Abstract
Comment on: Xu R, et al. Cell Cycle 2010; 9:4315-4322.
The biochemical and structural composition of the extracellular matrix (ECM) provides contextual information that guides tissue morphogenesis and controls cellular function. The specific processes by which specific signals from the ECM control cellular phenotype have been studied extensively in mammary epithelial cells (MEC). Isolated from the lactating mammary glands and plated on conventional tissue culture plastic, MEC rapidly lose mammary-specific characteristics and stop synthesizing milk proteins.Citation1 However, when provided with lactogenic hormones and cultured in laminin-111 (LN1)-rich 3-dimensional (3D) gels, isolated MEC can be induced to form polarized acinar structures and to generate milk proteins, markers of functional differentiation,Citation2 showing that specific signals from the ECM are responsible for the maintenance of the differentiated state. While subsequent experiments have provided much information about the many different mechanisms induced by LN1, many aspects of the signaling pathway connecting extracellular LN1 to the activation of milk protein gene transcription remain unknown.
LN1 is a major component of the basement membrane, a specialized ECM that surrounds the epithelial cells in the mammary gland. Studies using transgenic models have elucidated the critical role of the LN1 receptors, dystroglycan and β1-integrin, and their downstream signaling pathways in the control of glandular development, tissue organization, and cellular differentiation.Citation3,Citation4 Parallel studies using cell culture models have helped to define the specific signals activated by cell association with LN1, showing that dystroglycan and β1-integrin serve both to physically connect LN1 molecules in the basement membrane to the cytoskeleton and also to function as activators of key signaling pathways.Citation3,Citation4
Prolactin is a lactogenic hormone mainly produced by the pituitary gland. Canonical prolactin signal transduction is initiated by binding of the hormone to the prolactin receptor, which activates JAK2-mediated phosphorylation and nuclear translocation of the transcription factor STAT5.5 However, LN is required for this pathway to be functional in MEC, as cells cultured on plastic do not show sustained phosphorylation of STAT5 in response to prolactin.Citation6,Citation7 In a previous study, Xu et al set out to determine why prolactin fails to activate STAT5 in 2D culture, finding that the prolactin receptor is mainly basolaterally localized, and in 2D culture, apically localized prolactin could not bind to and activate the basolateral receptor, preventing robust activation of STAT5.Citation8 The authors also found that exposure to LN1 led to MEC reorganization into polarized acini, in which the prolactin receptor was oriented toward the basolateral surface where it could be more effectively activated by soluble hormone.Citation8 In the study, Xu et al. identified phosphoinositide-3 kinase (PI3K) as the key signaling molecule linking LN1 and prolactin signaling pathways. They found that when cultured in plates coated with polyHEMA, which prevents adhesion to the plastic substratum, MEC form unpolarized cell aggregates that show only transient activation of STAT5 and no stimulation of β-casein gene expression in response to prolactin. By contrast, they found that LN1-induced structural polarization of the MEC was associated with functional activation of the prolactin signaling pathway: sustained activation of STAT5 and stimulation of β-casein gene expression. They evaluated PI3K as a potential mediator of this effect due to its role as a key mediator of integrin-induced signaling pathways, and found that inhibition of PI3K blocked LN1-mediated activation of prolactin signaling and also blocked LN1-mediated activation of Rac1, a key downstream effector of PI3K which was previously found to be crucial for STAT5 activation and functional differentiation of MECs.Citation9
Now that PI3K has been identified as the key signaling integrator of LN1 and prolactin signaling pathways, it will be important to determine exactly how LN1 stimulates PI3K during MEC functional differentiation. Xu et al. speculate that β1-integrin or dystroglycan could be leading to activation of PI3K through stimulation of focal adhesion kinase (FAK) or integrin-linked kinase (ILK). Another possibility could be an indirect activation of cell surface signaling receptors that can activate PI3K such as EGFR. Another important question would be to determine whether LN1-induced PI3K activation is also involved in the structural polarization that appears to be a prerequisite for functional differentiation. As such pathways play critical roles in both mammary gland development and in tumor progression, these studies may provide key insight into the differential response of normal and cancer cells to ECM and soluble signaling factors, and better understanding of why the former respond by arresting growth and functionally differentiating, while the latter continue to proliferate and fail to differentiate. The elegant cell culture system described by Xu et al. their recent study should make these questions much more tractable.
Acknowledgements
D.C.R. is supported by National Institutes of Health Grant CA122086 and by support from the Mayo Clinic Breast Cancer Specialized Program of Research Excellence (SPORE) Grant CA116201 (to James Ingle).
