Abstract
In response to DNA damage, cells activate a complex, kinase-based signaling network that consist of two components - a rapid phosphorylation-driven signaling cascade that results in immediate inhibition of Cdk/cyclin complexes to arrest the cell cycle along with recruitment of repair machinery to damaged DNA, followed by a delayed transcriptional response that promotes cell cycle arrest through the induction of Cdk inhibitors, such as p21. In recent years a third layer of complexity has emerged that involves post-transcriptional control of mRNA stability, splicing, and translation as a critical part of the DNA damage response. Here, we describe recent work implicating DNA damage-dependent modification of RNA-binding proteins that are responsible for some of these mRNA effects, highlighting recent work on post-transcriptional regulation of the cell cycle checkpoint protein/apoptosis inducer Gadd45a by the checkpoint kinase MAPKAP Kinase-2.
Post-transcriptional Control of Gene Expression is Essential for a Functional DNA Damage Response
In response to DNA damage, cells activate a complex interconnected signaling network that alters cell metabolism, temporarily or permanently halts cell cycle progression, activates and recruits DNA repair machinery and controls the decision of whether to initiate some type of cell death program.Citation1 Many of these changes occur as a consequence of post-translational modifications of proteins within the DNA damage signaling network through phosphorylation, ubiquitylation or sumoylation.Citation2 In addition, the pattern of mRNA expression also undergoes significant changes after DNA damage. In a study of human lymphoblastoid cell lines from healthy adults, for example, Chu and colleagues found that thousands of mRNAs were up or downregulated after cells were exposed to ionizing radiation (IR) or ultraviolet light.Citation3 Similarly, as much as 20% of the genes in budding yeast showed a two-fold or greater change in mRNA levels in response to MMS treatment or ionizing radiation.Citation4 However, it would seem intuitively obvious that the last thing one would expect cells to do shortly after incurring significant DNA damage is to start transcribing new genes. First, there is the real probability that the genetic information in the DNA strand itself is damaged. Second, the transcription process is both energy intensive (synthesis of a message with n bases requires at least n NTP molecules) and slow from the point of view of creating a final protein product if it was needed for cell cycle arrest or DNA repair. In agreement with this, a number of studies have shown that DNA damage triggers a transient repression of gene transcription in eukaryotic cells.Citation5,Citation6 How, then, do cells accomplish DNA damage-induced changes in mRNA expression, when transcriptional activity is globally suppressed?
One possible explanation lies in manipulating the post-transcriptional regulatory circuits that control mRNA splicing, stabilization and translation. All three of these processes depend on specific RNA-binding proteins (RBPs) which modulate mRNA half-life, spliceosome targeting, nuclear export and translatability through direct interactions with their client mRNAs.Citation7–Citation11 Importantly, recent data from several laboratories now suggests that these RBPs are direct targets of the DNA damage response (DDR), and are likely to be critically important in controlling the phenotypic response in cells experiencing genotoxic stress. In a mass spectrometry-based proteomic screen for binding partners of the DNA damage response molecule/tumor suppressor 14-3-3σ, for example, we found that the largest classes of interacting proteins were molecules involved in mRNA splicing or translation.Citation12 Matusoka et al. performed a phospho-proteomic mass-spectrometry screen looking for substrates of the dedicated DDR kinases ATM, ATR and DNA-PK in response to IR.Citation13 Surprisingly, the largest subset of ATM/ATR/DNA-PKcs substrates they identified were proteins linked to nucleic acid metabolism, and in particular proteins involved in posttranscriptional mRNA regulation. Cimprich and colleagues conducted a genome-wide RNAi screen looking for spontaneous γH2AX formation following gene knock-down.Citation14 In that screen, mRNA-binding and processing factors emerged as the largest subset of ‘hits’. Finally, in a study of alterations in gene expression that directly compared changes in mRNA abundance with changes in gene transcription, Gorospe and colleagues found that over 50% of the observed changes in steady state mRNA levels in non-small cell lung carcinoma H1299 cells exposed to UV irradiation were directly attributable to mRNA turnover.Citation15 Taken together, these observations, from very diverse experimental approaches, highlight the emerging importance of post-transcriptional regulatory mechanisms in the context of DDR signaling. What, then, are the molecular signals emerging from damaged DNA that are responsible for controlling these post-transcriptional events?
