Abstract
Autophagy is a conserved cellular pathway responsible for the sequestration of spent organelles and protein aggregates from the cytoplasm and their delivery into lysosomes for degradation. Autophagy plays an important role in adaptation to starvation, in cell survival, immunity, development and cancer. Recent evidence in mice suggests that autophagic defects in hematopoietic stem cells (HSCs) may be implicated in leukemia. Indeed, mice lacking Atg7 in HSCs develop an atypical myeloproliferation resembling human myelodysplastic syndrome (MDS) progressing to acute myeloid leukemia (AML). Studies suggest that accumulation of damaged mitochondria and reactive oxygen species result in cell death of the majority of progenitor cells and, possibly, concomitant transformation of some surviving ones. Interestingly, bone marrow cells from MDS patients are characterized by mitochondrial abnormalities and increased cell death. A role for autophagy in the transformation to cancer has been proposed in other cancer types. This review focuses on autophagy in human MDS development and progression to AML within the context of the role of mitochondria, apoptosis and reactive oxygen species (ROS) in its pathogenesis.
Autophagy
Autophagy, a lysosomal/vacuolar-mediated catabolic pathway for degradation of cytoplasmic constituents, has approximately 30 identified direct molecular players.Citation1 Autophagy takes place constitutively at basal levels which can be induced to higher levels in response to multiple physiological conditions, including starvation, hormonal imbalance and oxidative stress.
Autophagy is initiated by the Atg1–Atg13 protein complex, which, in mammals, is activated by the absence of signaling from the nutrient-sensing kinase mammalian target of rapamycin (mTOR). Next, the class III phosphoinositide-3-kinases (PI3K) (hVps34 and hVps35)—Beclin 1 (mammalian homolog of Atg6) complexes are key to the nucleation step, which results in the formation of an isolation membrane. Elongation of the isolation membrane is then mediated by two ubiquitin-like conjugation systems; one where Atg7 (E1-like) and Atg10 (an E2-like protein) act to conjugate Atg5 to Atg12, and another in which the Atg5–Atg12 conjugate (which functions as an E3-like protein) acts in concert with Atg7 and Atg3 (an E2-like protein) to conjugate Atg8 to phosphatidylethanolamine in the membrane of the growing autophagosome. Finally, autophagy culminates in the fusion of the autophagosome with lysosomal compartments and the degradation of autophagosomal contents by lysosomal hydrolases.Citation2
Autophagic Processes in Cancer
Autophagy has been shown to play several important roles in cancer.Citation3 Indeed, multiple autophagy genes, including Beclin 1 and UVRAG, have now been characterized as tumor suppressor genes.Citation4 Autophagy is implicated in cancer due to its extensive crosstalk with apoptosis, a programmed cell death pathway that is often impaired during tumorigenesis. Autophagy also has a protective role through controlling oxidative stress as well as the build-up of potentially DNA damaging waste.Citation3,Citation5,Citation6
Myelodysplastic Syndrome and Acute Myeloid Leukemia
MDS comprises a group of anemic disorders of uncertain etiology characterized by abnormal cell morphology in the bone marrow (BM) and peripheral blood cytopenias. Increased caspase-dependent apoptosis, reactive oxygen species (ROS) and mitochondrial damage are observed in BM cells from MDS patients.Citation7 The classification of MDS subtypes and their underlying mechanisms has remained contentious;Citation8 several systems for classifying cases of this disorder have been devised based on morphology and/or prognostic parameters. The World Health Organization (WHO) recognizes seven main MDS subtypes, primarly based on refractory anemias/cytopenias: (1) refractory cytopenias with unilineage dysplasia (RCUD), (2) refractory anemia with ring sideroblasts (RARS), (3) refractory cytopenia with multilineage dysplasia (RCMD), (4 and 5) refractory anemia with excess blasts (RAEB-1 and RAEB-2), (6) MDS-unclassified (MDS-U) and (7) MDS associated with isolated del (5q).Citation9 Although classification issues make prevalence hard to determineCitation8 recent studies have estimated MDS to be more common than previously thought, with a prevalence of 2/1,000 above the age of 65 in the US and Canada.Citation10 MDS has an overall transformation rate of 30% to acute myeloid leukemia (AML).