The continuous battle between a homeostatic organism and the chaotic environment is epical, and stress response mechanisms lie at the front line of survival. Which role does age play here? In human and animal populations, it is known that very young and old individuals are most sensitive to environmental stresses. Although in older animals stress sensitivity results from a decrease in fitness and the onset of disease, the increased mortality in very young individuals is still puzzling. A recent study suggests that this relationship between age and stress resistance is conserved also in the budding yeast S. cerevisiae,Citation1 a genetically tractable unicellular organism that ages.
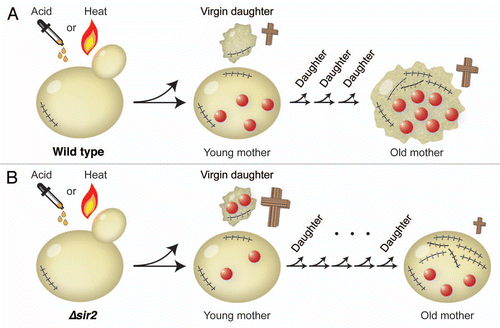
After exposing yeast cells to acetic acid or heat shock, the authors noticed that the daughter cells died more frequently than their mothers. By carefully analyzing the relationship between the total number of divisions and the cell death under stress, the researchers reported a U-shaped dependence: The virgin daughters had a higher incidence of death, which decreased for the first three divisions of young mothers, and increased again from the fourth division onwards. This held true for both pH and temperature shocks, suggesting that the relationship between age and stress survival is independent from the origin of stress.
Is damage segregation an important player here? It is generally thought that a dividing mother retains most of the deleterious components, segregating to the daughter “fresh” structures, thereby producing damage-free progeny. Studies in both prokaryoticCitation2 and unicellular eukaryoticCitation3,Citation4 organisms have revealed that the asymmetric inheritance of a higher fraction of the total protein damage (carbonylated and aggregated proteins) results in aging of the mother cell. Furthermore, higher eukaryotes also accumulate protein aggregates during aging.Citation5 In budding yeast, sir2 deletion has been shown to cause an overproduction of toxic rDNA circlesCitation6 and disrupt the asymmetric segregation of damaged proteins.Citation3 Knorre et al.Citation1 observed an increase in daughter death frequency following stress in a Δsir2 strain compared to wild type, probably arising from a higher level of inherited damage. Concomitantly, the older mother cells in the Δsir2 strain died less frequently than in wild type, presumably because Δsir2 mothers retain less damage than the wild-type mothers. Similarly to the Δsir2 mutant, mother-daughter protein damage distribution is symmetric in a Δbud6 mutant,Citation4 but in this mutant the levels of rDNA circles are, unlike in Δsir2, similar to wild type. Hence it would be interesting to test if the stress resistance is higher in older Δbud6 cells compared to those in a Δsir2 strain.
Interestingly, it has recently been shown that S. cerevisiae cells that have undergone a low number of divisions (1–6) fail to enter meiosis in response to nutrient depletion and pheromone signaling. The same effect is observed for older cells, here due to the absence of expression of the meiotic transcription factor Ime1.Citation7 This suggests that the relationship between age and the resistance to enter meiosis, which may be considered as a response to nutrient stress, is also U-shaped.
Knorre et al.Citation1 propose that the high stress sensitivity of small virgin daughters results from the shortage of energy due to their high surface-to-volume ratio. A testable prediction would be that large virgin daughters, which may be generated by blocking the cell cycle, are less sensitive to stress than small ones. An alternative explanation could reside on the requirement for cell growth and de novo biogenesis of mitochondria, since a new daughter is born smaller and usually inherits a small fraction of the mother mitochondria.Citation8 In this scenario, another replicative cycle would be necessary to achieve the full stress resistance. Further divisions would result in accumulation of damage, which contributes to the stress sensitivity observed in older mothers.
A yeast population seems to be composed of three distinct cell types: the virgin daughters (50%) whose objective is to mature in the current environment, the young mothers (47%, with 2–5 divisions) that are able to adapt to changing environmental conditions, and older cells (3%) which are competent to undergo meiosis and at the same time less resistant to stress. The ability to have a stratification of cell types may allow for a combination of different survival strategies simultaneously, although this remains to be addressed in future studies.
Figures and Tables
Figure 1 Virgin daughters are more sensitive to acetic acid or heat stress than young mothers in (A) wild type and (B) Δsir2 strain. Crosses represent stress-induced death, where the size of the cross correlates with the death rate. Each scar depicts a division event. The red circles represent the total amount of damage, which is in wild type retained by the mother cell and in the Δsir2 strain distributed between the mother and the daughter. Thus, virgin Δsir2 daughters are more sensitive to stress than virgin wild-type daughters. Old Δsir2 mothers, on the other hand, are less sensitive to stress than old wild-type mothers, because Δsir2 mothers accumulate less damage.
