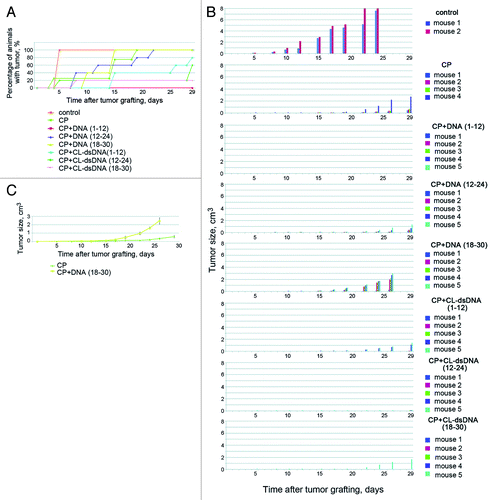
CP+dsDNA-treated animals with ascites tumor grafts were used to collect ascites at selected time points after dsDNA injection. After washes, cells were grafted intramuscularly into right hips of congenic mice. Experiments were repeated 5 times using different cell numbers per graft (). We found that treatment of ascites-engrafted mice with DNA preparation 1–12 h post CP injection had a dramatic effect on tumorigenic properties of the cells, and no proliferation of the transplant was detected even when 1.5 × 106 cells were inoculated, which is in contrast to the rapid tumor formation upon administration of just 5 × 104 ascites cells (see previous section). Our experiments also show that treatment of mice with CL-dsDNA (across all treatment regimens) inhibits tumorigenic properties of the graft, yet fails to entirely eliminate them. One surprising finding was that CP+dsDNA (18–30 h) treatment had a positive effect on tumorigenic properties of the grafted cells (). DsDNA- or CP-only injections did not affect the engraftment potential of ascites cells (data not shown).
Figure 5. Analysis of CP or CP+DNA treatments on tumorigenic efficiency of Krebs-2 tumor cells. (A and B) In vivo analysis. Groups of mice were i/m grafted with ascites cells collected from mice treated with CP or CP+DNA (different regimens). Plots show dynamics of tumor formation and transplant growth. In a group of CP+DNA (1–12 h), Krebs-2 transplant failed to proliferate in any of the animals. (C) Comparison of tumor graft growth in mice treated with CP or CP+DNA (18–30 h). CP+DNA (18–30 h) treatment schedule displays pronounced positive effect on transplant proliferation.
In this experimental series, the choice of specific injection schedules was based on our previously published data on exogenous DNA internalization by mouse bone marrow cells. These data suggested that presence of dsDNA inside bone marrow cells while they are trying to repair CP-induced ICLs interferes with this repair, resulting in a severely compromised pluripotency of hematopoietic stem cells.Citation15,Citation16 It was suggested that dsDNA fragments internalized into nuclei of tumor-forming cells (in the context of Krebs-2 ascites) may similarly interfere with the repair process. We thus performed a series of experiments that allowed us to explore the interplay between elimination of tumorigenic cells and phases of ICL repair process.
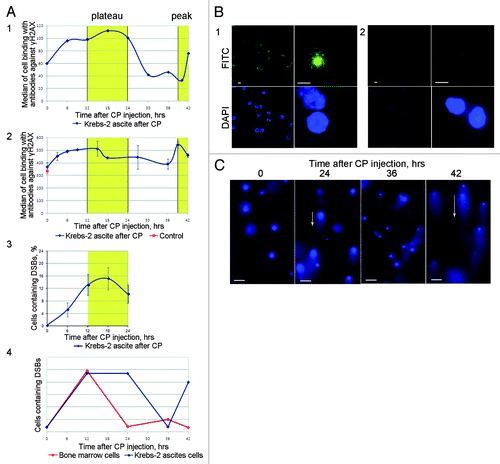
Ascites-engrafted mice were injected with CP. Next, every 3–6 h ascites cells were assayed for double-strand break (DSB) repair foci formation. Presence of DSBs was independently confirmed using a comet assay.Citation22,Citation23 Our data suggest that in ascites, the repair of DSBs formed as intermediates of ICLs is temporally distinct from that observed in bone marrow cells. The phase when DSBs become accumulated is very similar between the cell types (0–12 h, in both ascites and bone marrow cells). The major difference is that in bone marrow cells DSB repair begins immediately after DSBs have accumulated, whereas ascites cells show an extended period, when the number of DSBs stays constant (12–24 h), and only during the period of 24–36 h does the DSB repair occur. The duration of this repair phase is the same in ascites and bone marrow cells ().
Figure 6. Analysis of DSB formation and repair in ascites cells of Krebs-2 tumor; CP treatment was used to induce DSBs in experimental animals. (A) Results of three independent experiments (1, 2, and 3) to quantify γН2AХ-positive repair foci found at DSBs. FACS-based (1 and 2) and fluorescence microscopy-based (3) quantifications were performed. 4, schematic diagram comparing formation and repair dynamics of DSBs in bone marrow cells (red)Citation15 and in ascites cells Krebs-2 (blue). (B) Representative image of ascites Krebs-2 cells immunostained with γН2AХ-specific antibodies (1). 2, control, incubation with primary antibodies was omitted from the protocol. DAPI, chromatin staining; FITC, γН2AХ. Bars correspond to 10 µm. (C) Comet assay showing formation and disappearance of DSBs in ascites cells Krebs-2. Note the presence and absence of comet tails matches the timepoints with maximal and minimal DSBs shown on plot 4. Bars correspond to 50 µm.