AML is thought to occur due to the transformation of an immature hematopoietic stem or progenitor cell (HSPC), leading to myeloid leukemia. The dysregulated production and proliferation of myeloid cells in the BM interferes with normal hematopoiesis resulting in a reduction in red blood cell (RBC), platelet and leukocyte numbers. The WHO classifies AML into: (1) AML with characteristic genetic abnormalities (translocations and inversions), (2) AML with multilineage dysplasia (progressed from MDS), (3) therapy-related AML and (4) AML not otherwise categorized. AML is the most common myeloid leukemia, with a prevalence of 3–8 cases per 100,000 which rises to 17–19 per 100,000 in those 65 and older. It is often associated with common mutations and chromosomal abnormalities; as with many cancers a multiple genetic-hit model for its initiation has been proposed.Citation11
Here, we will review the accumulating evidence for an important role of autophagy in MDS and AML. Recent evidence arising from mouse models deficient for autophagy in the hematopoietic system will be reviewed, followed by an assessment of the cellular hallmarks of the human disease with possible links to autophagy. Finally, we will present our in-silico analysis of existing studies on genomic alterations in autophagy genes found in AML cells.
Autophagy, Mitophagy and Stem Cell Maintenance
When autophagy is induced by nutrient deprivation, autophagic substrate selection is thought to be mostly random; on the other hand, in nutrient-rich conditions, cargo-selective autophagy takes place to remove superfluous, damaged organelles and protein aggregates. The selective removal of mitochondria by autophagy (mitophagy) has been particularly well studied.Citation12,Citation13
Some of the pathways leading to mitophagy have now been elucidated.Citation12 In yeast, several of the main autophagy players, such as ATG1, ATG5, ATG6, ATG8, ATG9, ATG12 and ATG13, have been found important for mitophagy.Citation14 Based on knockout models of several core autophagy genes, there is good evidence that mitophagy also utilizes some of the classical autophagic proteins in mammalian cells.
Within three years of the term “autophagy” being introduced by Christian de Duve in 1963,Citation15 autophagosomes were described engulfing mitochondria in human RBCs.Citation16 Yet, it was only recently that, thanks to studies of autophagy gene knockout models, mitophagy was confirmed to be essential for erythroid development.Citation14,Citation17–Citation19 In mammals, erythrocytes mature from HSC-derived early progenitors, primarily in the BM, starting as erythroblasts before losing their nuclei and becoming reticulocytes. Newly formed reticulocytes develop further into erythrocytes in peripheral circulation, where they complete protein synthesis, acquire a biconcave shape and get rid of all their remaining organelles, primarily mitochondria.Citation20 Mitophagy is essential for this developmentally regulated mitochondrial degradation. It has also been found to play an important role in T lymphocytes, in which the regulation of mitochondrial load is essential for their development and survival.Citation21–Citation23
Mice deficient for Atg7 in the hematopoietic system develop severe anemia, lymphopenia and atypical myeloproliferation.Citation13,Citation23 Importantly, these symptoms are all hallmarks of MDS and/or AML. These mice also displayed an impaired self-renewal capacity of their HSCs, as assessed by serial replating of colony-forming unit assays and in vivo reconstitution assays, leading to the conclusion that Atg7 and autophagy are vital to HSC maintenance. Moreover, mice lacking Atg7 in the hematopoietic system, despite the near complete loss of true HSCs, were found to have an expanded Lin−Sca+c-kit+ (LSK) compartment, normally containing hematopoietic stem and progenitor cells. The expanded LSK compartment in the absence of Atg7 showed increased mitochondrial content, higher levels of mitochondrial ROS, DNA damage, an enhanced proliferation rate and higher apoptosis levels. In other words, autophagy-deficient hematopoietic progenitor cells show evidence of mitochondrial damage and ROS build-up along with increased rates of