References
- Knorre DA, et al. Cell Cycle 2010; 9:4501 - 4505
- Lindner AB, et al. Proc Natl Acad Sci USA 2008; 105:3076 - 3081
- Aguilaniu H, et al. Science 2003; 299:1751 - 1753
- Liu B, et al. Cell 140:257 - 267
- David DC, et al. PLoS Biol 8:e1000450
- Sinclair DA, et al. Cell 1997; 91:1033 - 1042
- Boselli M, et al. Dev Cell 2009; 16:844 - 855
- Cerveny KL, et al. Dev Cell 2007; 12:363 - 375
Successful progression through the cell cycle is the culmination of many orderly events, such as the condensation of chromosomes. They must be packaged to a high order to ensure they are compact enough to be efficiently separated at anaphase and their condensation begins as they enter mitosis.
It has long been known that chromosome condensation is spatially and temporally concurrent with the phosphorylation of the amino tail of histone H3 on serine 10.Citation1 However phosphorylation of H3Ser10 does not seem to be an absolute requirement for chromosome condensation.Citation2 More recent studies have implicated the Aurora Kinases in phosphorylation of this residue as they can bind the histone H3 amino tail and phosphorylate Ser10 both in vitro and in vivo.Citation3 Indeed, H3Ser10 phosphorylation is the hallmark phosphorylation target of Aurora B; knockdown of Aurora B removes H3Ser10 phosphorylation and adversely affects chromosome morphology.Citation2
Aurora kinases are a well studied, highly conserved group of serine-threonine kinases that regulate multiple, crucial events throughout the cell cycle. Three mammalian Auroras have been identified and clearly defined roles have been assigned to Auroras A and B in mitosis.Citation4 Recent findings suggest that Aurora C may substitute for Aurora B in meiosis.Citation5 Aurora B functions within the chromosomal passenger complex (CPC), which localizes to the centromeres during prophase, becoming more concentrated there until metaphase, then abruptly translocating to the spindle midzone at anaphase.Citation6 This matches the pattern of H3Ser10 phosphorylation in mitosis, which begins in pericentromeric chromatin during G2/prophase and then spreads throughout the chromosomes as they condense.Citation1 Loss of H3Ser10 phosphorylation begins in anaphase and is complete by telophase,Citation1 coinciding with the CPC translocation to the spindle midzone.
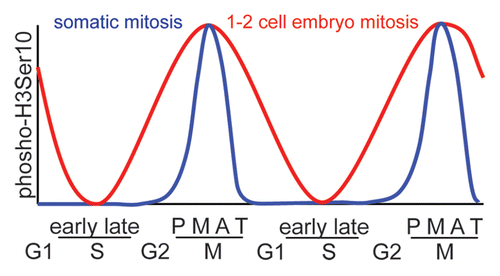
The study by Teperek-Tkacz et al. in a previous issue studied the importance of the hallmark Aurora B phosphorylation target, histone H3 serine 10, during the first and second mitotic divisions in the newly-formed mouse embryo.Citation7 These are the first divisions to be had with the maternal and paternal chromosomes in a common cytoplasm, and chromatin regulation at this stage of development is crucial. Teperek-Tkacz et al. find that H3Ser10 is phosphorylated earlier than in somatic cell mitosis, commencing in late S phase. Interestingly the phosphorylation observed during G1 phase of the two embryonic cell divisions is not merely lingering phosphorylation from the previous meiosis/mitosis brought about by a lack of phosphatase activity, but is due to kinase activity, specifically that of the aurora kinases.
Teperek-Tkacz et al. also discovered that the phosphorylation of H3Ser10 persists through anaphase and telophase of the first mitotic division, somewhat resembling the pattern seen in meiosis.Citation8 These results show significant differences from somatic cell mitosis () and demonstrate similarities with the meiotic divisions. In meiosis the pattern of H3Ser10 phosphorylation is maintained in pericentromeric chromatin during anaphase and telophase.Citation8 This was thought to be due to the fact that uniquely in meiosis I, sister cohesion must be maintained in this region, additionally there is no intervening S phase between chromosome divisions. The same reasoning cannot however be applied to anaphase or telophase of the second meiotic division nor the first mitotic division, therefore the importance of this meiosis-like H3Ser10 regulation remains to be found. One commonality is that it is occurring when the division is under the control of maternal gene transcription, as embryonic genome activation regulation does not commence until the second mitotic cell cycle.Citation9
Figures and Tables
Figure 1 Schematic showing the differing levels of H3Ser10 phosphorylation during the various stages of mitosis in somatic cells (blue) and in the first two embryonic divisions (red). (P, prophase; M, metaphase; A, anaphase; T, telophase).
