 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Reduced fecundity has been associated with some alleles that enhance longevity in invertebrate and mammalian models. This observation has been suggested to support the antagonistic pleiotropy theory of aging, which predicts that alleles of some genes promoting fitness early in life have detrimental effects later in life that limit survival. In only a few cases, however, has the relative fitness of long-lived mutants been quantified through direct competition with the wild type genotype. Here we report the first comprehensive analysis of longevity/fitness trade-offs by measuring the relative fitness of 49 long-lived yeast variants in a direct competition assay with wild type cells. We find that 32 (65%) of these variants show a significant defect in fitness in this competition assay. In 26 (81%) of these cases, this reduction in fitness can be partially accounted for by reduced maximal growth rate during early life, usually resulting from a G0/G1-specific cell cycle defect. A majority of the less fit longevity-enhancing variants are associated with reduced mRNA translation. These findings are therefore consistent with the idea that enhanced longevity often comes with a fitness cost and suggest that this cost is often associated with variation in a subset of longevity factors, such as those regulating mRNA translation, growth, and reproduction.
Introduction
The antagonistic pleiotropy model of aging posits that aging results from the action of genetic variants that are beneficial early in life, but detrimental later.Citation1 Natural selection favors those variants that promote survival to reproductive age or enhance reproductive output, but the effect of these same variants on post-reproductive survival is relatively unimportant, since the force of natural selection is no longer significant late in life. Evidence consistent with this model has been provided by the observation that many mutations associated with increased life span also cause a reduction in fecundity. Several examples of this “trade-off” have been noted in different species, including bacteria,Citation2 nematodes,Citation3–Citation5 fliesCitation6,Citation7 and mice.Citation8 In particular, a number of fruit fly experiments were designed to increase lifespan through artificial selection and mutants which resulted from this selection were also shown to have reduced rates of larval development or egg laying.Citation9–Citation13 This relationship is not universal, however, since other longevity enhancing alleles have been isolated that do not significantly reduce fecundity.Citation14,Citation15 Reproduction under optimal conditions is not the only measure of fitness that determines evolutionary success, however and it may be that longevity-associated variants without fecundity defects reduce evolutionary fitness in other ways.
One way to quantify fitness is to allow different genetic variants to compete against each other under conditions that mimic the natural environment. A few experiments along these lines have been performed in invertebrate organisms to test the prediction that mutations causing slower aging also reduce fitness. For example, Jenkins et al. showed that C. elegans mutants with decreased function of the insulin-like-receptor DAF-2 resulted in decreased fitness when co-cultured with wild type animals.Citation16 There are also cases where long-lived mutants only have reduced fitness under certain conditions. For example, Walker et al. examined the relative fitness of long-lived age-1(hx546) mutant nematodes, which also have reduced insulin-like signaling, and found that under standard laboratory conditions age-1(hx546) mutant animals were not outcompeted by wild type animals.Citation17 However, when a cyclic period of starvation was introduced into the experimental design, the wild type animals outcompeted the age-1(hx546) mutants, although the mechanistic basis for this was not determined. A similar observation was made using long-lived Indy mutant fruit flies which only showed reduced fitness when food was calorie poor,Citation18 although it should be noted that the longevity and fitness phenotypes of these flies is likely not due to mutation at the Indy locus.Citation19 Despite the plethora of longevity alleles known in biology, very few additional studies have directly addressed the prevalence or mechanistic basis of fitness trade-offs among long-lived variants via direct competition assays.
The budding yeast Saccharomyces cerevisiae is an important model system for aging-related research, complementing the other invertebrate and mammalian model organisms.Citation20 The replicative life span (RLS) of a yeast mother cell is defined as the number of daughter cells produced prior to senescence, and can be measured by physical separation of the daughter cells away from the mother cell. Many different cellular processes have been implicated in RLS determination, including ribosomal DNA stability, nutrient response signaling, mitochondrial function, oxidative stress resistance and mRNA translation.Citation20,Citation21 As part of an ongoing genome-wide analysis of yeast aging, we have reported more than 50 genetic variations that cause increased RLS in one strain background.Citation22–Citation28 Each of these variants is a single-gene deletion mutant contained in the yeast ORF deletion collection, with one exception: overexpression of the Sir2 protein deacetylase.Citation29–Citation31 This collection of long-lived deletion mutants provides a unique opportunity to systematically test the prediction that genetic variants associated with longevity result in reduced fitness.
Here we describe the first large scale examination of the fitness costs associated with enhanced longevity by directly quantifying the relative fitness of 49 replicatively long-lived yeast variants in direct competition assays with the parental wild type strain. While our competition assay does not account for every challenge experienced by yeast in nature, such as the need for sexual reproduction and sporulation, the fitness assay nonetheless tests the strains under common conditions of rapid growth followed by periods of starvation.Citation32 Of the 49 long-lived mutants, more than half were found to be significantly less fit than the wild type strain. In most cases, the longevity-associated fitness defect could be attributed to an elongated cell division cycle under optimal growth conditions, a measure of fecundity. Interestingly, this fecundity defect could largely be attributed to a delay in the G0/G1 phase of the cell cycle, further defining the molecular nature of the trade-off between longevity and fitness in yeast.