Clearly, the Matsuoka et al. data suggest that ATM and ATR are likely to play a direct role. In addition to the ATM-Chk2 and ATR-Chk1 modules, however, genotoxic stress also activates the p38MAPK-MAPKAP2 Kinase-2 (MK2) pathway downstream of ATM and ATR,Citation9,Citation16–Citation22 and the function of this pathway is particularly critical for cell cycle arrest and prevention of apoptosis in cells with defective p53 function. We recently discovered that major targets of the p38MAPK/MK2 pathway are RNA-binding proteins involved in upregulating the cell cycle checkpoint protein Gadd45α.Citation23
Gadd45α is Post-Transcriptionally Regulated in Response to DNA Damage
Gadd45α is a known component of the DDR that has been mechanistically linked to numerous cellular processes, including cell cycle arrest and apoptosis.Citation24,Citation25 Gadd45α expression is rapidly induced after genotoxic stress both in a p53-dependent and -independent fashion.Citation23,Citation26–Citation33 In addition to transcriptional control, Gadd45α mRNA levels are post-transcriptionally stabilized in response to UV or MMS exposure.Citation34 Recent work from Gorospe and co-workers identified the RNA-binding proteins (RBPs) AUF1 and TIAR as critical negative regulators of Gadd45α mRNA at the post-transcriptional level.Citation35 In resting cells, AUF1 and TIAR are found in ribonucleo-protein (RNP) complexes with Gadd45α mRNA. AUF1 targets Gadd45α mRNA for degradation, while TIAR prevents the association of Gadd45α mRNA with translating polysomes. The combined effect of these two inhibitory mechanisms is a potent repression of Gadd45α protein biosynthesis.
Upon exposure to UV or treatment with MMS, Lal et al. showed that AUF1 and TIAR rapidly dissociate from Gadd45α mRNA, correlating with a substantial increase in Gadd45α mRNA stability and increased Gadd45α protein accumulation.Citation35 The nature of the molecular signals responsible for the DNA damage-induced dissociation of AUF1 and TIAR remained enigmatic, as did the potential involvement of other damage-induced RBPs in this process. In an attempt to understand why p53-defective cells required p38MAPK/MK2 activity for cell cycle checkpoint function, we recently made the surprising discovery that this pathway is a critical regulator of RBPs mediating posttranscriptional stabilization of Gadd45α mRNA in response to genotoxic stress.Citation23 Specifically, we discovered three novel p38MAPK/MK2-dependent posttranscriptional mechanisms of Gadd45α mRNA control. First, we found that RNAi-mediated MK2 depletion prevented the accumulation of Gadd45α mRNA and protein in response to doxorubicin and identified the known MK2 substrate hnRNP A0 as a novel Gadd45α mRNA-binding protein.Citation23 We and others showed that MK2 directly phosphorylates hnRNP A0 on Ser-84.Citation23,Citation36 This specific phosphorylation event was critical for hnRNP A0:Gadd45α mRNA RNP complex formation, which in turn was required to stabilize Gadd45α mRNA in response to DNA damage. Second, we found that MK2 directly phosphorylates Poly-(A) ribonuclease (PARN) on Ser-557 in response to doxorubicin. PARN phosphorylation at this site was required for prolonged Gadd45α mRNA and protein expression after doxorubicin. The molecular details of this apparent inhibition of Gadd45α mRNA degradation remain somewhat unclear; despite our best efforts, we have not been able to observe any changes in PARN activity in vitro, following MK2-mediated phosphorylation on Ser-557. Finally, we could show that the MK2-activating kinase p38MAPK directly phosphorylates TIAR after doxorubicin and that p38MAPK inhibition completely prevented the doxorubicin-mediated dissociation of TIAR:Gadd45α mRNA RNP complexes.