References
- Hendzel MJ, et al. Chromosoma 1997; 106:348 - 360
- Adams RR, et al. J Cell Biol 2001; 153:865 - 880
- Crosio C, et al. Mol Cell Biol 2002; 22:874 - 885
- Carmena M, et al. Curr Opin Cell Biol 2009; 21:796 - 805
- Yang KT, et al. Mol Biol Cell 2010;
- Murata-Hori M, et al. Mol Biol Cell 2002; 13:1099 - 1108
- Marta Teperek-Tkacz MM, et al. Cell Cycle 2010; 9:4674 - 4687
- Wang Q, et al. Cell Cycle 2006; 5:1974 - 1982
- Wang QT, et al. Dev Cell 2004; 6:133 - 144
Cell cycle deregulation is one of the hallmarks in human cancer; cell cycle kinases such as cyclin-dependent kinases, polo-like kinases, and Aurora kinases have been implicated in tumor progression.Citation1 Members of the NIMA-related kinase (Nek) family have also been shown to be involved in tumorigenesis through up-regulation of mitotic progression. NIMA-related kinase 6 (Nek6) is activated in the G2/M transition of cell cycle; knockdown of Nek6 function either by over-expression of a dominant negative form of Nek6 or by siRNA depletion of endogenous Nek6 leads to mitotic arrest in G2/M and abnormal chromosome segregation which subsequently induce cell apoptosis.Citation2,Citation3 More recent studies have shown that Nek6 is over-expressed in a variety of cancer cell lines and tumor samples from patients, and that this overexpression is able to promote tumorigenesis.Citation4,Citation5 Furthermore, down-regulation of Nek6, either by siRNA knockdown of endogenous Nek6 or over-expression of kinase inactive Nek6, blocks cancer cell transformation and inhibits tumor growth in a xenograft mouse model.Citation4 These data clearly indicate that Nek6 plays a critical role in the promotion of tumor progression, presumably through cell cycle control; however, the precise molecular mechanism remains unclear.
Cellular senescence is characterized by the inability of cells to proliferate and can be induced by stress growth condition, DNA damage, cell aging, and oncogenic mutation. Of these, oncogenic mutation is an important mechanism to limit tumor progression.Citation6 For example, restoration of tumor suppressers such as PTEN and p53 can induce senescence and tumor regression.Citation7,Citation8 A report by Jee et al. in a previous issue of Cell Cycle linked Nek6 function to p53-induced senescence in tumor cells and may shed light on how Nek6 regulates tumor progression through cell cycle control.Citation9 The recent study by Jee et al. reveals that Nek6 expression is decreased in the presence of p53-induced senescence in human tumor cells. Moreover, ectopic expression of wild-type Nek6 but not kinase inactive Nek6 is able to reverse p53-induced senescence by up-regulating not only the G2/M events of cell cycle but by expression of cyclin B and cdc2 as well. These results confirm that Nek6 promotes tumor progression by controlling the G2/M transition and implicate Nek6 involvement in tumorigenesis through reversal of p53-induced senescence.
Together with previous studies, the work by Jee et al. provides compelling evidence that targeting Nek6 is likely to inhibit tumor growth through cell cycle blockade and senescence, and that a Nek6 inhibitor may have clinical benefit for cancer patients. It is important to further investigate the molecular mechanism(s) regulating senescence that may be driven by the interplay of Nek6 and cyclin B/cdc2, to identify the Nek6 direct substrate(s) in order to establish the position of Nek6 in regulation of the cell cycle cascade, and to determine whether Nek6 is able to suppress tumor cell senescence that is triggered by p53-independent events such as DNA damage or oncogene related deregulation. Results from these further studies will help guide clinical strategies in selection of appropriate patients based on their p53 or tumor suppressor/oncogene status as well as provide insight into combination therapies for a potential Nek6 inhibitor with various cytotoxic agents.
References
- Malumbres M, et al. Curr Opin Genet Dev 2007; 17:60 - 65
- Yin MJ, et al. J Biol Chem 2003; 278:52454 - 52460
- Roig J, et al. Genes Dev 2002; 16:1640 - 1658
- Nassirpour R, et al. Mol Cancer Res 2010; 8:717 - 728
- Jeon YJ, et al. J Biol Chem 2010; 285:28126 - 28133
- Evan GI, et al. Curr Opin Genet Dev 2009; 19:25 - 31
- Chen Z, et al. Nature 2005; 436:725 - 730
- Xue W, et al. Nature 2007; 445:656 - 660
- Jee HL, et al. Cell Cycle 2010; 9:4703 - 4710
Increasing evidence suggests that idiosyncratic mitoses, such as the first and second cell cycles of embryonic development, are controlled by many of the same regulators but in different ways. In a previous issue of Cell Cycle Teperek-Tkacz et al., found that the timing of Aurora kinase activity is different between the embryonic and other cell cycles suggesting that early embryonic mitoses are regulated differently.Citation1 Furthermore, one-cell embryos cultured in an Aurora kinase inhibitor (ZM447439) arrest in a different phase of the first cell cycle than two-cell embryos cultured in the same inhibitor, suggesting that Aurora kinase activity acts differently between the first and second cycle.
