Abstract
Comment on: Kabacik S, et al. Cell Cycle 2011; 10:1152-61.
Over the past decades it has become increasingly clear that the DNA damage response (DDR) plays a crucial role in the prevention of cancer. Our DNA is extremely vulnerable to genotoxic stress. Genotoxic insults commonly arise from within as well as from without the organism. From within, genotoxic stress can be imposed by toxic byproducts of cellular metabolism, such as reactive oxygen species. From without, genotoxic stress can be imposed by numerous chemicals as well as by irradiation, such as exposure to ionizing radiation in medical practice. The estimated number of DNA lesions is as high as 105 per cell per day.Citation1 To ensure genomic stability, it is critical that an appropriate DNA damage response (DDR) is mounted. The DDR is a complex signaling network and involves detection of the lesion, induction of transient cell cycle arrest to allow time for and activation of repair and the execution of a cell fate decision.Citation2 Different cell fates include resumption of cell cycle progression, induction of irreversible cell cycle arrest (cellular senescence) or induction of cell death (apoptosis). Numerous factors can modulate cell fate decision, including the efficiency of DNA repair and the degree and nature of persistent lesions.
DNA double-strand breaks (DSBs) are genotoxic lesions that can be induced by exposure to ionizing radiation. Activation of the threonine kinase ataxia-telangiectasia mutated (ATM) comprises a first step in the DDR to DSBs. ATM can directly phosphorylate and stabilize the tumor suppressor p53 and indirectly regulate p53 phosphorylation by activating the cell cycle regulator CHEK2.Citation3 Components of the DDR signaling pathway are mutated in several cancers, suggesting that the DDR must be overcome during the process of tumorigenesis. Moreover, previous work has demonstrated that germline mutations in genes encoding components of the DDR are at the basis of several human hereditary cancer-prone syndromes, including mutations in ATM [which are at the basis of ataxia telangiectasia (AT)]Citation4 and mutations in TP53, which are at the basis of Li-Fraumeni syndrome (LFS)Citation5. In the population at large, analysis of genetic variation in the DDR pathway in relation to cancer susceptibility has been a topic of much interest. In case of sporadic cancers, several common polymorphisms in components of the DDR have been associated with enhanced cancer susceptibility. However, cancer susceptibility will be influenced by the combined and often subtle effects of individual genetic variations in the DDR pathway, as well as in interacting pathways. Moreover, epigenetic variations and post-translational modifications will also affect the activity of the DDR pathway. In the April 1 2011 issue of Cell Cycle, Kabacik and colleagues propose to focus on an intermediate phenotype instead of genetic variations in the DDR pathway for individual cancer risk assessment. Kabacik et al. argue that, since most functions of the DDR pathway are attributable to its role as transcriptional activator in response to DNA damage, assessment of ionizing radiation (IR)-induced changes in the transcription of key p53 target genes could provide a simple readout of DDR pathway activity.
The study by Kabacik et al. convincingly shows, using mouse strains with different copy numbers of the DDR pathway components Atm, Trp53 (p53) or Check2 that the IR-induced changes in the transcription of the p53 target genes p21, Puma and Sens2 are strongly dependent on the DDR pathway component copy number. As expected, for the mouse strains with different copy numbers of the Trp53 gene, cancer incidence is also strongly dependent on the p53 copy number. Across the mouse strains with different copy numbers of the DDR pathway components, the most robust associations between gene copy number and IR-induced changes in transcription were observed for the p53 target gene Puma (p53 upregulated modulator of apoptosis), which plays a key role in execution of the cell fate apoptosis. Interestingly, the mean IR induced upregulation of Puma in fresh blood from wild-type mice was comparable to the mean IR induced upregulation of PUMA in fresh blood from healthy humans. Moreover, similarly to the DDR component copy number dependent increase in IR-induced Puma expression that was observed in mice, a linear increase in IR-induced expression of PUMA was observed across mitogen-stimulated T lymphocyte cultures from a human AT case, a group of AT heterozygotes and LFS heterozygotes and a group of healthy donors. Most importantly, in a limited sample of healthy donors, a range of IR induced PUMA upregulation was observed, suggesting that considerable variation of IR-induced upregulation of PUMA is present among healthy humans. Future studies, including large prospective studies on cancer incidence in the population at large should be directed at unraveling the potential functional significance of the observed variation in IR-induced PUMA upregulation to assess the value of the minimally invasive assay for individual assessment of the ATM/CHEK2/p53 pathway activity proposed by Kabacik for individualized prediction of human cancer susceptibility.