Results
Increased replicative life span is associated with reduced fitness in yeast.
As an initial attempt to explore whether enhanced RLS is associated with decreased fitness, we competed 49 long-lived mutant strains against wild type cells in a fitness assay designed to mimic three cycles of growth and starvation (see Materials and Methods). Each cycle lasted for one week and was accomplished by growing mixed cultures containing wild type and one long-lived mutant through exponential phase into stationary phase in rich media. Roughly equal numbers of wild type and mutant cells were inoculated into the mixed culture at the initial time point, and relative viable cell number in each culture was quantified at the initial time point and weekly thereafter for three iterations. The strains chosen for this study were comprised from 52 previously reported long-lived yeast strains in the BY4742 genetic background,Citation22–Citation28 51 of which are single-gene deletion mutants plus one overexpression strain (SIR2-OX) (). Three strains were excluded from the study because our flow cytometric analysis indicated that these strains from our MATα haploid ORF deletion collection were, in fact, diploid (described in Materials and Methods).
We used a measure of relative fitness (RF) defined as the log base 2 ratio of mutant to wild type relative to the initial ratio, such that RF = 0 indicates no change in the ratio of mutant to wild type, an RF = 1 corresponds to twice as many mutant cells as wild type cells relative to the initial ratio, while an RF = −1 corresponds to twice as many wild type cells as mutant cells. Those strains for which no mutant cells were counted (ones which were completely outcompeted by wild type) at a non-zero time point were assigned an RF value of −7.
As an additional control, we compared the RF of wild type cells (BY4742) to a second ‘wild type’ BY4742 strain engineered with a single additional selectable marker at its endogenous locus (LYS2). The LYS2 selectable marker allows for quantitation of the ratio of cells at each time point (see Materials and Methods). As expected, there was no significant difference in RF at the end of this control competition experiment between BY4742 and the LYS2 strain (RF = −0.11 ± 0.12), demonstrating relatively low intrinsic variance in the assay. Following three weeks of competition, 32 out of 49 long-lived strains showed a significant fitness defect compared to the LYS2 control ().
In order to determine whether the RF of long-lived mutants differs from randomly selected deletion strains, we also determined RF for 20 randomly selected single-gene deletion mutants using identical assay conditions (Sup. Fig. 2b and Sup. Table 1). Based on a rank-sum comparison of final RF values, the set of long-lived mutants had significantly greater fitness defects than the randomly selected strains (p = 8.41E-5). The difference in magnitude of the defect can also be seen from the fact only that 2/20 (10%) randomly selected deletion strains have an RF <−3, whereas, 25/49 (51%) in the long lived set reach an RF <−3. In addition, a g-test shows these data are from a sufficiently large sample size to show enrichment for competitive defect (p = 7.12E-4).
The findings from the competitive fitness analysis indicate that single gene deletions causing enhanced RLS in yeast result, on average, in a more severe fitness defect than randomly selected gene deletions. The distribution of RF values for the two sets of gene deletions is noteworthy, however. The randomly selected gene deletions show a distribution of RF values that is normal (By Anderson-Darling test, p = 0.223), while the long-lived mutants show a non-normal distribution of RF values that appears to be bimodal (p = 3.17E-5, ). This distribution is suggestive of two types of long-lived mutants: one set that has a fitness profile similar to randomly selected deletion mutants and a second set that has a severe fitness defect.
Replicative longevity is associated with reduced maximal growth rate.
A common trait associated with longevity enhancing mutations or interventions in higher eukaryotes is a reduced rate of reproduction or reduced maximal reproductive potential (reviewed in ref. Citation33 and Citation34). This observation has been interpreted to suggest that there is a trade-off between reproduction and somatic maintenance.Citation35 Since RLS is defined as the number of daughter cells an individual mother cell is capable of producing under optimal growth conditions, the doubling time of the cell is a measure of reproductive fitness under identical conditions. Variants with a shorter doubling time (faster growth rate) will produce more genetically identical progeny in a given period of time, relative to slower growing variants.
To assess whether slower growth rate is associated with reduced fitness in long-lived yeast strains, the doubling times of all 49 long-lived mutants were quantified using a Bioscreen C MBR machine (see Materials and Methods). Strikingly, 26 out of 32 (81%) of the long-lived mutants with significant fitness defects also exhibited a significant (p < 0.05) increase in doubling time relative to the parental wild-type strain ( and ). Among all of the long-lived strains tested, 28 out of 49 (57%) were slow growing. For comparison, only 14 out of 50 (28%) randomly selected single gene deletion strains showed a significant growth rate defect, and most defects were much lower in magnitude. These data indicate that reduced maximal growth rate, analogous to reduced fecundity of multicellular eukaryotes, is more common in long-lived variants compared to random deletion mutants and likely contributed to reduced fitness among a subset of these variants. Not all long-lived yeast strains are slow growing, however, nor are all slow-growing yeast strains long-lived. Thus, slow growth is neither necessary nor sufficient for enhanced replicative longevity.