The first and second cell cycles of a mouse embryo differ from subsequent cell cycles and from one another, with the first having long G1 and short G2 phases, and the second having a short G1 and long G2 phases; the second also occurs in conjunction with the maternal-to-zygotic transition. Fertilized eggs complete meiosis II and enter G1 of the first mitotic cell cycle; thus, there is a unique transition from a haploid meiotic to diploid mitotic cell. During this cycle, paternal and maternal pronuclei exist separately from one another, only merging at the prolonged mitosis phase. Because the pronuclei are separate from one another, DNA replication, chromatin condensation and the first minor wave of zygotic gene transcription can occur asynchronously. During the second cell cycle, a major wave of transcription occurs during an unusually long G2 phase.
Mice contain three Aurora kinase homologs termed AURKA, B and C. AURKA and B are expressed in all tissues whereas AURKC is germ-cell specific. AURKA localizes to centrosomes where it regulates the centrosome cycle and spindle assembly.Citation2 AURKB is a component of the chromosome passenger complex that regulates microtubule attachment to chromosomes and subsequent segregation.Citation3 Although little is known about AURKC, in cells depleted for AURKB, ectopic expression of AURKC can rescue the mitotic defects, suggesting that they are functionally similar.Citation4
To follow AURK activity during the embryonic cell cycles, the authors detect phosphorylation of serine 10 in histone H3 (H3S10Ph), a known substrate for AURKB in somatic cells.Citation5 In all somatic cells studied so far, H3S10Ph occurs during G2 phase and is dephosphorylated at anaphase onset. During the early embryonic divisions, however, the authors found that H3S10Ph occurs in late S phase and persists until late in G1 of the following cell cycle. The functional consequence of H3S10Ph is unclear because some reports indicate that it is required for chromosome condensation whereas others argue it regulates gene expression during interphase.Citation6
To assess the requirement for AURK activity, the researchers cultured one- and two-cell embryos in medium containing ZM447439. Interestingly, different phenotypes were observed. One-cell embryos cultured in the presence of the inhibitor failed to cleave. Although chromatin condensation was normal, the embryos contained perturbed chromosome alignment at metaphase, had multipolar spindles and attempted to execute anaphase in the absence of cytokinesis. In contrast, two-cell embryos arrested in interphase and a lower percentage of the embryos contained condensed chromatin suggesting a different requirement for AURK activity.
Because H3S10Ph is thought to regulate gene expression it is possible that AURK is a positive regulator of zygotic gene activation. Further exploration of the different requirements of AURK activity and identification of their substrates during these cell cycles will help elucidate critical cell-cycle events in the early embryo. Although the ZM447439 inhibitor has higher specificity for AURKB than it does for the other two AURKs, the authors note that they required a higher concentration to observe the reported defects than levels previously used for meiotic or somatic cells.Citation7–Citation9 One possible explanation for this difference is that AURKC and/or AURKA also need to be inhibited to observe the cell cycle arrest phenotypes. Because AURKC can compensate for loss of AURKB in somatic cells, it is possible that maternal AURKC protein regulates these cell cycles. Alternatively, if AURKC has meiosis-specific functions and if the first mitotic cell cycle is more meiosis-like, it is possible that AURKC's unique meiotic functions are used as a bridge into the first cell cycle. Understanding why mammals contain a germ-cell specific AURK will likely shed light on how idiosyncratic cell cycles are regulated and why they are more prone to chromosome segregation mistakes often detrimental to embryo survival.
References
- Teperek-Tkacz M, et al. Cell Cycle 2010; 9:4674 - 4687
- Gopalan G, et al. J Cell Biol 1997; 138:643 - 656
- Hauf S, et al. J Cell Biol 2003; 161:281 - 294
- Sasai K, et al. Cell Motil Cytoskeleton 2004; 59:249 - 263
- Hsu JY, et al. Cell 2000; 102:279 - 291
- Perez-Cadahia B, et al. Biochem and Cell Biol 2009; 87:695 - 709
- Swain JE, et al. Mol Hum Reprod 2008; 14:291 - 299
- Shuda K, et al. Mol Reprod Dev 2009; 76:1094 - 1105
- Ditchfield C, et al. J Cell Biol 2003; 161:267 - 280
After prolonged replication or upon exposure to particular stresses, most cells stop proliferating and enter an irreversible cell cycle arrest known as senescence. This is a well-defined state characterized by altered morphology, enzyme activity and gene expression that can be experimentally monitored. Importantly, although senescence is thought to contribute to the aging process, it is also a vital tumor suppressive mechanism that can halt the progression of emerging cancer cells.Citation1 Deciphering the pathways that regulate senescence is therefore key to understanding how tumors evade this protective event and which regulators might be therapeutically targeted to induce senescence in a tumor.Citation2
In a previous issue of Cell Cycle, Jee and colleagues identified the serine/threonine protein kinase, Nek6, as a novel senescence regulator.Citation3 They found not only that endogenous Nek6 levels decrease upon replicative senescence, but also that ectopic expression of active Nek6 blocks the premature senescence that is induced by expression of the tumor suppressor gene, p53. Specifically, Nek6 prevented the changes in cell cycle distribution and production of reactive oxygen species that normally occur in senescent cells. However, apart from upregulation of the Cdk inhibitor, p21, which blocks cell cycle progression, it is not well understood how p53 induces premature senescence. And in cells overexpressing Nek6, p21 expression was still elevated in response to p53-induced senescence suggesting that Nek6 was not blocking the transcriptional activity of p53.