proliferation and apoptosis, which are all frequently observed in human MDS patient samples. Mice lacking autophagy in the hematopoietic system died within 12 weeks and post-mortem analysis of their tissues showed myeloid blast infiltrates in multiple organs including the thymus, liver, spleen, intestines, skin, heart, pancreas and kidney. Histopathological observations revealed the presence of over 20% myeloid blasts in the BM. This, combined with significantly increased numbers of myeloid CD11b+Gr1+ cells, strongly suggests that these mice developed a myeloproliferative/myelodysplastic overlap disease resembling human AML of the acute myelomonocytic subtype. Moreover, transplantation of the entire marrow or the selected expanded Atg7−/− LSK compartment into immunocompromised mice replicated this atypical myeloproliferation.Citation13
FIP200, part of the Ulk1 (mammalian Atg1 homolog) autophagy initiation complex, was also recently studied in hematopoiesis. It was conditionally deleted by Liu et al. (2010),Citation24 in early hematopoietic cells using a Tie2-Cre system, which lead to perinatal lethality. Similar to the conditional Atg7 knockouts in the hematopoietic system, hematopoietic FIP200 knockout animals displayed severe anemia and depletion of the HSC compartment. Moreover, FIP200-deficient fetal HSCs, like adult Atg7−/− HSCs, lost the ability to repopulate lethally irradiated hosts and displayed abnormally increased proliferation along with increased mitochondrial mass and ROS levels. Interestingly, the parallels between the two conditional hematopoietic autophagy gene knockouts did not end there. Despite their early lethality, FIP200 null mice also displayed aberrant myeloproliferation, with a 17-fold increase in the frequency of myeloid cells in the blood of knockout embryos. Although the FIP200 phenotype is associated with fetal hematopoiesis rather than adult hematopoiesis as was the case in the Vav-iCre driven Atg7 knockouts, these data support the idea that autophagy/mitophagy is responsible for the two phenotypes described, rather than an autophagy-independent function of either the FIP200 or Atg7 genes, and that removal of autophagy/mitophagy genes results in anemia along with abnormal myeloproliferation.
Mitochondrial Abnormalities in MDS/AML: An Indication of Defective Mitophagy?
The phenotypes in mice with impaired autophagy in the hematopoietic system raise the question of whether autophagy/mitophagy may also contribute to hallmarks of human MDS and its development and progression to AML. If so, one might expect abnormalities in mitochondria and mitochondrial-linked processes to be associated with those diseases. Multiple studies have observed structurally abnormal mitochondria associated with MDS and enlarged mitochondriaCitation25 have been described in pro-erythroblasts, basophilic erythroblasts and, less commonly, in more mature erythroblastsCitation26 of MDS patients. As mitochondria are the main regulators of the caspase-dependent apoptosis pathway, it is not surprising that, in addition to structurally abnormal mitochondria, increased apoptosis is commonly observed in MDS.Citation25 The anemia that is part of the MDS diagnosis is often described as the result of an imbalance between proliferation and apoptosis of HSCs. Consistent with this, murine Atg7−/− erythrocytes and hematopoietic progenitors showed increased susceptibility to caspase 3-dependent apoptosis.Citation23 In a study by Lin et al. (2002), the higher than normal proliferation observed in MDS was accompanied by significantly increased apoptosis as measured by caspase-3 activity, especially in the HSC-containing CD34+ BM population. This was in contrast to the AML samples tested, where the CD34+ population showed significantly lower apoptosis as compared to normal controls. Similarly, Encomopoulou et al. (2010), found caspase-3 activity to be positively correlated with the S-phase of the cell cycle, an indication of proliferation, as well as being correlated with cyclin-dependent kinase (CDK) expression and apoptosis. Thus, MDS is characterized by mitochondrial abnormalities, implying possible defects in mitochondrial homeostasis as a consequence of defective mitophagy.