About 90% of pancreatic ductal adenocarcinomas (PDACs) harbor oncogenic mutations in the K-ras oncogene, but genetic mouse models demonstrated that this genetic alteration alone cannot drive malignant tumor growth. Additional mutations in the p53 tumor suppressor gene strongly enhance progression towards invasive and metastatic PDACs.Citation1 The role of p53 in controlling crucial cellular processes are well described, and genetic alterations resulting in its loss of function or creating a dominant oncogenic version affect the control of cell cycle progression, apoptosis, senescence and DNA repair.Citation2 However, how p53 mutations are inducing an invasive and metastatic potential, which requires increased cellular motility for tumor cell dissemination as well as stem cell properties for colonization at the metastatic site, is less well understood.
A study by Pinho et al.Citation3 indicates potential underlying mechanisms by linking loss of p53 function to the induction of an epithelial-mesenchymal transition (EMT) and stemness properties. EMT is a key program in embryonic development and is conferred by so-called EMT activators, transcriptional repressors, such as Snail, and ZEB family members, which suppress expression of epithelial genes. Its aberrant activation in cancer cells induces malignant tumor progression, invasion, dissemination and finally metastasis, due to acquisition of an abnormal cellular motility. Recently, EMT was also linked to an activation of stemness properties, thereby conferring a combined stemness and motility phenotype to cancer cells.Citation4 Now, Pinho et al. have shown that if cultivated under stress conditions, pancreatic epithelial cells derived from p53-/- mice, but not those from p16-/- and p21-/- mice, not only display enhanced proliferation, but also show features of stemness, such as increased sphere-forming capacities and expression of pancreatic multipotent progenitor and stemness markers, like Bmi1. Moreover p53-/- cells loose their epithelial differentiation and undergo an EMT, characterized by a high expression of Vimentin and of the EMT inducers Snai1, Snai2, Twist, ZEB1 and ZEB2. These data not only have impact on understanding how normal p53 can control the homeostasis of exocrine pancreatic cells under stress conditions, like in chronic pancreatitis, but also indicate new mechanisms by which mutant p53 might induce malignant tumor progression.
How does p53 control EMT and stemness properties? Another recent publication, describing similar effects of p53 in breast epithelial and breast cancer cells, extends the findings by Pinho et al., showing that p53 activates the transcription of the miR-200c gene.Citation5 Belonging to the miR-200 family, miR-200c was shown to suppress translation of stemnessassociated factors, such as Bmi1.Citation6,Citation7 Moreover, miR-200 is a strong inducer of epithelial differentiation, inhibiting expression of the EMT-inducers ZEB1 and ZEB2.Citation8 In the publication by Pinho et al., exactly these factors, Bmi1, ZEB1 and ZEB2, were shown to be upregulated in p53-/- cells, indicating that miR-200 could also be the transmitter of the respective p53 functions in pancreatic epithelial cells.
The results of both studies investigating different tissues suggest that the identified molecular link might be a general mechanism for p53's control of epithelial differentiation and homeostasis and that mutated p53 triggers invasion and metastasis in epithelial cancers of different origins. They further indicate that p53 is crucial to assure proper re-differentiation of pancreatic epithelial cells under stress conditions, particularly during chronic pancreatitis, and that p53 mutations, if occurring in such conditions, might initiate and drive tumor progression.