Abrogation of p53-induced senescence did not occur upon expression of inactive Nek6. This implies that senescence is inhibited as a result of Nek6-dependent phosphorylation events. Identification of physiological substrates of Nek6 relevant to the senescent phenotype will therefore be essential to understand how Nek6 function impinges on the senescence programme, and whether this is a direct or indirect effect of upregulated Nek6 activity. Interestingly, Nek6 was recently reported to phosphorylate the oncogenic transcription factor, STAT3, suggesting that this may be one potential candidate through which senescence might be regulated.Citation4
These findings are intriguing as Nek6 is thought to function primarily, if not exclusively, in mitotic progression.Citation5 Here, it promotes both bipolar spindle formation and cytokinesis, with depletion of Nek6 leading to spindle assembly checkpoint activation, mitotic arrest and apoptosis.Citation6,Citation7 Of course, this raises a number of questions about how a mitotic regulator can act to block senescence. First and foremost, does this relate to its mitotic functions or is upregulated Nek6 also active and playing additional roles in interphase? Secondly, as Nek6, and a closely related kinase, Nek7, are activated in mitosis by another related kinase, Nek9, then does overexpression of active Nek7 or Nek9 also interfere with senescence?
From a clinical viewpoint, this study provides an important clue as to how Nek6 expression might contribute to tumorigenesis. Nek6 is upregulated in a variety of cancers where, in theory, it could be acting to prevent the onset of senescence.Citation8 However, elevated Nek6 expression can also induce changes consistent with cellular transformation suggesting that it could promote cancer progression through multiple pathways.Citation4,Citation9 Encouragingly, RNAi-mediated depletion of Nek6 inhibits not only proliferation of cancer cell lines but also growth of tumor xenografts in nude mice.Citation9 It will be exciting to determine whether this may, at least in part, be due to induction of senescence. Ultimately, though, it will of course be necessary to study the effects of Nek6 expression and the response to Nek6 depletion or inhibition in real tumors. Nevertheless, these data add to the growing evidence that Nek6 could act as an effective target for novel cancer therapeutics.
References
- Campisi J, et al. Nat Rev Mol Cell Biol 2007; 8:729 - 740
- Ewald JA, et al. J Natl Cancer Inst 2010; 102:1536 - 1546
- Jee HJ, et al. Cell Cycle 2010; 9:4703 - 4710
- Jeon YJ, et al. J Biol Chem 2010; 285:28126 - 28133
- O'Regan L, et al. Cell Division 2007; 2:25
- O'Regan L, et al. Mol Cell Biol 2009; 29:3975 - 3990
- Yin M-J, et al. J Biol Chem 2003; 278:52454 - 52460
- Capra M, et al. Cancer Res 2006; 66:8147 - 8154
- Nassirpour R, et al. Mol Cancer Res 2010; 8:717 - 728
During metazoan development life, multipotent stem cells produce the differentiated cell types present in a tissue while maintaining a stem cell population.Citation1,Citation2 They do so by dividing asymmetrically to produce one daughter that adopts a differentiated fate, and one daughter that maintains the stem cell fate; however stem cells can also divide in a proliferative manner to generate two stem cells. Although the nematode C. elegans does not require adult somatic stem cells during its short life, there is a population of multipotent cells that divide in a stem cell-like manner during larval development.Citation3 The lateral epidermal seam cells generally divide asymmetrically in each of the four larval stages to generate an anterior daughter that differentiates as an epithelial cell, and a posterior daughter that retains the seam cell fate. In addition, a subset of seam cells undergo a proliferative division in the second larval stage to generate the full complement of seam cells. After their division in the fourth larval stage, the seam cells exit the cell cycle and terminally differentiate; fusing together and secreting cuticular ridges called alae.