Many actively proliferating cell populations in mice and humans contain subpopulations of cells capable of internalizing extracellular dsDNA fragments
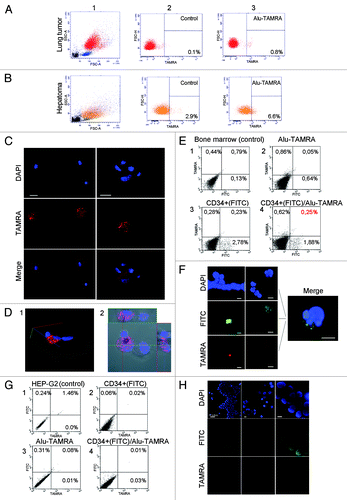
What would be the biological meaning of this property of a cell to internalize DNA? To indirectly address this question, one could explore it in other cells showing active proliferation or well-described stem-like phenotypes. We hypothesized that any actively proliferating cell communities invariable have a subpopulation of cells that can internalize dsDNA, and that this ability is a general property of poorly differentiated cells of various origin having stem-cell like potential. In view of this, we tested the ability of several primary murine and human cell lines showing stem-like properties to internalize dsDNA fragments. We observed that the percentage of such cells is 0.7% in ascites form of mouse lung tumor line and 3.7% in ascites form of mouse hepatoma cells (). One percent of mouse bone marrow cells (or 40% of CD34+ cells) are capable of internalizing dsDNA.Citation16 In humans, 0.25% of bone marrow cells (or 11.7% of CD34+ cells) and 2–5% mesenchymal stem cells show evidence of dsDNA internalization (). In contrast, HEP-G2 immortalized cell line lacking a tumorigenic phenotype in NOD/SCID mice,Citation24,Citation25 as well as mouse and human fibroblast cell linesCitation26 completely fail to internalize dsDNA fragments (). These observations are highly suggestive of the fact that subpopulations of cells capable of internalizing dsDNA are specific for meristem-like cell communities in mammals, and that a number of immortalized cell cultures lack such a cell subpopulation.
Figure 7. Cytofluorescence and flow cytometry analyses of TAMRA-Alu DNA internalization by various cellular communities. Field view is shown for microscopy imaging. DNA is stained with DAPI, FITC denotes CD34+ staining, and TAMRA corresponds to TAMRA-Alu DNA. (A) FACS profiles showing localization of TAMRA-labeled DNA in mouse lung tumor ascites. (B) FACS profiles showing localization of TAMRA-labeled DNA in mouse hepatoma ascites. (C) Internalization of TAMRA-labeled DNA by CD34+ human mesenchymal stem cells (short-term culture). (D) 3D reconstitution image of TAMRA-DNA internalization by human mesenchymal stem cells showing cytoplasmic localization of the probe. (E) FACS profiles showing localization of TAMRA-labeled DNA in CD34+ human bone marrow cells (short-term culture). (F) Cytofluorescence analysis of human bone marrow stem cells showing TAMRA-DNA labeling in CD34+ cells. (G and H) Non-tumorigenic human HEP-G2 cells grown in immunocompromised mice display no TAMRA-DNA signal in cellular compartments on either FACS profiles (G) or upon cytofluorescence analysis (H).
Discussion
One general finding of this work is that Krebs-2 ascites cells and human glioma cells are capable of dsDNA internalization and show some features of TISCs. This establishes several prominent features of tumor-initiating stem cells from Krebs-2 ascites and human glioma.
First, these cells can internalize dsDNA of various origins including PCR-synthesized or genomic double-stranded DNA fragments, as well as pUC19 and pEGFP-N1 plasmid DNA in either supercoiled or linear form.
Next, such dsDNA-internalizing cells constitute a very limited and permanent sub-population. Using ascites Krebs-2 cells, we demonstrate that this property is cell cycle-independent, that it is not shared by the entire population of proliferating ascites cells, and that it is not restricted to a particular timepoint in the cell cycle. Should cell cycle be a contributing factor, this would have been observed in our cell-cycle profiling experiments as characteristic shifts in flow cytometry peaks. Cell cycle profiles of sorted TAMRA+ cells are indistinguishable from those of the total ascites cell populations; furthermore, percentage of dsDNA-internalizing cells does not match the percentage of cells in the S phase. Also, if DNA internalization was restricted to a specific short period of cell cycle, prolonged cell incubation with TAMRA-labeled DNA would be expected to result in a progressive increase in TAMRA+ cells. However, our experiments showed that upon longer incubation the percentage of TAMRA+ cells plateaued. When internalization by neurospheres was continuously monitored in real-time, saturation was achieved within one hour of incubation and no evidence of concurrent DNA substrate release by the cells was observed. Thus, it appears very unlikely that at a very specific time point in the cell cycle the tumor cells would start internalizing dsDNA and simultaneously pump it out, thereby keeping the percentage of TAMRA+ cells constant. Rather it is more parsimonious to suggest that the ability to internalize dsDNA is a feature of a small population of cells with very specific characteristics.
In our direct engraftment experiments with Krebs-2 ascites, we show that TAMRA+ cells that are also largely CD34+, induce more rapid tumor formation than TAMRA– cells. Very similar data were obtained upon reciprocal engraftment of sorted CD34+ cells (of which some are DNA-internalizing), vs CD34– cells. Notably, CD34+ cells induced tumor growth similarly time-wise to the control ascites. When engrafting TAMRA+ cells, we observed delayed onset of transplant growth as compared with the control. One can speculate that within CD34+ population of cells, the cells capable of internalizing DNA (which constitute 90% of CD34+ cells in these experiments) are responsible for the tumorigenic phenotype of CD34+ cells. The difference between the onset of graft proliferation by CD34+ and TAMRA+ cells (both inter se and relatively to the control) is due to the presence of DNA in internal cell compartments, which may negatively affect their survival. It must be noted that tumorigenic potential of TAMRA+ cells is superior to that of TAMRA– grafts and persists at this level for more than 10 d.