Reduced maximal growth rate explains a majority of the observed fitness defects in long-lived mutants.
In order to determine whether growth rate is sufficient to account for the observed RF defects of long-lived mutants, we performed in silico simulations of the competition assay in which the cell ratio of mutant to wild type was calculated as a function of the number of cell division cycles, taking into account maximal growth rate and RLS of each cell type. The cell count for wild type and mutant at each cell division round can be estimated by the following two formulas:
and
where Cwt(d) and Cmut(d) are the absolute cell counts for wild type and mutant at each wild type cell division (d), RLSwt and RLSmut are the experimentally determined mean RLS values for wild type and mutant, and Δwt and Δmut are the experimentally determined doubling times (maximal growth rates) for wild type and mutant.
An initial characterization of the global behavior of this simulation demonstrates that growth rate is a much more substantial determinant of fitness than is RLS in populations of dividing cells ( and B). We estimate that in our competitive fitness assay, the initial cells in each outgrowth phase are able to complete approximately 13 cell divisions prior to reaching saturation. Since three periods of growth and quiescence were used in the competitive fitness assay, we estimate a total of approximately 40 cell division cycles occurred during the course of each experiment. Even if we assume that a mutant strain has a wild type RLS of 52, which is approximately double that of the wild type strain, the effect on RF after 40 divisions is negligible as long as maximal growth rate of the mutant and wild type strains are equal [RLSmut = 52, RLSwt = 26, (Δwt = 90, Δmut = 90)]:
Indeed, based on this simulation, the effect of life span extension will never have a significant positive effect on RF of the mutant over the first 40 population doublings, and a reduced life span will only result in an RF <−3 when RLS of the mutant falls below 3, an approximately 90% reduction in mean RLS.
In contrast to the negligible effect of RLS on RF, if the maximal growth rate of the mutant strain is 10 minutes slower than the wild type [RLSmut = 26, RLSwt = 26, (Δwt = 90, Δmut = 100)], our simulation predicts that it would result in a significant decrease in RF over the course of the competitive fitness assay:
This simulation strongly supports the assertion that maximal growth rate, rather than replicative potential, is an appropriate measure of fecundity in this system, since replicative potential is essentially free from the force of natural selection.
We next compared the predicted RF values from our in silico simulation with the experimentally observed RF values for two mutants with a significant reduction in maximal growth rate. For both sch9Δ and dbp3Δ, the RF values predicted by our algorithm closely parallel the actual RF values observed in the competitive fitness assay, based solely on the experimentally observed doubling times and RLS values ( and Sup. Fig. 1). These data further support the idea that, at least for a subset of long-lived mutants, the observed fitness defect can be primarily attributed to reduced fecundity.
Reduced maximal growth rate is most apparent early in the replicative lifespan.
An important aspect of the antagonistic pleiotropy theory is that natural selection maximizes vigor early in life at the expense of vigor late in life.Citation1 A logical extension of this prediction is that life span extending alleles may reduce vigor specifically during early life stages. To test this possibility, we examined the relative growth rate of mother cells over the course of their RLS. This was accomplished by micromanipulation of daughter cells away from mother cells and quantification of the number of daughter cells removed from each mother cell at each time-point during a standard RLS assay. Relative to wild type mother cells, the 32 long-lived mutants for which a competitive defect was observed show a significant decrease in the number of daughter cells produced during the first time-point, which corresponds to the first 1–2 budding cycles of the cell life span (). This defect in vigor is also observed, although reduced in magnitude, at the second time-point of the life span experiment, corresponding to budding cycles 2–4 of the mother cell life span. By the third time-point, there is no significant difference between wild type and long-lived mutants. This early life loss of vigor in some long-lived mutants is further illustrated by quantifying the cumulative number of daughter cells produced as a function of time over the entire life course for dbp3Δ, rpl31aΔ and sch9Δ mother cells, which have a substantial reduction in competitive fitness, relative to wild type cells (). Importantly, these long-lived mutants show improved vigor relative to wild type cells late in life. These data suggest that the reduced vigor associated with life span extension in these mutants is specific for the early stage of life and that late life vigor is enhanced, consistent with the antagonistic pleiotropy theory of aging.
Long-lived, slow growing mutants tend to have a G0/G1-specific cell cycle delay.
We further explored the nature of the growth defect observed for the slow-growing, long-lived variants by examining their cell cycle profiles by flow cytometry ( and Sup. Fig. 1). Interestingly, the growth defects associated with extended life span could be attributed almost exclusively to a delay in the G0/G1 phase of the cell cycle (). Many long-lived variants in yeast are known to have reduced levels of global mRNA translation,Citation36 which can result in an elongated G0/G1 phase of the cell cycle due to delayed accumulation of G1 cyclins.Citation37–Citation39 Consistent with this idea, all of the slow-growing translation-related mutants examined, with the exception of tor1Δ, showed a significant enrichment in the percentage of cells in the G0/G1 phase of the cell cycle ( and ). These long-lived variants also showed evidence of reduced cell size, as assayed by forward scatter in the flow cytometer (Sup. Fig. 3), although this may be attributed in part to a greater percentage of smaller G0/G1 cells in the population.