References
- Hingorani SR, et al. Cancer Cell 2005; 7:469 - 483
- Vousden KH, et al. Cell 2009; 137:413 - 431
- Pinho AV, et al. Cell Cycle 2011; 10:1312 - 1321
- Thiery JP, et al. Cell 2009; 139:871 - 890
- Chang C-J, et al. Nat Cell Biol 2011; 13:317 - 323
- Shimono Y, et al. Cell 2009; 138:592 - 603
- Wellner U, et al. Nat Cell Biol 2009; 11:1487 - 1495
- Brabletz S, et al. EMBO Rep 2010; 11:670 - 677
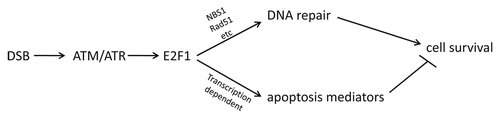
E2F1 is best known as the founding member of the E2F transcription factor family, which (together with retinoblastoma family proteins) regulates the expression of a large number of genes involved in DNA synthesis, cell cycle progression, DNA repair and apoptosis.Citation1,Citation2 E2F1 has also been implicated in responses to DNA damage, and E2F1 is posttranslationally modified and stabilized in response to DNA damage and recruited to DNA strand breaks (DSBs). Previous studies have shown that E2F1 promotes nucleotide excision repair of ultraviolet-induced DNA damage, contributing to enhanced cell survival post-exposure.Citation3,Citation4 A recent report by Chen et al provides mechanistic teeth to previous associations between E2F1 and responses to DSBs by demonstrating a critical and direct role for E2F1 in the recruitment of repair factors to DSBs and subsequent repair, apparently independent of E2F1 roles in transcriptional regulation.Citation5 Mutation or knockdown of E2F1, in both mouse and human cells, is shown to substantially reduce NBS1 phosphorylation, Rad51 accumulation and the formation of DNA damage foci containing Rad51, NBS1 and RPA. Importantly, the repair of DSBs is substantially delayed and reduced in cells with reduced E2F1. The formation of γH2AX foci is actually enhanced, because the resolution of the foci is delayed in E2F1-deficient cells following DNA damage. Thus, E2F1 appears to be required for the repair but not the recognition of DSBs.Citation5 It remains to be determined how E2F1 mediates the recruitment of repair factors to DSBs, and the authors speculate that E2F1-dependent chromatin remodeling could contribute to effective repair.
Thus, in addition to its ability to promote apoptosis in response to DNA damage, E2F1 can also mediate DNA repair, which should promote clonogenic survival (). Whether by promoting the repair or the elimination of cells with DNA damage, E2F1 should contribute to the maintenance of genomic integrity, limiting the propagation of cells with potential DNA mutations. It will be important to better understand how E2F1 contributes to the cellular decision to repair or die following DNA damage and what role E2F1 plays in tissue maintenance and carcinogenesis following genotoxic insults. In some ways, this role for E2F1 is reminiscent of p53's divergent cell fate impacts following DNA damage, where it can either play a pro-survival role, by instituting a temporary cell cycle arrest to allow for repair, or pro-death/senescence roles, preventing the clonogenic maintenance of the cells.Citation6 In addition, p53 may also play a direct role in the repair of DNA strand breaks, independent of its role in regulating transcription.Citation7 Given the known relationship between E2F1 and p53, it will be interesting to tease out how the interplay between these factors can control cell fate decisions, contributing to the “greater good” of the tissue: repair and retain or eliminate from the replicative cell pool.
Figures and Tables
Figure 1 E2F1 can either promote either DNA repair or apoptosis in response to DNA damage, which should exert opposing effects on cell survival.