The limitations inherent to the present research preclude experimental analysis of why the control population (total ascites) starts growing sooner than the sorted TAMRA+ cells.
Yet, these observations may be explained by the synergistic effects of cell populations in the grafted samples composed of residual tumorigenic, proliferating cancer and stromal cells, which secrete some cellular factors (cytokines, microvesicles, etc.). Stroma is known to provide a physiological niche for cancer stem cells,Citation27-Citation29 clearly then, when these cell populations become physically separated upon sorting, appropriate signaling is lost which affects the proliferative characteristics of tumorigenic cancer stem cells. In this respect, it appears most appropriate to perform pair-wise comparison of TAMRA+ and TAMRA− cells, sorted as individual cell populations and grafted in identical conditions. It is comparison of grafting potentials, rather than the property of the whole ascites to induce tumor growth (used here merely as a control of cell viability and cancer features), that should adequately mirror the true potential of sorted cell populations: upon cell sorting, the cells lose their microenvironment, and show their true character (in contrast to the use of the whole ascites graft).
Experiments on engraftment of CD34+ cells also had a separate important goal. One could think that higher tumorigenicity of TAMRA+ subpopulation was caused by DNA internalization in cellular compartments. However, the observations that up to 90% of CD34+ cells are TAMRA positive (in these experiments), and that CD34+ cells show tumorigenic potential similar to that of TAMRA+ cells, argue that DNA internalization is not causing increased tumorigenicity, but rather it marks the cells with this feature. Growth of TAMRA– grafts is explained by the fact that not all TISCs can efficiently internalize dsDNA at a given moment of time. This conclusion follows from the analysis of the ability of ascites cells to internalize TAMRA-labeled DNA, presence of CD34 marker and CD34+ cell engraftment, which clearly shows that both markers label the same TISC subpopulation. Overlap between these two markers as well as their contributions to graft formation are incomplete and unequal. This means that when sorting for one of these markers, the depleted cell population will still contain TISCs that will ultimately induce transplant engraftment (although somewhat delayed).
Engraftment experiments of human glioma cells demonstrate that two cell populations from the same cell culture display distinct tumorigenic properties. Adherent cell fraction devoid of dsDNA-internalizing cells, fails to engraft. Neurosphere cell population, of which ~20% cells can internalize dsDNA, induces formation of intracerebral graft. Taken together with the data of engraftment of TAMRA-positive Krebs-2 ascites, one can suggest that tumorigenicity of glioma cell fractions is largely attributable to the cells capable of dsDNA internalization.
Elimination or alteration of cancerous potential/phenotype of Krebs-2 TISCs and abrogation of their tumorigenic potential are likely due to the participation of internalized dsDNA fragments in the repair of ICLs. The details of ICL repair process are now well-documented.Citation30 It can be subdivided into nucleotide excision repair (NER) and homologous recombination (HR) phases. NER begins immediately after ICL formation, takes 12 h to complete and is characterized by DSB accumulation. HR also lasts for 12 h, and involves complete DSB repair. We observed that the period 1–12 h post-CP injection (NER) is crucial for the subsequent elimination or for voiding the tumorigenic potential of CD34+/TAMRA+ ascites cells. In contrast, when human dsDNA is internalized during the HR stage, an opposite effect is observed, and tumor growth is further stimulated. These facts support the idea that cancer cells are no exception to the rule, and are sensitive to the presence of human dsDNA during both NER and HR phases of ICL repair. DsDNA may participate in NER at the gap repair step, this gap originating from excision of an adduct and involvement of the homologous sequence. One can speculate that dsDNA fragments outcompete the homologous sequences, and this prolonged failure to resolve the intermediates or incorrect resolution of intermediates may underlie the elimination of TISCs or abrogation of TISC functions.Citation31 Importantly, the opposite may hold true for the repair proceeding via HR pathway, as extracellular RNA fragments may in fact contribute to the correct DSB repair.Citation32
Our approach to target TISCs is essentially a Trojan horse strategy, and it is fundamentally different from the existing methods of targeted TISC ablation that exploit specific cell markers and factors of stromal niches, or inhibit specific signaling pathways such as Hedgehog, Wnt, and Notch.Citation33,Citation34 The opportunity to use extracellular DNA as a factor interfering with repair process allows neutralization of the tumorigenic potential of TISCs. Importantly, this occurs without affecting the functional signaling molecules whose downregulation is frequently compensated by other pathways; rather, it is a physical interference taking place during random chaotic cell reaction, which involves irreparable damage to chromatin structure.
To conclude, we show that dsDNA internalization is an immanent and inherent property of Krebs-2 and glioma cells. We further demonstrate that dsDNA internalization is a property of stem cells and of many stem-like cell lineages, including tumor-initiating cancer stem-like cells. Finally, we provide experimental evidence for an opportunity to eliminate TISCs or abrogate their tumorigenic potential by interfering with their repair process via dsDNA internalization.
Materials and Methods
Lab animals
We used 2- to 3-mo-old CBA/Lac and NOD/SKID mice strain bred in the SPF-animal facility at the Institute of Cytology and Genetics, SB RAS. Animals were grown in groups of 5–10 mice per cage with free access to food and water. All experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Institute of Cytology and Genetics.