While G0/G1 delay could account for slow growth in a majority of long-lived variants, three mutants showed abnormal cell cycle profiles without a substantial enrichment in G0/G1 cells (Sup. Fig. 1). Only the long-lived ELP4 deletion strain has an increase in S-phase cells, consistent with an S-phase delay. Elp4 is subunit of the Elongator complex, a histone acetyltransferase component of the RNA polymerase II holoenzyme. Cells lacking ALG12 or SPT4 were enriched for G2 phase cells. Like Elp4, Spt4 also functions in transcriptional elongation by RNA polymerase II; however, spt4Δ cells are known to also have a high rate of chromosome missegregation, which could account for the observed polyploidy and the G2 enrichment. Alg12 adds a mannose group to asparagine residues in the endoplasmic reticulum, and its role in the cell cycle is unclear. Two other mutants (pmr1Δ, sps1Δ) had increased doubling times, but had no specific cell cycle defect that we could detect.
Among the randomly selected deletion strains, bud22Δ showed a strong growth defect that was accompanied by a significant increase in G0/G1 cells (). Deletion of BUD22 failed to increase RLS, however, demonstrating that not all mutants with elongated G0/G1 are long-lived (). Similar to bud22Δ cells, deletion of the small ribosomal subunit protein RPS0B also leads to an increase in G0/G1 cells but does not increase RLS (). This data is consistent with our prior report that depletion of the large ribosomal subunit, but not the small ribosomal subunit, is associated with increased RLS by a mechanism that is partially dependent on the stress- and nutrient-responsive transcription factor Gcn4.Citation25 Among translation-related longevity mutants, there is a trend toward correlation of enrichment for G0/G1 cells with the magnitude of replicative lifespan extension (). This trend is not strong, however, suggesting that the magnitude of the translation defect (and hence G0/G1 delay) is not the sole factor determining longevity.
Discussion
In this study, we have shown that over half the known long-lived variants in the BY4742 background have a significant fitness defect relative to the parental strain. In other words, the wild type allele of each of these genes functions to promote fitness at the expense of longevity. This represents, by far, the most extensive analysis of the relationship between fitness and longevity in any model system to date and provides experimental data consistent with antagonistic pleiotropy. Among the long-lived variants with detectable fitness defects, a majority contain mutations that impair global mRNA translation. The overwhelming majority of these variants show a similar defect in maximal growth rate, a reduction in cell size and a specific delay in cell cycle progression resulting in an accumulation of G0/G1 phase cells.
We have suggested here that maximal growth rate is an appropriate measure of fecundity for the replicative aging model in Saccharomyces cerevisiae. The logic behind this assertion is that in a mixed population of asymmetrically dividing cells, the primary determinant of relative fitness is growth rate, not replicative potential. To demonstrate this, in silico simulations were used to estimate the relative importance of RLS and maximal growth rate in a competitive fitness assay based solely on exponential growth. While this simulation does not reflect the importance of survival during quiescence, it does adequately recapitulate the experimentally observed RF values for slow growing mutants, suggesting that, at least in these cases, maximal growth rate is the primary determinant of fitness. Importantly, extended RLS plays essentially no role in determining fitness, even when the simulation is carried out to more than 100,000 population doublings (Sup. Fig. 5). We note that this lack of selection on replicative capacity adequately reflects the reduction in selection on post-reproductive longevity of chronologically aging multicellular organisms. Thus, in a replicative aging paradigm, growth rate is an appropriate measure of fecundity while replicative potential is not.
Our observations suggest that while decreased RF is generally associated with reduced fecundity, this need not always the case. For example, idh1Δ, idh2Δ, sam1Δ, sok1Δ, tma19Δ and ure2Δ have normal doubling times, but were outcompeted by wild type in the fitness assay (). One possibility is that these mutants are outcompeted specifically during the phase of the fitness competition in which cells are not growing exponentially (i.e., entry into or survival during stationary phase). Indeed, idh1Δ and idh2Δ cells are respiratory-deficient and yeast cells rely heavily on respiratory metabolism during the shift from logarithmic growth to stationary phase (Sup. Fig. 1). Such mutants may be akin to the long-lived age-1 mutants in C. elegans, which are outcompeted by wild type animals specifically during starvation periods.Citation17
It is noteworthy that 17 out of 49 (35%) long-lived variants showed no significant effect on fitness. While it may be that our assay was not sufficiently sensitive to detect more subtle defects, at least two additional explanations for this observation exist. First, it may be that these mutants are not less fit than wild type cells and it is possible to slow aging without large fitness costs. Alternatively, it may be that the competition assay employed did not sample a sufficient spectrum of conditions experienced by yeast in the natural environment to detect the fitness defects associated with these mutants. For example, variants defective for sporulation would be substantially less fit in a natural context, but would not necessarily show a fitness defect in the assays used here. Consistent with this idea, 14 out of 49 long-lived strains are annotated to have reduced sporulation efficiency, compared to only 4 out of 50 randomly selected strains. Altered resistance to temperature fluctuation, susceptibility to environmental toxins or reduced ability to utilize alternative carbon sources could also lead to substantially reduced fitness in the natural environment and were not queried here. Further studies will be required to adequately address this question.