In additional experiments, we showed that Gadd45α operates as part of a positive feedback loop that is necessary to maintain MK2 activity at late times, but not at early times, after DNA damage. RNAi-mediated depletion of Gadd45α specifically prevented the phosphorylation and activation of MK2 from ∼12 hrs onwards after DNA damage, likely through a late loss of p38 activity. These data suggest that the initial activation of MK2 after genotoxic stress does not depend on Gadd45α, but subsequent p38/MK2-dependent stabilization of Gadd45α, through phosphorylation of TIAR, PARN and hnRNP A0, becomes essential for maintaining MK2 activity at late times.Citation23
Why is this late MK2 activity important? We and others had shown previously that MK2 acts as a cell cycle checkpoint effector kinase that is activated downstream of ATM and ATRCitation18,Citation37 much like Chk1 and Chk2. And just like Chk1 and Chk2, MK2 phosphorylates Cdc25B and C to create a 14-3-3-binding site, sequestering Cdc25B/C in the cytoplasm to prevent CyclinB/Cdk1 activation. In our recent experiments, we found that nuclear-localized Chk1 is responsible for initiating an early G2/M checkpoint, but this arrest is lost after about 12 hours. In cells that lack p53 function, MK2 activity, primarily in the cytoplasm, is required to then maintain this arrest.Citation18,Citation23 Intriguingly, we could show that MK2 rapidly exits the nucleus after DNA damage and remains active in the cytoplasm for at least 30 hrs following the genotoxic insult. MK2 depletion resulted in premature nuclear re-entry of CDC25B/C followed by checkpoint collapse and a catastrophic attempt to undergo mitosis. Importantly, the time at which we observed premature CDC25B/C nuclear re-entry in MK2-depleted cells corresponded perfectly to the time when MK2 activity had dropped to baseline levels in Gadd45α-depleted cells that were treated with doxorubicin. These data strongly suggest that the positive feedback loop involving MK2-dependent stabilization of Gadd45α and Gadd45α-dependent maintenance of MK2 activity, are essential for prolonged cell cycle arrest through cytoplasmic Cdc25B/C sequestration in response to doxorubicin. Together, our data indicate that a feed forward loop consisting of p38MAPK, MK2 and Gadd45α is critical for allowing cells time to recover from doxorubicin-induced genotoxic damage before entering the next mitotic cell division ().
Concluding Remarks
The DNA damage response has classically been thought of as a signaling network consisting of rapid phosphorylation-driven signal transduction on one hand and a delayed transcriptional response mediated through transcription factors, such as p53. In addition to these well-established branches of DNA damage signaling, posttranscriptional regulatory mechanisms are now beginning to emerge as critical components of the DNA damage response. Recent mechanistic studies are pinpointing at a number of RNA-binding proteins, such as HuR, TIAR, AUF1, hnRNP A0 and PARN as well as protein kinases, such as ATM, ATR, Chk2, p38MAPK and MK2 that control the function and localization of these RNA-binding proteins.Citation9,Citation11,Citation13,Citation14,Citation23,Citation35,Citation38 These reports strongly suggest that cells employ complex regulatory circuits that control mRNA transcript splicing, stability and translatability in response to genotoxic stress. The major challenges in this emerging area of research will be identifying the relevant RBPs, mapping the transcripts that are post-transcriptionally regulated by specific RBPs, and determining whether individual RBPs control a subset of transcripts with related function in the DNA damage response a so-called RNA operon.Citation39 New technologies, such as genome-wide RNAi screening and next generation sequencing of cell lines and primary tumor samples should allow the systematic search for novel transcripts and RNA-binding proteins that are critical regulators of the DNA damage response.
Figures and Tables
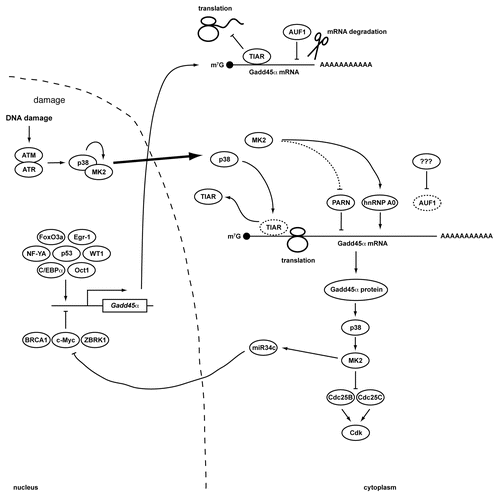
Figure 1 Transcriptional and post-transcriptional regulation of Gadd45α. A simplified schematic overview of regulatory mechanisms that control the expression of Gadd45α. In resting cells, the expression of Gadd45α is transcriptionally suppressed through c-Myc and a repressive complex consisting of ZBRK1 and BRCA1. In addition, translation of existing Gadd45α transcripts is suppressed by TIAR, while AUF1 binding to Gadd45α mRNA targets it for degradation. In response to DNA damage, both Gadd45α transcription and translation are upregulated. The Gadd45α transcriptional inhibitor c-Myc is translationally repressed through miR-34c. While miR-34c can be induced by p53 following genotoxic stress, miR-34c expression in p53-deficient cells depends on the p38/MK2 signaling complex.Citation40 Gadd45α transcription is induced by a variety of damage- and stress-activated transcription factors including p53, WT1, Oct1, NF-YA, FoxO3a, Egr-1 and C/EBPα. Furthermore, both TIAR and AUF1 dissociate from the Gadd45α mRNA after genotoxic stress. The disruption of the TIAR:Gadd45α mRNA RNP depends on p38-mediated TIAR phosphorylation. Gadd45α mRNA degradation is prevented by MK2-dependent Ser-557 phosphorylation of PARN, while Gadd45α mRNA is actively stabilized by binding to the MK2-dependent phospho-Ser-84 form of hnRNP A0. The resulting accumulation of Gadd45α protein functions within a positive feedback loop that maintains p38/MK2 activity at late times following DNA damage. Prolonged MK2 activity in turn is required to maintain Cdc25B and C in an inactive state sequestered in the cytoplasm.