Tumor model
Murine Krebs-2 ascites tumor model was used. To obtain ascites, Krebs-2 cells were diluted 1:10 in 200 µL of normal saline and inoculated intraperitoneally (i.p.) (~2 × 106 cells). To obtain solid tumors, Krebs-2 cells were engrafted intramuscularly into the right hind leg in 100 µL of RPMI-1640 or PBS. In course of experiments, as soon as tumors became palpable (about 7 d following the inoculation), they were measured every 1–2 d with calipers, and tumor volume was calculated as follows: volume = length × width × height.
Injection of CP and exogenous DNA preparations
I.p. injections of a cytostatic drug cyclophosphamide (CP) were administered to mice at a dose of 200 mg/kg body weight. Next, depending on the specific experimental design, the preparations of fragmented human DNA (hereafter, “DNA”) or nitrogen mustard-treated DNA (“CL-dsDNA”) were given as 0.5 mg/injection. The following injection schedules were used: hourly 1–12 h, 12–24 h, and 18–30 h after CP injection (i.e., a total of 6 mg DNA prep/mouse). As a control, an equal volume of normal saline (200 µL/injection) was used.
TAMRA labeling of DNA probes
PCR labeling of human Alu repeat DNA
DNA was labeled using PCR. PCR template was human Alu repeat material cloned in pUC19, this repeat encompassed the start and the end of tandemly repeated AluJ and AluY (NCBI: AC002400.1, 53494–53767). Standard M13 primers were used for amplification. PCR purification was done by standard phenol-chloroform extraction followed by ethanol precipitation using ammonium acetate as a salt. DNA concentration and incorporation of dUTP-TAMRA were measured using Nanodrop (Eppendorf) and calculated by comparing the signal before and after PCR re-precipitation.
Klenow labeling of total human DNA
Total human DNA was isolated from placentas of healthy donors using a phenol-free extraction method and fragmented down to the average size of 0.3–6 kb using a sonifier set at 22 kHz. DNA thus prepared is a pharmacopeial drug (registration certificate [Medical Drugs of Russia] 004429/08 of 09.06.2008) and is owned and produced by Panagen LLC. Template DNA was boiled on a water bath for 5 min and quickly placed on ice. Klenow labeling was done in a reaction volume of 50 µL. The reaction mix contained 0.5–1 µg template DNA, 1× Klenow buffer (50 mM TRIS-HCl, pH 7.5; 10 mM MgCl2; 1 mM DTT; 0.5 mg/mL BSA; 50 mM EDTA, pH 8.0), 0.1 mM dATP, dCTP, dGTP, 65 µM dTTP and 35 µM TAMRA-dUTP, 0.1 mM random primer and 5 U Klenow fragment. Reactions were kept at room temperature for 1 h and stopped by addition of 1 µL 0.5 M EDTA. DNA was ethanol-precipitated, washed, and dissolved in water.
Culturing of Krebs-2 ascites with exogenous DNA
Delivery of exogenous 32P-labeled DNA into ascites cells
Analysis of in vitro delivery of exogenous DNA into tumor cells was done using ascites form of Krebs-2 cell line. Delivery of DNA into bone marrow cells was used as a well-described control.Citation15 Ascites cells were collected from an untreated mouse or from a mouse 18 h post CP injection. Bone marrow cells were taken from an untreated mouse and separated on a ficoll-urographin density gradient. All cell samples were washed trice in PBS and split as 107 ascites cells per well and 5 × 106 bone marrow cells per well. Each sample was incubated with either 0.75 µg 32P-labeled GFP DNA fragment (~700 bp), or 0.77 µg pEGFP-N1 plasmid DNA linearized with HindIII. Cells were kept in a humidified CO2-incubator (5% CO2, 95% air humidify at 37°C) and collected 1, 2, 4, or 8 h following addition of DNA. Cells were washed three times with PBS, resuspended in 30 µL PBS, mixed with 40 µL 1.5% low-melt agarose (Low Melt Ultra-Pure DNA Grade Agarose, Bio-Rad) and embedded in plugs. Agarose plugs were then transferred into 500 µL of buffer (1% sarcosyl, 50 mM EDTA, 1 mg/mL proteinase K) and incubated for 12 h at 58°C. Next, 1% agarose gel was prepared and low-melting agarose plugs were placed in the gel. Upon electrophoretic DNA separation, the gels with 32P-labeled samples were dried and imaged using a Molecular Imager FX Pro+ (Bio-Rad). To exclude the possibility that the signals are derived from the degradation of the labeled probe inside the cells followed by the resynthesis from the labeled monomers, and to safely conclude that Krebs-2 ascites cells have internalized exactly the DNA that was added to the medium, samples incubated with pEGFP-N1/HindIII DNA were run on the gels, Southern-blot transferred on Zeta-Probe membranes (Bio-Rad) and hybridized with the labeled GFP probe. Signal quantification was similarly monitored using PhosphoImager.