In summary, we have identified 32 long-lived yeast mutants with significant defects in fitness through direct competition assays. This is, by far, the most comprehensive analysis of the prevalence of longevity-associated fitness trade-offs in any model system, expanding by roughly an order of magnitude the number of such mutants identified. These less fit, long-living variants tend to be defective for regulating cell size, growth rate and mRNA translation. Taken together, these data support the idea that enhanced life span is often associated with an early life fitness cost and suggest that such costs may be more likely to result from mutations in genes that modulate specific molecular processes related to growth control and mRNA translation.
Materials and Methods
Yeast strains used.
Unless otherwise indicated, all yeast strains were derived from the MATα ORF deletion collection and are isogenic to the parental BY4742 strain.Citation30,Citation31 The LYS2 strain was generated by sporulation of diploid cells from a cross between a BY4741 MATα deletion strain and wild type BY4742. The resulting spore clone was verified as MATα his3Δ leu2Δ LYS2 met15Δ ura3Δ. The SIR2-OX strain is a tandem two copy SIR2 strain with a URA3 marker generated by integration of a second copy of SIR2 in BY4742 as previously described in reference Citation29. A few strains (adh1Δ, rom2Δ and rpl20bΔ) were excluded from our analysis upon finding the strains had undergone spontaneous diploidization (Sup. Fig. 4a). A remade rpl20bΔ strain was generated by replacing the RPL20B open reading frame with HIS3 and was verified to have an extended replicative lifespan (Sup. Fig. 4b). All gene disruptions were verified by PCR.
Competition assay.
In an effort to simulate a periodic growth and starvation cycle similar to what yeast might experience in the natural environment and analogous to what has been previously studied in Caenorhabditis elegans,Citation17 we developed a competition assay consisting of inoculating yeast into fresh YPD followed by intermediate periods of starvation. Test strains were first inoculated into 150 µL YPD in individual wells of a 96 well plate and incubated at 30°C for two days. 8 µl of these cultures were then serially diluted in 150 µl YPD twice and then 8 µl of this dilution was inoculated into 150 µL of fresh YPD in a single well of a new 96 well plate. An equal volume of BY4742 was similarly inoculated into the same well, providing a roughly equal number of mutant and wild type cells in each culture. This is defined as the initial time-point for each experiment. Every 7 days the individual cultures were serially diluted in an identical fashion into a well containing fresh YPD in a 96 well plate. The procedure allowed for about 50,000 cells to be re-inoculated into fresh culture, thereby preventing any bottlenecks, and to divide 13 times before the next round of fresh YPD.
Each long-lived mutant is marked with the selectable KanMX marker (except the SIR2-OX strain, which is URA3 marked), allowing convenient quantification of the ratio of mutant to wild type cells in the mixed culture. For each weekly time point, the competition culture was serially diluted four times (8 µl of culture into 150 µl YPD) and then all 158 µl was plated onto YPD agar, yielding 150–300 total colony forming units (CFUs) per plate. After 2 days of growth at 30°C, the total number of CFUs per plate was counted, and then replica plated onto YPD supplemented with G418 to select for the KanMX marker (or SD-LYS media for the LYS2 strain, or SD-URA media for the SIR2-OX strain). The number of CFUs on each selective media plate was then counted after 1 day of incubation. The ratio of mutant to wild type cells in the mixed culture can then be defined as the number of CFUs grown on selective media divided by the number of CFUs unable to grow on selective media (calculated by CFUWT = CFUYPD − CFUselective media).
Relative fitness was calculated using the following equation: Relative fitness = log2[(CFUxmutant/CFUxWT)/(CFU°mutant/CFU°WT)]
Where CFUx is the CFU count at the end of week x and CFU° is the CFU count at initial inoculation (day zero). This allowed us to normalize for changes between cultures in the mutant/wild type ratio depending on initial inoculation. While most cultures were inoculated near the 50/50 ratio, we confirmed that even intentionally large changes in the starting ratio did not significantly change the results of the assay (Sup. Fig. 2a). Statistical significance was determined by a two-tailed Student's t-test at the end of week three, comparing relative fitness of long lived mutant cultures to the LYS2 control competition cultures for a minimum of triplicate biological replicates for each strain.
Growth rate analysis.