Acknowledgements
We apologize to our colleagues for the omission of many important contributions to the field, and their references, due to space limitations. We thank the members of the Yaffe and Reinhardt laboratories for helpful discussions. This work was supported by the National Institutes of Health (GM68762, CA112967, ES015339 to M.B.Y.), the Deutsche Forschungsgemeinschaft (RE2246/1-1, RE2246/2-1 and SFB-832 to H.C.R.), the Deutsche Nierenstiftung (to H.C.R.), the Austrian Science Fund (FWF, Erwin-Schroedinger-Fellowship to S.M.), the Anna Fuller fund of New Haven (to I.G.C.) and the David H. Koch Fund (H.C.R. and M.B.Y.).
References
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 1078
- Harper JW, Elledge SJ. The DNA damage response: Ten years after. Mol Cell 2007; 28:739 - 745
- Rieger KE, Chu G. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res 2004; 32:4786 - 4803
- Gasch AP, Huang M, Metzner S, Botstein D, Elledge SJ, Brown PO. Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol Biol Cell 2001; 12:2987 - 3003
- Rockx DA, Mason R, van Hoffen A, Barton MC, Citterio E, Bregman DB, et al. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci USA 2000; 97:10503 - 10508
- Vichi P, Coin F, Renaud JP, Vermeulen W, Hoeijmakers JH, Moras D, et al. Cisplatin- and UV-damaged DNA lure the basal transcription factor TFIID/TBP. EMBO J 1997; 16:7444 - 7456
- Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Posttranscriptional gene regulation by RNA-binding proteins during oxidative stress: implications for cellular senescence. Biol Chem 2008; 389:243 - 255
- Lunde BM, Moore C, Varani G. RNA-binding proteins: Modular design for efficient function. Nat Rev Mol Cell Biol 2007; 8:479 - 490
- Lafarga V, Cuadrado A, Lopez de Silanes I, Bengoechea R, Fernandez-Capetillo O, Nebreda AR. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol Cell Biol 2009; 29:4341 - 4351
- Mazan-Mamczarz K, Galban S, Lopez de Silanes I, Martindale JL, Atasoy U, Keene JD, et al. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc Natl Acad Sci USA 2003; 100:8354 - 8359
- Wang W, Furneaux H, Cheng H, Caldwell MC, Hutter D, Liu Y, et al. HuR regulates p21 mRNA stabilization by UV light. Mol Cell Biol 2000; 20:760 - 769
- Wilker EW, van Vugt MA, Artim SA, Huang PH, Petersen CP, Reinhardt HC, et al. 14-3-3sigma controls mitotic translation to facilitate cytokinesis. Nature 2007; 446:329 - 332
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160 - 1166
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 2009; 35:228 - 239
- Fan J, Yang X, Wang W, Wood WH 3rd, Becker KG, Gorospe M. Global analysis of stress-regulated mRNA turnover by using cDNA arrays. Proc Natl Acad Sci USA 2002; 99:10611 - 10616
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, et al. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001; 411:102 - 107
- Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, Yaffe MB. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol Cell 2005; 17:37 - 48
- Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007; 11:175 - 189
- Hirose Y, Katayama M, Berger MS, Pieper RO. Cooperative function of Chk1 and p38 pathways in activating G2 arrest following exposure to temozolomide. J Neurosurg 2004; 100:1060 - 1065
- Hirose Y, Katayama M, Stokoe D, Haas-Kogan DA, Berger MS, Pieper RO. The p38 mitogen-activated protein kinase pathway links the DNA mismatch repair system to the G2 checkpoint and to resistance to chemotherapeutic DNA-methylating agents. Mol Cell Biol 2003; 23:8306 - 8315