Delivery of human TAMRA-labeled Alu DNA into ascites Krebs-2 cells
On day 6–7 after engraftment, Krebs-2 ascites cells were collected from a mouse. Cells were spun for 5 min, 400 g at 4 °C and washed with RPMI-1640. Cells were counted on a hemocytometer. A total of 106 cells were incubated in 200 µL of RPMI-1640 mixed with 0.2 µg TAMRA-labeled Alu DNA for 1 h at room temperature. Cells were centrifuged, washed with a small volume of cell culture medium, and resuspended in a final volume of 500 µL. Next, the cells that incorporated the labeled DNA probe were analyzed by either FACS (BD FACSAria, Becton Dickinson) or by fluorescence microscopy. In FACS analysis, the percentage of TAMRA-positive cells was counted using untreated cells as a control. For fluorescence microscopy, cell suspensions were spun onto glass slides using cytospin (1000 rpm for 1 min). Cells were then covered with a droplet of Antifade DABCO supplemented with 0.5 µg/mL DAPI, covered with a coverslip and imaged using a fluorescence microscope Axioskop 2 Plus (Zeiss) and AxioVision software.
Treatment of ascites form of Krebs-2 tumor with pUC19 plasmid DNA
Following incubation with pUC19 plasmid DNA, samples of Krebs-2 tumor ascites were processed to isolate total DNA. Briefly, cells were washed twice with 1 mL PBS and centrifuged for 5 min at 400 g, 4 °C. After the last wash (supernatant S1), the cells were resuspended in 1 mL PBS supplemented with 10 mM MgCl2. Next, DNase I was added to a final concentration of 50 µg/mL and incubated for 1 h at 37 °C. Cells were washed again two times with 1 mL PBS and spun down. The last supernatant (S2) was saved, and cells were resuspended in 50 mM EDTA. Next, SDS and proteinase K were added to a final concentration of 1% and 400 µg/mL, respectively. Cell lysis proceeded for 2 h at 58 °C. DNA was phenol-chloroform extracted, precipitated with 0.6 volumes of isopropanol and dissolved in 30 µL water. Chemically competent XL10-Gold E. coli cells were used for transformation. When bacteria were transformed with total genomic DNA isolated from cells incubated with supercoiled plasmid DNA, we obtained ~500 transformants that were identical to the original plasmid DNA. In contrast, when linear DNA was used, only 2 transformants identical in sequence to the original plasmid were obtained. This seemingly simple experiment shows that within 60 min of incubation, linear DNA fragments fail to self-circularize and likely undergo nuclease degradation (data not shown).
Incubation of a solid form of Krebs-2 tumor with TAMRA-labeled Alu DNA
Incubation of tissue pieces with Alu-TAMRA DNA, preparation and microscopy analysis of tissue sections
Solid Krebs-2 tumor was excised from a hind limb of a mouse. A piece of tissue about 0.5 cm3 in size was placed into 500 µL of RPMI-1640 medium supplemented with 0.5 µg Alu-TAMRA DNA and incubated for 1 h at room temperature. Then, the sample was washed several times RPMI-1640 and frozen at −20 °C. Ultramicrotome Leica Ultracut EM UC6 was used to obtain tissue sections that were transferred to the slides. Each slide was covered with ~10 µL Antifade DABCO with DAPI (0.5 µg/mL), covered with a coverslip and analyzed under the laser scanning microscope LSM 780 NLO (Zeiss).
Incubation of physically dissociated solid form of Krebs-2 tumor with Alu-TAMRA DNA
Piece of a solid form of Krebs-2 tumor was dissociated by homogenization in a small volume of RPMI-1640 medium. Cell suspension was filtered through a mesh filter (40 µm pore size), spun for 5 min at 400 g, 4 °C, and resuspended in a small volume of RPMI-1640. Cell were counted and incubated with Alu-TAMRA DNA exactly as it was described for the ascites form of Krebs-2 tumor (see above).
Analysis of DSB formation in ascites Krebs-2 cells
Mice engrafted with the ascites form of Krebs-2 tumor, were injected with CP (200 mg/kg). Next, ascites cells were collected every 6 h. Cell samples were washed in PBS, and a small aliquot of cells was placed on clean de-greased slides, another aliquot was fixed in 2% formaldehyde in PBS. Cells on the slides were fixed for 15 min in 2% formaldehyde in PBS and washed with 0.5% BSA in PBS.
Subsequent sample processing was performed either on glass slides in a humidified chamber (for microscopy), or in solution (for FACS analysis). Cells were permeabilized for 2 min with PBS+0.1% Triton X-100, and washed with PBS+0.5% BSA. Cells were incubated with rabbit anti-phospho-histone γH2AX antibodies (Sigma) (1:1000 dilution in PBS, pH 7.4, 0.15% glycine [Sigma] and 0.5% BSA) for one hour at room temperature. Cells were washed in PBS+0.5% BSA and incubated with goat-anti-rabbit IgG FITC conjugates (Sigma) diluted 1:500 in the same buffer as the primary antibodies for 1 h at room temperature. All samples were protected from light during incubations and washes. Cells were washed with PBS+0.5%BSA and either resuspended in a small volume of PBS and FACS-analyzed on BD FACSAria instrument (Becton Dickinson), or overlaid with a ~10 µL droplet of Antifade DABCO, covered with a coverslip and imaged using fluorescence microscope Axioskop 2 Plus (Zeiss) and AxioVision software.