References
- Johnson DG, et al. Curr Mol Med 2006; 6:731 - 738
- Stevens C, et al. DNA Repair (Amst) 2004; 3:1071 - 1079
- Berton TR, et al. Oncogene 2005; 24:2449 - 2460
- Guo R, et al. J Biol Chem 2010; 285:19308 - 19315
- Chen J, et al. Cell Cycle 2011; 10:1287 - 1294
- Bensaad K, et al. Trends Cell Biol 2007; 17:286 - 291
- Gatz SA, et al. Cell Death Differ 2006; 13:1003 - 1016
The vertebrate pancreas is a complex organ comprised of both exocrine and endocrine compartments. Within these compartments, acinar, ductal and islet cell types appear to be extremely long-lived, with half-lives measured on the order of months and perhaps even years. Under normal conditions, the gradual replacement of endocrine and exocrine cells appears to be accomplished predominantly through the generation of “like-from-like,” involving the proliferation of already differentiated cells.Citation1,Citation2 However, recent studies have also highlighted a dramatic ability of differentiated pancreatic cell types to undergo trans- and de-differentiation following either transcription factor or injury-induced reprogramming. In addition to providing a potential source of replacement β cells in diabetes, understanding the basis for this plasticity may also provide insights into the pathogenesis of common pancreatic diseases, including chronic pancreatitis and pancreatic ductal adenocarcinoma (PDAC).
In particular, pancreatic acinar cells appear to be especially susceptible to reprogramming in response to injury and other stimuli, with a documented ability to generate either endocrine or exocrine derivatives.Citation3,Citation4 Further evidence of acinar cell plasticity is provided by the “ductal” neoplasia initiated following acinar cell-specific activation of oncogenic KrasCitation5 as well as the induction of epithelial-mesenchymal transition (EMT) and adipocyte differentiation following acinar cell-specific c-Myc deletion.Citation6 In two recent manuscripts, including one in the April 15 issue of Cell Cycle, Real and colleagues have significantly advanced our understanding of the molecular events underlying acinar cell plasticity, demonstrating that dedifferentiation of adult murine acinar cells in suspension culture involves resumption of an embryonic pancreatic progenitor-like phenotype.Citation7 This includes upregulated expression of the progenitor markers Pdx1, Ptf1a, Cpa1, Sox9 and HNFb, accompanied by activation of a senescence-like program involving elevated levels of p53, p21 and p16. This in vitro behavior appears to have in vivo correlates, with similar changes in acinar cell gene expression observed in the setting of experimental chronic pancreatitis following either pancreatic duct ligation or cerulean administration in mice.
In their recent report, Real and his group directly assessed the functional role of p53 activation in dedifferentiated acinar cells. The authors convincingly showed that dedifferentiated acinar cells isolated from p53-/- mice exhibit enhanced proliferative activity, reflected by larger sphere formation in primary culture and an apparent capacity for unlimited expansion. In contrast, acinar cells isolated from mice deficient in either p21 or p16 behave in a manner similar to wild type. While wild-type dedifferentiated acinar cells retain expression of the pancreatic progenitor markers listed above, dedifferentiated p53-/- acinar cells lose expression of pancreas-specific transcription factors and activate markers of pre-pancreatic endoderm as well as markers of pluripotency, including Bmi1, Klf4, cMyc and Abcg2. Most remarkably, in the absence of p53, dedifferentiated acinar cells also appear to lose their epithelial identity, reflected by loss of E-cadherin and activated expression of Vimentin, Twist, Snai1, Snai2, ZEB1 and ZEB2. Using genetic lineage tracing experiments involving Ptf1a:Cre; R26-LSL-YFP; p53-/- mice, the authors clearly demonstrate a pancreatic epithelial origin for these mesenchymal-like cells. These results indicate that in the absence of p53, dedifferentiated acinar cells display an enhanced susceptibility to stress-induced reprogramming, losing their pancreatic identity and undergoing an effective epithelial-mesenchymal transition (EMT). In this regard, p53 appears to restrain acinar cell reprogramming, reminiscent of its ability to restrain the reprogramming of somatic cells to induced pluripotent stem (iPS) cells.Citation8