Previous work has characterized two pathways regulating the behavior of the seam cells. First, the proteins and microRNAs in the “heterochronic pathway” function to regulate the timing of the L2 proliferative division and seam cell terminal differentiation in the late L4.Citation4 In “precocious” heterochronic gene mutants the seam cells carry out stage-specific events at an earlier time, while in “retarded” mutants such events are delayed. Second, the asymmetric divisions of two seam cells, V5.p and T, is regulated by Wnt signaling.Citation5 Wnt signals are known to control the division of adult stem cell populations in vertebrates.Citation6
The work of Banerjee et al further examines the regulation of seam cell behavior by the Wnt and heterochronic pathways.Citation7 They characterize the C. elegans CKIα (casein kinase Iα) homolog KIN-19. In vertebrates, CKIα is as a negative regulator of the “canonical” Wnt pathway in which the transcription factor β-catenin interacts with TCF/LEF transcription factors to upregulate target genes in the presence of Wnt ligand. In the absence of ligand, β-catenin is bound by a “destruction” complex containing the kinases CKIα and GSK-3, which phosphorylate β-catenin and target it for degradation.Citation8 C. elegans kin-19 was shown previously to act during embryonic development as a positive regulator of a variant C. elegans Wnt signaling pathway called the “Wnt/β-catenin asymmetry” pathway.Citation9 In this pathway, the activation of target genes is regulated not by degradation of β-catenin, but by Wnt signaling-dependent alterations in the nuclear ratio of the TCF homolog POP-1 and the β-catenin SYS-1.Citation5
The authors use RNA interference (RNAi) to show that kin-19 reduction causes seam cells to divide symmetrically in the L4 stage and generate two seam cell daughters, leading to an increase in adult seam cell numbers. They show that RNAi for other members of the “Wnt/β-catenin asymmetry” pathway (eg., apr-1/APC, pry-1/Axin, lit-1/MAPK, wrm-1/β-catenin, pop-1/TCF) also causes production of too few or too many seam cells, in a manner consistent with the current model for this pathway.Citation5 KIN-19 clearly functions as a negative regulator of Wnt signaling in this process, unlike its role during embryonic development.Citation10 Curiously, the gsk-3 homolog does not appear to function in this process. Equally curious, no role was found for any Wnt or Wnt receptor genes, leading the authors to suggest the Wnt pathway may act cell autonomously in these cells. Recent work by Gleason and Eisenmann shows similar results, including a negative role for kin-19, lack of function of a GSK homolog, and lack of effect upon reduction of Wnt gene activity.Citation10 These data suggest that a Wnt pathway acts in the regulation of most or all seam cell asymmetric cell divisions during late larval life, and that Wnt regulation of stem cell-like asymmetric cell divisions is an evolutionarily-conserved process also found in nematodes.
Banerjee et al also look for an interaction between the Wnt and heterochronic pathways. Late larval heterochronic mutants did not affect adult seam cell number, and there were no unexplained effects of Wnt pathway alterations on the timing of stage-specific seam cell events, suggesting the Wnt and heterochronic pathways act in parallel to regulate seam cell behavior. However, kin-19(RNAi) could suppress the precocious terminal differentiation phenotypes of late larval heterochronic pathway mutants, and enhance the retarded phenotype of another. These results suggest that KIN-19 acts in late larval seam cells in two pathways; as a negative component of the Wnt pathway regulating asymmetric cell divisions, and as a positive regulator of seam cell terminal differentiation in the heterochronic pathway. Recent work by Ren and Zhang also reports that mutations in several Wnt/β-catenin asymmetry pathway components can suppress retarded heterochronic mutant phenotypes, however these authors suggest that the Wnt pathway acts upstream of the heterochronic genes in late larval life.Citation3 Unresolved questions about the method of activation of the Wnt pathway, the regulation of CKIα in each pathway, and the manner in which the Wnt and heterochronic pathways interact to control asymmetric seam cell division and differentiation along the temporal axis, will be fruitful avenues for further work.