Comet assay to quantify DSB-positive cells
Cells engrafted with ascites form of Krebs-2 tumor were injected with CP (200 mg/kg). Then, 0, 24, 30, and 42 h post CP injection, ascites cells were collected, washed in PBS and embedded in low-melting agarose plugs (Low Melt Ultra-Pure DNA Grade Agarose, Bio-Rad) in 80 µL PBS at a density of 1000 cells per block. Next, agarose blocks were immersed in a lysis buffer (50 mM EDTA, 1% N-Lauroylsarcosine [Serva], 1 mg/mL proteinase K) for 30 min at 50 °C, which was replaced with 0.5 M EDTA. Agarose-embedded cells were then washed in TE buffer (10 mM TRIS-HCl, pH 7.6; 1 mM EDTA) for 15–30 min. The blocks were similarly oriented and subjected to electrophoresis (30 min, 1 V/cm). DAPI staining was done by placing the blocks into 500 µL of DAPI solution (0.5 µg/mL), transferred onto the glass slides and analyzed under the fluorescence microscope Axiovert 40 Inverted Microscope (Zeiss).
Quantification of CD34+ cells in the ascites form of Krebs-2 tumor using FACS and indirect immunofluorescence microcopy
Ascites cells collected were washed in PBS and spun for 5 min at 4 °C, 400 g. The cells were resuspended in PBS + 0.1% NaN3, 1% FBS. Four microliters of antibodies (FITC Rat anti-Mouse CD34, BD PharMingen) or 4 µg isotype control (FITC Rat IgG2a, κ Isotype Control, BD PharMingen) were added per one million cells, incubated for 40 min at 4 °C. The percentage of CD34+ cells was calculated using BD FACSAria instrument and isotype control, alternatively, the cells were washed in PBS and centrifuged onto the glass slides using cytospin (1000 rpm, 1 min). Next, ~10 µL Antifade DABCO was added, and coverslips were placed on the specimens. Data acquisition and imaging were performed using Axioskop 2 Plus (Zeiss) and AxioVision software.
Dual analysis of CD34 positivity and TAMRA-Alu incorporation by ascites form of Krebs-2 tumor
Krebs-2 ascites were incubated with TAMRA-labeled Alu DNA as described above. Then, the cells were washed twice with RPMI-1640 and fixed in methanol:glacial acetic acid mix (3:1) for 1 h at 4 °C. The cells were pelleted (400 g, 5 min), washed in PBS, and incubated with CD34-specific antibodies. Microscopy analysis was performed as above. At a small magnification, several fields were first imaged on DAPI and FITC filters, then the presence of Alu-TAMRA label and CD34 surface marker was analyzed at a greater magnification. A total of 787 cells were analyzed.
Pathomorphology of mouse tissues and organs
Pieces of organs were fixed in 4% formaldehyde, dehydrated in a graded series of alcohols, cleared in xylol, and embedded in paraffin. Five micrometer thick paraffin sections were stained with hematoxylin and eosin. AxioImager ZI microscope (Zeiss) was used for imaging.
Establishment of human glioblastoma culture
Histology description of surgical material
The tumor is composed of non-uniform clusters of polymorphic anaplastic astrocytes with gemystocytic and astroblast-like forms diffusely scattered along the swollen glioreticulum showing the features of glial cell dystrophy and Rosenthal fibers. Cell nuclei are polymorphic, sometimes hyperchromic, with rare abnormal mitoses and apoptosis. Necrotic foci are present, showing perifocal glial-mesenchymal reaction and proliferation of small blood vessels with hyperplastic endothelium. The tumor has well-developed vasculature composed of large blood vessels with thickened walls. Isolated regions of the tumor display xanthomatosis, diffuse focal sclerosis and interstitial lymphoid infiltrations. Pathology diagnosis: Glioblastoma Gr.4. 01.02.2013.
Isolation of a primary human glioma culture
Primary glioblastoma cell culture was established by mechanically shearing a piece of the tumor sample that was surgically resected from a patient with histology-confirmed grade IV glioblastoma, followed by culturing at 37 °C in 5% CO2, in α-MEM medium supplemented with 0.3 mg/mL l-glutamine, 5 mM HEPES-buffer, 10−4 M β-mercapthoethanol, 100 µg/mL gentamycin and 10% FBS. Three days later, detached cells were removed, and a fraction of adherent cells was kept in culture for about 1 mo until 80–90% confluence was achieved. Cell passaging was performed by adding 0.25% trypsin solution (Sigma) for 5–10 min. FACSCalibur flow cytometer was used to analyze cell phenotypes from the first passage. Tumor cells were negative for CD3 and HLA-DR markers, but expressed “stromal markers” CD73 (38%), CD90 (18%), and CD105 (17%). Additionally, 3% of cells were CD133+, which is a characteristic marker of glial stem cells. The cells were then cultivated in complete culture medium until a monolayer was formed. After two more passages (1 mo later), in addition to adherent cells, we observed spontaneous formation of neurosphere-like detached clusters of 20–90 cells. These neurosphere-like cell subpopulation was isolated and cultured separately at 37°C and 5% CO2 in α-MEM medium supplemented with 7% FBS. Following mechanical dissociation, neurosphere-forming cells displayed pronounced proliferative activity and were capable of forming secondary neurospheres.
Engraftment of primary human glioma cells into NOD/SCID immunocompromised mice
Primary human glioblastoma cell cultures were separated into adherent cell population and free-floating neurospheres. Adherent cells were incubated with 2% trypsin for 10 min and recovered from the plastic flasks. Cells were counted using hemocytometer and centrifuged at 400 g, 4 °C. Cell pellet was resuspended in α-MEM medium to a concentration of 75 000 cells/µL. Cell proliferation assay was performed using orthotopic glioblastoma xenograft into immunocompromised NOD/SCID (Charles River) mice housed under SPF conditions. Animals were grown in SPF animal facility at the Institute of Cytology and Genetics (Novosibirsk, Russia), in OptiMice system IVC units (Animal Care Systems), in standard conditions with free access to Ssniff complete feeds and water. Three hundred and seventy-five thousand cells in a volume of 5 µL were injected intracerebrally into the left hemispheres of NOD/SCID mice. Neoplastic progression was monitored 26 d later using a T2-weighted high field (11.7 T) MRI system BioSpec 117/16USR (Bruker). Of 10 mice used, 4 were injected with adherent cell fraction, and 6 were received injections of neurosphere cells.