The work obviously poses a number of additional questions, some of which are addressed by the authors. First, what is the mechanism by which inactivation of p53 in pancreatic epithelial cells leads to acquisition of the mesenchymal phenotype? Based on similar work done in human mammary epithelia,Citation9 the authors hypothesize that loss of p53-dependent miR-200c expression activates the EMT program by derepressing ZEB1. Second, it remains to be established whether the phenotypic changes observed in dedifferentiated p53-/- acinar cells are unique to the in vitro context, or if they also occur in vivo, in the setting of p53 mutation. Future studies will be necessary to determine if acinar cells in p53-/- mice undergo similar reprogramming in the context of either chronic pancreatitis or PDAC. Third, to what degree is can loss of epithelial identity in p53-/- acinar cells be reversed? The authors allude to preliminary data suggesting that acinar cell-derived mesenchymal cells have an ability to re-epithelialize when placed within an appropriate 3D matrix; further studies will obviously be necessary to determine the efficiency of this process as well as relevant cell autonomous and non-autonomous regulators of redifferentiation.
While p53's more lofty functions as “guardian of the genome” and as a more general “guardian of reprogramming”Citation8 may represent the primary sources of its ongoing fame, Real and colleagues have further defined p53 as an effective guardian of pancreatic epithelial identity, a role that may play a critical role in the initiation and progression of pancreatic disease.
References
- Desai BM, et al. J Clin Invest 2007; 117:971 - 977
- Dor Y, et al. Nature 2004; 429:41 - 46
- Zhou Q, et al. Nature 2008; 455:627 - 632
- Baeyens L, et al. Biol Chem 2009; 390:995 - 1001
- Guerra C, et al. Cancer Cell 2007; 11:291 - 302
- Bonal C, et al. Gastroenterology 2009; 136:309 - 319 e9
- Pinho AV, et al. Gut 2010; 60:958 - 966
- Menendez S, et al. Cell Cycle 2010; 9:3887 - 3891
- Chang CJ, et al. Nat Cell Biol 2011; 13:317 - 323
- Pinho AV, et al. Cell Cycle 2011; 10:1312 - 1321
The E2F1 transcription factor plays a multifaceted role in cell proliferation and survival and can alternatively behave as tumor suppressor gene or an oncogene, depending on biological context.Citation1 E2F1's role as a tumor suppressor can be explained by its ability to induce apoptosis; however, at least three emerging lines of evidence suggest that E2F1 may have a role in maintaining genomic integrity through direct participation in the DNA damage response. Firstly, E2F1 is regulated by multiple DNA damage-induced signaling cascades. For one example, ATM can phosphorylate E2F1 on Ser 31 in response to ionizing radiation (IR), leading to E2F1 protein stabilization.Citation2 Secondly, recent work has shown that E2F1 deficiency can impair various DNA damage response processes, such as nucleotide excision repair.Citation3 Finally, E2F1 is found physically associated with DNA regions undergoing repair, which is consistent with a direct role in the assembly of DNA repair complexes.Citation3,Citation4
Given these multiple lines of evidence implicating E2F1 in the maintenance of DNA integrity, Chen et al.Citation5 recently asked if E2F1 deficiency would affect genome stability. They utilized the phosphorylation of histone variant H2AX, which occurs in chromatin that flanks a double-stranded break, as an indirect marker of DNA damage. They observed that primary adult fibroblasts derived from mice lacking E2F1 had an increase in the number of cells with spontaneous γH2AX foci as well as a dramatic increase the number cells having more than six foci. Likewise, when the E2F1-deficient cells were challenged with ionizing radiation (IR), there was evidence of significantly increased double-stranded breaks, as measured by single cell gel electrophoresis assay.