Yeast growth rates were analyzed with a Bioscreen C MBR machine (Growth Curves USA) as previously described using the Yeast Outgrowth Data Analyzer (YODA).Citation40 Reported doubling times in 30°C YPD are taken from interval readings from the OD420–580 = 0.2–0.6 range of maximum growth rate. Significance was tested with a two-tailed Student's t-test of mutant doubling times compared to wild type doubling times. Error bars are shown as standard error of the mean (SEM). Each experiment was performed in biological triplicates per run (three cultures, three Bioscreen C Honeycomb Plate wells) and the runs were performed on at least three biological replicate cultures (independently grown from different colonies on different days) for each mutant ().
Flow cytometry.
Samples were prepared following a protocol modified from that previously described in reference Citation41. The modifications were as follows: no pepsin digestion was used and SYTOX® Green (Invitrogen, S7020) was utilized at a 2 µM final concentration. All yeast strains were inoculated into fresh YPD from an overnight culture with an initial OD540 <0.05. Cells were allowed to grow logarithmically for at least two cell cycles and were harvested with a final OD600 between 0.4 and 0.6 in 70% ethanol. All strains shown, unless otherwise indicated, were grown, harvested, processed, stained and analyzed for at least three biological replicates from independent cultures grown and analyzed on different days. The flow cytometer used was a BD Biosciences Influx Cell Sorter. The relative percentages of cells in G1, S and G2 phase were calculated using WinCycle. Forward scatter was analyzed in FCS Express and mean values were used to compare relative cell size.
Replicative lifespan.
Replicative lifespan assays were performed as previously described in references 23 and 42. 2% glucose YPD plates were used, and statistical significance was determined by a Wilcoxon Rank-Sum test (MATLAB “rank-sum” function). RLS values shown in are derived from prior studies.Citation22–Citation28 Growth curves from replicative lifespans were constructed by averaging across all mother cells the sum of daughter cells that had budded before each time point during the lifespan assay ().
Figures and Tables
Figure 1 Direct competition reveals a fitness defect in 32 long-lived yeast strains. (A) Representative example of colony forming units (CFUs) from a fitness competition. Cells were plated from a mixed culture of control (BY4742) and a mutant (rpp2bΔ is a long lived mutant example, LYS2 is a wild type mimetic control example) onto YPD medium to ascertain a total CFU count and then replica plated onto selective media to gather a mutant CFU count (SD-LYS top part, YPD + G418 bottom part). (B) Relative fitness of LYS2 control and rpp2bΔ compared to wild type (BY4742) during the competition experiment. (C) Histogram showing the observed relative fitness values at the end of the assay for long-lived deletion strains and randomly selected deletion strains.
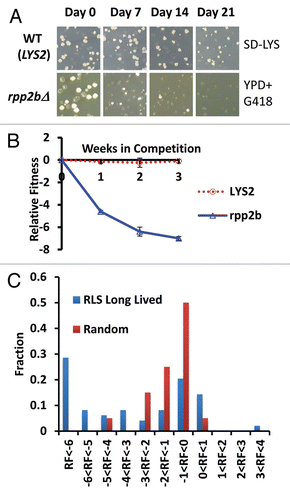
Figure 2 Long-lived yeast are enriched for slow growth. (A) Percent increase in maximal doubling time relative to wild type cells for 50 randomly selected deletion strains and 49 long-lived strains. Each bar represents one strain. (B) Comparison of the long lived mutants' doubling time to the magnitude of their respective relative fitness defects. Strains with a fitness defect are shown.
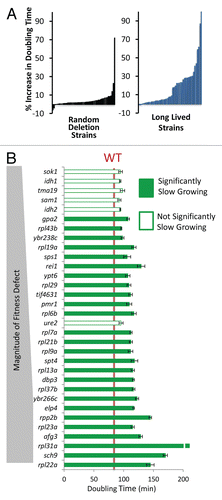
Figure 3 Simulation of the relative importance of growth rate and replicative potential on fitness of yeast cells after 40 wild type cell division cycles. (A) The replicative life span of a mutant has relatively little effect on fitness, except in cases where life span is reduced to less than about 3 generations. Replicative life span for wild type is set at 26 generations. Doubling time for mutant and wild type is defined as equal (90 minutes). (B) Doubling time of the mutant has a relatively large effect on relative fitness. Wild type doubling time is set at 90 minutes. Replicative life span for mutant and wild type is defined as equal (26 generations). (C) Comparison of the in silico predicted relative fitness for dbp3Δ and sch9Δ cells based on experimentally measured replicative life span and doubling time values with the experimentally measured relative fitness of these mutants. Comparisons of the remaining mutants to in silico calculations can be found in Supplemental Figure 1.