Analysis of GFP expression in glioblastoma populations of adherent cells and neurospheres
To analyze pEGFP-N1 plasmid DNA internalization by human glioblastoma cells, adherent and neurosphere cells were cultivated in FCS-free α-MEM medium and 1 × 106 cells were incubated with 10 µg of pEGFP-N1 DNA in a volume of 1 mL of α-MEM for 1 h at 37°C, 5% CO2. Cells were washed twice with α-MEM. After the last centrifugation at 400 g, cells were resuspended in 1 mL of α-MEM supplemented with 7% FBS and incubated under the same conditions for 24 h. Cell samples were analyzed using a laser scanning microscope LSM 780 NLO (Zeiss).
| Abbreviations: | ||
| CP | = | cyclophosphamide |
| DSB | = | double-strand break |
| dsDNA | = | double-stranded DNA |
| ICL | = | interstrand crosslink |
| HR | = | homologous recombination |
| NER | = | nucleotide excision repair |
| TAMRA | = | fluorescent dye (carboxy tetramethyl-rhodamine) |
| TISCs | = | tumor-initiating stem cells |
Additional material
Download Zip (416.8 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors are grateful to Dr Mikhail P Moshkin for organizing the work at SPF animal facility, Dr Petr M Illarionov and Dr Dmitry V Subbotin for technical support, Evgeniya A Vaskova for providing the HUES9 cell line, Dr Andrey Gorchakov and Dr Robert McKallip for translation of the paper.
The work was funded by the Novosibirsk Region Long-term Target Project “Development of SMEs in Novosibirsk region in 2012-2016” (286-16/27 of 21.10.2013) and by the Novosibirsk City Budgetary Subsidy 2014 for Young Scientists and Specialists Involved in Innovative Projects (No. 15-5).
References
- Dick JE. Stem cells: Self-renewal writ in blood. Nature 2003; 423:231 - 3; http://dx.doi.org/10.1038/423231a; PMID: 12748623
- Dick JE. Breast cancer stem cells revealed. Proc Natl Acad Sci U S A 2003; 100:3547 - 9; http://dx.doi.org/10.1073/pnas.0830967100; PMID: 12657737
- Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene 2004; 23:7274 - 82; http://dx.doi.org/10.1038/sj.onc.1207947; PMID: 15378087
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66:9339 - 44; http://dx.doi.org/10.1158/0008-5472.CAN-06-3126; PMID: 16990346
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367:645 - 8; http://dx.doi.org/10.1038/367645a0; PMID: 7509044
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997; 3:730 - 7; http://dx.doi.org/10.1038/nm0797-730; PMID: 9212098
- Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods 2009; 347:70 - 8; http://dx.doi.org/10.1016/j.jim.2009.06.008; PMID: 19567251
- Frisan T, Levitsky V, Masucci M. Limiting dilution assay. Methods Mol Biol 2001; 174:213 - 6; PMID: 11357647
- Cao L, Zhou Y, Zhai B, Liao J, Xu W, Zhang R, Li J, Zhang Y, Chen L, Qian H, et al. Sphere-forming cell subpopulations with cancer stem cell properties in human hepatoma cell lines. BMC Gastroenterol 2011; 11:71; http://dx.doi.org/10.1186/1471-230X-11-71; PMID: 21669008
- Rajasekhar VK. Analytical methods for cancer stem cells. Methods Mol Biol 2007; 407:83 - 95; http://dx.doi.org/10.1007/978-1-59745-536-7_7; PMID: 18453250
- O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res 2010; 16:3113 - 20; http://dx.doi.org/10.1158/1078-0432.CCR-09-2824; PMID: 20530701
- Zhang W, Kai K, Choi DS, Iwamoto T, Nguyen YH, Wong H, Landis MD, Ueno NT, Chang J, Qin L. Microfluidics separation reveals the stem-cell-like deformability of tumor-initiating cells. Proc Natl Acad Sci U S A 2012; 109:18707 - 12; http://dx.doi.org/10.1073/pnas.1209893109; PMID: 23112172
- Lu J, Cui Y, Zhu J, He J, Zhou G, Yue Z. Biological characteristics of Rh123(high) stem-like cells in a side population of 786-O renal carcinoma cells. Oncol Lett 2013; 5:1903 - 8; PMID: 23833664
- Oates JE, Grey BR, Addla SK, Samuel JD, Hart CA, Ramani VA, Brown MD, Clarke NW. Hoechst 33342 side population identification is a conserved and unified mechanism in urological cancers. Stem Cells Dev 2009; 18:1515 - 22; http://dx.doi.org/10.1089/scd.2008.0302; PMID: 19260804
- Dolgova EV, Proskurina AS, Nikolin VP, Popova NA, Alyamkina EA, Orishchenko KE, Rogachev VA, Efremov YR, Dubatolova TD, Prokopenko AV, et al. “Delayed death” phenomenon: a synergistic action of cyclophosphamide and exogenous DNA. Gene 2012; 495:134 - 45; http://dx.doi.org/10.1016/j.gene.2011.12.032; PMID: 22227496