References
- Knoblich JA. Cell 2008; 132:583 - 597
- Morrison SJ, et al. Cell 2008; 132:598 - 611
- Joshi PM, et al. Dev Dyn 2010; 239:1539 - 1554
- Moss EG. Curr Biol 2007; 17:R425 - R434
- Mizumoto K, et al. Trends Cell Biol 2007; 17:465 - 473
- Reya T, et al. Nature 2005; 434:843 - 850
- Banerjee D, et al. Cell Cycle 2010; 9:4748 - 4765
- Clevers H, et al. Cell 2006; 127:469 - 480
- Peters JM, et al. Nature 1999; 401:345 - 350
- Gleason JE, et al. Dev Biol 2010; 348:58 - 66
- Ren H, et al. Dev Biol 2010; 345:144 - 155
Cdks and their cyclin partners provide the impetus that drives cell cycle transitions in eukaryotic cells. Despite over three decades of investigation, the details of how Cdks promote many aspects of the cell division cycle remain unclear. The mechanisms by which Cdks drive many developmental and differentiation programs remain particularly enigmatic owing to the difficulty in recapitulating these processes in vitro. It is clear that Cdk activity must accumulate at the right time and the right place to effectively promote progression through the cell cycle. Inappropriate accumulation of Cdk activity is deleterious as it disrupts the carefully orchestrated order of events required for successful cell division.Citation1
In a previous issue of Cell Cycle Szwarcwort-Cohen, et al.Citation2 report on their investigation of human Cdk2 (hCdk2) and the yeast meiosis-specific kinase Ime2 when these enzymes are expressed in the yeast Saccharomyces cerevisiae. The authors link these two kinases based upon the sequence similarity of their kinase domains. hCdk2 is capable of functioning as a Cdk in yeast as it rescues (albeit poorly) yeast temperature sensitive cdk1 mutants.Citation3 Over expression of hCdk2, or Ime2, in mitotically proliferating S. cerevisiae lead to cell cycle delay in G1-phase, delayed induction of the G1 transcriptional program, and reduced viability. Although hCdk2 over expression arrested DNA replication the cells displayed a significant proportion of budded cells suggesting that hCdk2 might be promoting other aspects of the division cycle. Similarly over expression of Ime2 in proliferating cells resulted in a G1-phase arrest. Why does over expression of these enzymes lead to cell cycle arrest? The authors propose that they interfere with the activity of yeast Cdk1, the Cdk that is tasked with regulating cell cycle progression.Citation4 This may be the case as hCdk2 could sequester or induce the degradation of the endogenous cyclins; however, Ime2 does not bind cyclins. It is just as likely that ectopic hCdk2 and Ime2 phosphorylate inappropriate substrates. This would not surprising in the case of Ime2 as this enzyme phosphorylates a consensus sequence distinct from the Cdk consensus.Citation5 There is evidence that cyclins determine the substrate specificity of Cdks.Citation6 If hCdk2 can bind yeast cyclins, should it not phosphorylate the correct substrates? One possibility is that hCdk2 rather than its cyclin partner has a greater influence on substrate selection than has previously been appreciated.
Ime2 is essential for meiosis in yeastCitation7 as is Cdk2 in mammalian cells.Citation8 A deeper investigation of the mitotic G1-arrest induced by over expression of these enzymes revealed that they induced the expression of meiosis-specific genes including genes encoding factors that function to induce the formation of DNA double strand breaks (DSBs) required for homologous recombination which occurs at high frequency during yeast meiosis. Clearly the inappropriate activation of the homologous recombination machinery has potential to be deleterious, particularly in a haploid cell. However, it is unclear whether this is the cause of inviability as deletion of SPO11 the DSB inducing endonuclease does not remedy the defect. The investigators did however find that cells with defects in the DNA damage checkpoint displayed elevated sensitivity to over expression of hCdk2 or Ime2, consistent with the notion that DNA damage is occurring in the over expressing strains. Surprisingly, Rad53 which is normally activated in response to DNA damage was not activated in the over expressing strains. Indeed these cells displayed a profound defect in Rad53 activation in response to DNA damage. This may in part be due to the accumulation of the over expressing strains in G1-phase where Rad53 not effectively activated. A more intriguing possibility is that Ime2 or hCdk2 actively suppress the activation of Rad53. DNA double strand breaks due to recombination form as yeast progress through meiosis. Yet, these do not appear to trigger checkpoint arrest. Perhaps Ime2 functions to suppress checkpoint activation, allowing unfettered progression through meiotic differentiation. Cdk2 may perform a similar function in mammalian gametogenesis.
Szwarcwort-Cohen, et al.Citation2 fail to conclusively determine the mechanism of toxicity induced by hCdk2 in yeast, but their investigation does reveal some interesting possibilities and opens the door to more extensive investigations of Cdk function through expression in different cellular environments.
References
- Malumbres M, et al. Nat Rev Cancer 2009; 9:153 - 166
- Szwarcwort-Cohen M, et al. Cell Cycle 2010; 9:4711 - 4719
- Ninomiya-Tsuji J, et al. Proc Natl Acad Sci USA 1991; 88:9006 - 9010
- Reed SI, et al. Proc Nat Acad Sci USA 1985; 82:4055 - 4059
- Sedgwick C, et al. Biochem J 2006; 399:151 - 160
- Loog M, et al. Nature 2005; 434:104 - 108
- Y oshida M, et al. Mol Gen Genet 1990; 221:176 - 186
- Ortega S, et al. Nat Genet 2003; 35:25 - 31