Next, Chen et al.Citation5 investigated the mechanism underlying the increase of double-stranded breaks in E2F1-deficient cells. NBS1 is one of the first proteins to be recruited to double-stranded breaks as a component of the MRN (Mre11-Rad50-NBS1) complex. Using indirect immunofluorescence, they demonstrated a profound decrease in the number of NBS1 containing foci in E2F1-deficient cells compared to WT cells following IR. In contrast, ?H2AX foci were increased in E2F1-deficient cells following irradiation. While the total levels of NBS1 protein were not affected by E2F1 deficiency, NBS1 phosphorylation in response to DNA damage was reduced in cells lacking E2F1.
Noting that E2F1 has been observed to be a physical component of foci representing sites of double-stranded breaksCitation3,Citation4 and that E2F can physically interact with NBS1 near origins of replication,Citation6 Chen et al. proposed that E2F1 and NBS1 might physically associate in response to DNA damage. This hypothesis was clearly supported by co-immunoprecipitation experiments following IR.
Double-stranded breaks are accurately repaired by homologous recombination. This process involves formation of single-stranded DNA at the breakage sites, stabilized initially by association with RPA (replication protein A). RPA is later displaced by the Rad51 recombinase in a BRCA2-dependent process. Chen et al. again used immunofluorescence to examine foci formation in WT and E2F1-deficient cells treated with IR. They discovered that E2F1 deficiency reduced the formation of both RPA- and Rad51-containing foci induced by IR. E2F1 deficiency did not affect total RPA protein expression levels, but reduced levels of the Rad51 protein by a post transcriptional mechanism. E2F1-defiency did not affect the formation of 53BP1-containing foci.
At this point there is insufficient evidence to predict how E2F1 influences the recruitment of NBS1, RPA and Rad51 to sites of DNA damage. Given recent work,Citation7 it is interesting to speculate that E2F may recruit chromatin-modifying enzymes, such as GCN5, to double-stranded breaks, thereby facilitating repair. The observation that E2F1 deficiency does not reduce γH2AX foci suggests that E2F does not affect the initial direct binding of the MRN complex and ATM (which phosphorylates H2AX) to the sites of DNA damage. More likely, E2F1 affects subsequent waves of NBS1 recruitment and DNA damage signal amplification. Perhaps E2F1 facilitates the recruitment of NBS1 by MDC1 (mediator of DNA damage checkpoint 1). The observation that E2F1 deficiency did not affect the formation of 53BP1-containing foci suggests that E2F1 does not affect MDC1 recruitment of the ubiquitin ligases, which mark histone H2A as 53BP1-binding sites.Citation8
Although much work remains to explain how E2F1 affects the assembly of DNA repair complexes, these findings clearly highlight the potential importance of E2F1 to the maintenance of genomic integrity. The work also leads to the very important question of whether E2F1 deficiency might contribute to human cancer. Although most clinical studiesCitation9 suggest increases in the activity of E2F1 and other E2Fs in human cancer, there are examples of E2F1 downregulation. It would be very interesting to examine clinical samples in which E2F1 is downregulated to determine whether these tumors would present evidence of increased genomic rearrangements compared to similar tumors expressing E2F1.
References
- Johnson DG, et al. Curr Mol Med 2006; 6:731 - 738
- Lin WC, et al. Genes Dev 2001; 15:1833 - 1844
- Guo R, et al. J Biol Chem 2010; 285:19308 - 19315
- Liu K, et al. Mol Cell Biol 2003; 23:3287 - 3304
- Chen J, et al. Cell Cycle 2011; 10:1287 - 1294
- Maser RS, et al. Mol Cell Biol 2001; 21:6006 - 6016
- Guo R, et al. Nucleic Acids Res 2011; 39:1390 - 1397
- FitzGerald JE, et al. Biochem Soc Trans 2009; 37:897 - 904
- Chen HZ, et al. Nat Rev Cancer 2009; 9:785 - 797