- Dolgova EV, Efremov YR, Orishchenko KE, Andrushkevich OM, Alyamkina EA, Proskurina AS, Bayborodin SI, Nikolin VP, Popova NA, Chernykh ER, et al. Delivery and processing of exogenous double-stranded DNA in mouse CD34+ hematopoietic progenitor cells and their cell cycle changes upon combined treatment with cyclophosphamide and double-stranded DNA. Gene 2013; 528:74 - 83; http://dx.doi.org/10.1016/j.gene.2013.06.058; PMID: 23911305
- Likhacheva AS, Nikolin VP, Popova NA, Dubatolova TD, Strunkin DN, Rogachev VA, Sebeleva TE, Erofeev IS, Bogachev SS, Yakubov LA, et al. Integration of human DNA fragments into the cell genomes of certain tissues from adult mice treated with cytostatic cyclophosphamide in combination with human DNA. Gene Ther Mol Biol 2007; 11:185 - 202
- Alyamkina EA, Dolgova EV, Likhacheva AS, Rogachev VA, Sebeleva TE, Nikolin VP, Popova NA, Orishchenko KE, Strunkin DN, Chernykh ER, et al. Combined therapy with cyclophosphamide and DNA preparation inhibits the tumor growth in mice. Genet Vaccines Ther 2009; 7:12; http://www.gvt-journal.com/content/7/1/12 http://dx.doi.org/10.1186/1479-0556-7-12; PMID: 19682353
- Nikolin VP, Popova NA, Sebeleva TE, Strunkin DN, Rogachev VA, Semenov DV, Bogachev SS, Iakubov LA, Shurdov MA. [Effect of exogenous dna injection on leukopoietic repair and antitumor action of cyclophosphamide]. [In Russian] Vopr Onkol 2006; 52:336 - 40; PMID: 17191708
- Ermakov AV, Konkova MS, Kostyuk SV, Smirnova TD, Malinovskaya EM, Efremova LV, Veiko NN. An extracellular DNA mediated bystander effect produced from low dose irradiated endothelial cells. Mutat Res 2011; 712:1 - 10; http://dx.doi.org/10.1016/j.mrfmmm.2011.03.002; PMID: 21392514
- Ermakov AV, Konkova MS, Kostyuk SV, Egolina NA, Efremova LV, Veiko NN. Oxidative stress as a significant factor for development of an adaptive response in irradiated and nonirradiated human lymphocytes after inducing the bystander effect by low-dose X-radiation. Mutat Res 2009; 669:155 - 61; http://dx.doi.org/10.1016/j.mrfmmm.2009.06.005; PMID: 19540246
- Olive PL, Wlodek D, Banáth JP. DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res 1991; 51:4671 - 6; PMID: 1873812
- Rojas E, Lopez MC, Valverde M. Single cell gel electrophoresis assay: methodology and applications. J Chromatogr B Biomed Sci Appl 1999; 722:225 - 54; http://dx.doi.org/10.1016/S0378-4347(98)00313-2; PMID: 10068143
- Ihrke G, Neufeld EB, Meads T, Shanks MR, Cassio D, Laurent M, Schroer TA, Pagano RE, Hubbard AL. WIF-B cells: an in vitro model for studies of hepatocyte polarity. J Cell Biol 1993; 123:1761 - 75; http://dx.doi.org/10.1083/jcb.123.6.1761; PMID: 7506266
- Mersch-Sundermann V, Knasmüller S, Wu XJ, Darroudi F, Kassie F. Use of a human-derived liver cell line for the detection of cytoprotective, antigenotoxic and cogenotoxic agents. Toxicology 2004; 198:329 - 40; http://dx.doi.org/10.1016/j.tox.2004.02.009; PMID: 15138059
- Likhacheva AS, Nikolin VP, Popova NA, Rogachev VA, Prokhorovich MA, Sebeleva TE, Bogachev SS, Shurdov MA. Exogenous DNA can be captured by stem cells and be involved in their rescue from death after lethal-dose γ-radiation. Gene Ther Mol Biol 2007; 11:305 - 14
- Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol 2006; 1:119 - 50; http://dx.doi.org/10.1146/annurev.pathol.1.110304.100224; PMID: 18039110
- Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 2010; 12:468 - 76; http://dx.doi.org/10.1038/ncb2048; PMID: 20418870
- McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013; 12:217 - 28; http://dx.doi.org/10.1038/nrd3870; PMID: 23449307
- De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol 2000; 20:7980 - 90; http://dx.doi.org/10.1128/MCB.20.21.7980-7990.2000; PMID: 11027268
- Tronov VA, Kramarenko II, Karpukhin AV. [Colorectal cancer: mismatch repair deficiency, genomic instability, tolerance to apoptosis and assessment of risk]. [In Russian] Vopr Onkol 2005; 51:159 - 66; PMID: 16222994
- Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA-templated DNA repair. Nature 2007; 447:338 - 41; http://dx.doi.org/10.1038/nature05720; PMID: 17429354
- Insan MB, Jaitak V. New approaches to target cancer stem cells: current scenario. Mini Rev Med Chem 2014; 14:20 - 34; http://dx.doi.org/10.2174/13895575113136660107; PMID: 24195662
- Ebben JD, Treisman DM, Zorniak M, Kutty RG, Clark PA, Kuo JS. The cancer stem cell paradigm: a new understanding of tumor development and treatment. Expert Opin Ther Targets 2010; 14:621 - 32; http://dx.doi.org/10.1517/14712598.2010.485186; PMID: 20426697