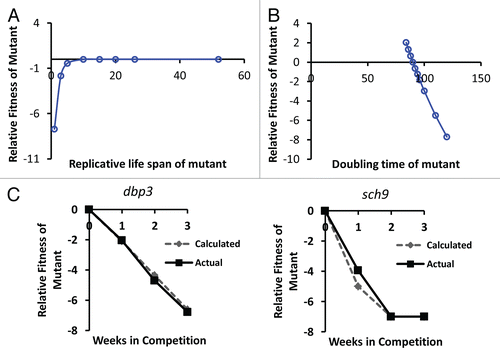
Figure 4 Reduced vitality of long-lived mutants occurs early in life. (A) The median number of daughters produced per mother cell over the first three time-points in a standard replicative life span assay is shown for experimentally-matched wild type (BY4742) and long-lived mutants (pooled data for 32 strains showing a significant defect in relative fitness). Each time point corresponds to removal of daughter cells by microdissection after 90–150 minutes of incubation at 30°C. *p < 0.05, **p < 0.01. (B) Cumulative daughter cell production per mother cell derived from a replicative lifespan experiment. Experimental time points are the same as described in (A).
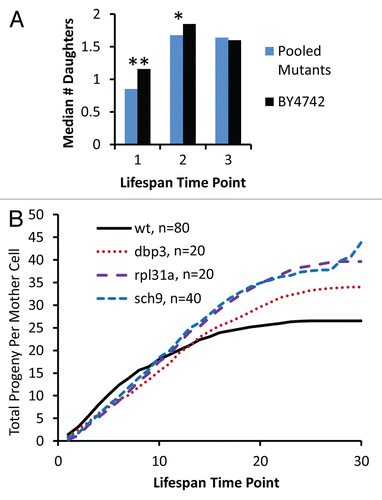
Figure 5 Slow growth is due to a G0/G1 delay in long-lived translation-related mutants. (A) Representative flow cytometry profiles of log phase yeast cells stained with SYTOX Green. Higher values on the x-axis indicate increased fluorescence. (B) Representative growth curves obtained from a Bioscreen C MBR machine. Cells were grown at 30°C in YPD. (C) Long-lived strains with a significant G0/G1 delay. (D) Relationship between G0/G1 delay and growth rate for all 49 long-lived strains.
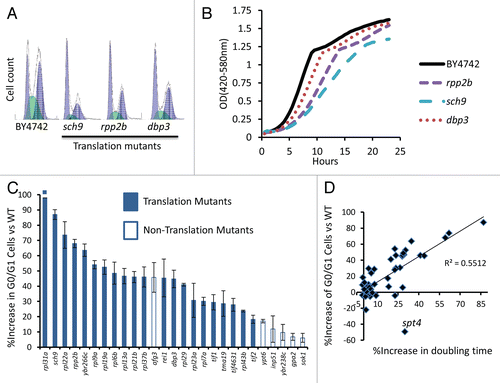
Figure 6 G0/G1 delay is not sufficient to increase lifespan. (A) Flow cytometry profiles bud22Δ and rps0bΔ log phase cells. Cells deleted for BUD22 were found to be the slowest growing of our 50 randomly selected strains while small ribosomal subunits have been previously published as not long lived,Citation25 and rps0b was chosen as a sample mutant. Higher values on the x-axis indicate increased fluorescence. (B) Replicative lifespan data of bud22Δ and rps0bΔ. The number of mother cells is shown in (), mean lifespan in []. (C) Comparison between G0/G1 delay and life span among translation-related long-lived mutants (filled diamonds) and the remaining long-lived mutants with no clear role in mRNA translation (open triangles).
![Figure 6 G0/G1 delay is not sufficient to increase lifespan. (A) Flow cytometry profiles bud22Δ and rps0bΔ log phase cells. Cells deleted for BUD22 were found to be the slowest growing of our 50 randomly selected strains while small ribosomal subunits have been previously published as not long lived,Citation25 and rps0b was chosen as a sample mutant. Higher values on the x-axis indicate increased fluorescence. (B) Replicative lifespan data of bud22Δ and rps0bΔ. The number of mother cells is shown in (), mean lifespan in []. (C) Comparison between G0/G1 delay and life span among translation-related long-lived mutants (filled diamonds) and the remaining long-lived mutants with no clear role in mRNA translation (open triangles).](/cms/asset/52e75f28-a799-4bc6-84c6-38a828febccc/kccy_a_10914457_f0006.gif)
Table 1 Relative fitness, maximal growth rate, and flow cytometry analysis of cell cycle progression for 49 long-lived mutants
Additional material
Download Zip (2.6 MB)Acknowledgements
This work was supported by NIH Grant R01AG025549. Flow Cytometry was performed through the University of Washington Nathan Shock Center of Excellence in the Basic Biology of Aging Imaging Core (NIH Grant P30AG013280). J.D. is supported by NIH Training Grant T32AG000057. M.K. is an Ellison Medical Foundation New Scholar in Aging.