- Raman M, Earnest S, Zhang K, Zhao Y, Cobb MH. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J 2007; 26:2005 - 2014
- Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2 and MK2. Curr Opin Cell Biol 2009; 21:245 - 255
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MATM, Wang X, et al. DNA Damage Activates a Spatially Distinct Late Cytoplasmic Cell-Cycle Checkpoint Network Controlled by MK2-Mediated RNA Stabilization. Molecular Cell 2010; 40:34 - 49
- Hildesheim J, Bulavin DV, Anver MR, Alvord WG, Hollander MC, Vardanian L, et al. Gadd45α protects against UV irradiation-induced skin tumors and promotes apoptosis and stress signaling via MAPK and p53. Cancer Res 2002; 62:7305 - 7315
- Wang XW, Zhan Q, Coursen JD, Khan MA, Kontny HU, Yu L, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci USA 1999; 96:3706 - 3711
- Jin S, Fan F, Fan W, Zhao H, Tong T, Blanck P, et al. Transcription factors Oct-1 and NF-YA regulate the p53-independent induction of the GADD45 following DNA damage. Oncogene 2001; 20:2683 - 2690
- Zhan Q, Chen IT, Antinore MJ, Fornace AJ Jr. Tumor suppressor p53 can participate in transcriptional induction of the GADD45 promoter in the absence of direct DNA binding. Mol Cell Biol 1998; 18:2768 - 2778
- Hirose T, Sowa Y, Takahashi S, Saito S, Yasuda C, Shindo N, et al. p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 2003; 22:7762 - 7773
- Takahashi S, Saito S, Ohtani N, Sakai T. Involvement of the Oct-1 regulatory element of the gadd45 promoter in the p53-independent response to ultraviolet irradiation. Cancer Res 2001; 61:1187 - 1195
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, et al. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 2002; 296:530 - 534
- Constance CM, Morgan JIt, Umek RM. C/EBPalpha regulation of the growth-arrest-associated gene gadd45. Mol Cell Biol 1996; 16:3878 - 3883
- Thyss R, Virolle V, Imbert V, Peyron JF, Aberdam D, Virolle T. NFkappaB/Egr-1/Gadd45 are sequentially activated upon UVB irradiation to mediate epidermal cell death. EMBO J 2005; 24:128 - 137
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009; 23:1895 - 1909
- Jackman J, Alamo I Jr, Fornace AJ Jr. Genotoxic stress confers preferential and coordinate messenger RNA stability on the five gadd genes. Cancer Res 1994; 54:5656 - 5662
- Lal A, Abdelmohsen K, Pullmann R, Kawai T, Galban S, Yang X, et al. Posttranscriptional derepression of GADD45alpha by genotoxic stress. Mol Cell 2006; 22:117 - 128
- Rousseau S, Morrice N, Peggie M, Campbell DG, Gaestel M, Cohen P. Inhibition of SAPK2a/p38 prevents hnRNP A0 phosphorylation by MAPKAP-K2 and its interaction with cytokine mRNAs. EMBO J 2002; 21:6505 - 6514
- Lopez-Aviles S, Grande M, Gonzalez M, Helgesen AL, Alemany V, Sanchez-Piris M, et al. Inactivation of the Cdc25 phosphatase by the stress-activated Srk1 kinase in fission yeast. Mol Cell 2005; 17:49 - 59
- Abdelmohsen K, Pullmann R Jr, Lal A, Kim HH, Galban S, Yang X, et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol Cell 2007; 25:543 - 557
- Keene JD, Tenenbaum SA. Eukaryotic mRNPs may represent posttranscriptional operons. Mol Cell 2002; 9:1161 - 1167
- Cannell IG, Kong YW, Johnston SJ, Chen ML, Collins HM, Dobbyn HC, et al. p38 MAPK/MK2-mediated induction of miR-34c following DNA damage prevents Myc-dependent DNA replication. Proc Natl Acad Sci USA 107:5375 - 5380