References
- Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution 1957; 11:398 - 411
- Nystrom T. Translational fidelity, protein oxidation and senescence: Lessons from bacteria. Ageing Res Rev 2002; 1:693 - 703
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature 1993; 366:461 - 464
- He KW, Shen LL, Zhou WW, Wang DY. Regulation of aging by unc-13 and sbt-1 in Caenorhabditis elegans is temperature-dependent. Neurosci Bull 2009; 25:335 - 342
- Van Raamsdonk JM, Hekimi S. Deletion of the mitochondrial superoxide dismutase sod-2 extends lifespan in Caenorhabditis elegans. PLoS Genet 2009; 5:1000361
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 2001; 292:104 - 106
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 2001; 292:107 - 110
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature 1996; 384:33
- Rose M, Charlesworth B. A test of evolutionary theories of senescence. Nature 1980; 287:141 - 142
- Partridge L, Prowse N, Pignatelli P. Another set of responses and correlated responses to selection on age at reproduction in Drosophila melanogaster. Proc Biol Sci 1999; 266:255 - 261
- Buck S, Vettraino J, Force AG, Arking R. Extended longevity in Drosophila is consistently associated with a decrease in developmental viability. J Gerontol A Biol Sci Med Sci 2000; 55:292 - 301
- Zwaan BJ, Bijlsma R, Hoekstra RF. Direct selection on lifespan in Drosophila melanogaster. Evolution 1995; 49:649 - 659
- Stearns SC, Ackermann M, Doebeli M, Kaiser M. Experimental evolution of aging, growth and reproduction in fruitflies. Proc Natl Acad Sci USA 2000; 97:3309 - 3313
- Giannakou ME, Goss M, Jacobson J, Vinti G, Leevers SJ, Partridge L. Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 2007; 6:429 - 438
- Liu X, Jiang N, Hughes B, Bigras E, Shoubridge E, Hekimi S. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev 2005; 19:2424 - 2434
- Jenkins NL, McColl G, Lithgow GJ. Fitness cost of extended lifespan in Caenorhabditis elegans. Proc Biol Sci 2004; 271:2523 - 2526
- Walker DW, McColl G, Jenkins NL, Harris J, Lithgow GJ. Evolution of lifespan in C. elegans. Nature 2000; 405:296 - 297
- Marden JH, Rogina B, Montooth KL, Helfand SL. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc Natl Acad Sci USA 2003; 100:3369 - 3373
- Toivonen JM, Gems D, Partridge L. Longevity of Indy mutant Drosophila not attributable to Indy mutation. Proc Natl Acad Sci USA 2009; 106:53
- Kaeberlein M. Lessons on longevity from budding yeast. Nature 2010; 464:513 - 519
- Jazwinski SM. Yeast longevity and aging—the mitochondrial connection. Mech Ageing Dev 2005; 126:243 - 248
- Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol 2004; 2:296
- Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005; 310:1193 - 1196
- Kaeberlein M, Kirkland KT, Fields S, Kennedy BK. Genes determining yeast replicative life span in a long-lived genetic background. Mech Ageing Dev 2005; 126:491 - 504
- Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, et al. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 2008; 133:292 - 302
- Smith ED, Tsuchiya M, Fox LA, Dang N, Hu D, Kerr EO, et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res 2008; 18:564 - 570
- Managbanag JR, Witten TM, Bonchev D, Fox LA, Tsuchiya M, Kennedy BK, et al. Shortest-path network analysis is a useful approach toward identifying genetic determinants of longevity. PLoS One 2008; 3:3802
- Tsuchiya M, Dang N, Kerr EO, Hu D, Steffen KK, Oakes JA, et al. Sirtuin-independent effects of nicotinamide on lifespan extension from calorie restriction in yeast. Aging Cell 2006; 5:505 - 514
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 1999; 13:2570 - 2580
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et al. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 1999; 285:901 - 906
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, et al. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998; 14:115 - 132
- R Mortimer MP. On the origins of wine yeast. Research in microbiology 1999; 150:199 - 204
- Kirkwood TB, Austad SN. Why do we age?. Nature 2000; 408:233 - 238
- Kirkwood TB. Evolution of ageing. Mech Ageing Dev 2002; 123:737 - 745
- Kirkwood TB, Holliday R. The evolution of ageing and longevity. Proc R Soc Lond B Biol Sci 1979; 205:531 - 546
- Kennedy BK, Kaeberlein M. Hot topics in aging research: protein translation 2009. Aging Cell 2009; 8:617 - 623
- Barbet NC, Schneider U, Helliwell SB, Stansfield I, Tuite MF, Hall MN. TOR controls translation initiation and early G1 progression in yeast. Mol Biol Cell 1996; 7:25 - 42
- Tyers M, Tokiwa G, Futcher B. Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. EMBO J 1993; 12:1955 - 1968
- Gallego C, Gari E, Colomina N, Herrero E, Aldea M. The Cln3 cyclin is downregulated by translational repression and degradation during the G1 arrest caused by nitrogen deprivation in budding yeast. EMBO J 1997; 16:7196 - 7206
- Olsen BMC, Kaeberlein M. YODA: Software to facilitate high-throughput analysis of chronological life span, growth rate and survival in budding yeast. BMC Bioinformatics 2010; 11
- Haase SB, Reed SI. Improved flow cytometric analysis of the budding yeast cell cycle. Cell Cycle 2002; 1:132 - 136
- Steffen KK, Kennedy BK, Kaeberlein M. Measuring replicative life span in the budding yeast. J Vis Exp 2009; In press