Another consequence of the accumulation of dysfunctional mitochondria would be increased levels of ROS. Multiple sources in the cell generate ROS, including the plasma membrane, peroxisomes and the cytosol. However, most cellular ROS (around 90%) are produced by mitochondria.Citation29 The primary generators of ROS within mitochondria are the electron transport chain cytochromes that create the largest potential energy flux: complexes I and to a lesser extent III.Citation29,Citation30 ROS have long been known to cause DNA mutations;Citation31 thus, as ROS levels increase with the mitochondrial load in the cell, they could begin to damage genetic material in both the mitochondrial and nuclear genomes. If severe enough, this could trigger apoptosis.Citation7 Indeed, multiple mitochondrial genes involved in the electron transport chain have been characterized as tumor suppressors which follow a two-hit model of deletionCitation32 highlighting the tumor-promoting potential of damaged mitochondria. While some studies have found increased incidence of mutations in electron transport chain proteins, such as cytochrome c oxidase I and II,Citation33 and other functionally relevant proteins in MDS patientsCitation34 other studies have found no such increased mitochondrial genome instability.Citation35 Therefore, the role that mitochondrial genome mutations play in MDS remains controversial. That being said, recent evidence of clonal expansion of cells with mtDNA mutations in MDS patients supports the idea of ROS/age-related damage to an erythroid precursor.Citation36
Mitochondrial iron accumulation, possibly due to ineffective hematopoiesis, is common in MDS cases,Citation36,Citation37 particularly in the refractory anemia with ring sideroblast (RARS) form. Importantly, excess cellular iron is hypothesized to catalyze ROS forming reactions.Citation38 This would mean that the already potentially pathological accumulation of ROS could be further exacerbated by the presence of excess iron in MDS patients.
Compounding this effect, evidence suggests that high levels of ROS can inhibit the activity of certain phosphatases, including the tumor suppressor PTEN,Citation39,Citation40 thus it is possible that ROS can have transformative influence independent of genetic damage. Finally, a recently proposed model highlights the role of oxidative stress in surrounding stromal support tissue as a mechanism of genetic instability and autophagic induction in tumors.Citation41 This also marks the autophagy-triggering role of ROS, as a possible response aimed at removing a major potential source: aging/damaged mitochondria.
Mitophagy in the Progression of MDS to AML
Recent work has shown that erythroid precursors from low-risk MDS patients (those less likely to progress to AML, as compared to high-risk patients) display increased numbers of mitochondria-laden autophagosomes.Citation42 The authors hypothesize that this increase could indicate intrinsic differentiation defects or could be a response to the higher levels of dysfunctional, iron-saturated mitochondria. However it may also be an indication of a positive feedback cycle consisting of mitophagy defects in hematopoietic stem or progenitor cells leading to improper degradation of targeted mitochondria, promoting increased ROS levels that cause DNA damage. This, combined with increased iron levels (which catalyze ROS formation), contributes to further metabolic defects as relevant genes are altered. This would continue until the abnormal mitochondria release enough cytochrome c for the cell to undergo apoptosis, unless the ROS-induced DNA damage occurs in such a way as to induce progression to AML (). Consistent with this, a study by Boultwood et al. (1996), found increased amounts of mtDNA in AML cells, which we hypothesize may be due to mitophagy defects leading to increased numbers of mitochondria in those cells. So, the high levels of mitochondria-laden autophagosomes observed in low-risk MDS patients may indicate that high levels of functional mitophagy protect these patients from the ROS build-up due to iron-damaged mitochondria until the cells undergo apoptosis from other causes, thus rendering the MDS low risk for transformation. In other words, functional autophagy may be required to prevent AML progression from MDS. The high prevalence of MDS in the aging population may be linked to the fact that autophagy levels decrease with age.Citation44
Similarly, there is indirect evidence that autophagy is involved in human AML. The PI3K/AKT/mTOR signaling pathway is constitutively active in AML patients,Citation45 a phenomenon shown to confer leukemogenic potential to mouse hematopoietic cells.Citation46 Indeed, mTOR inhibitors, such as Rapamycin, have been shown to reduce AML cell clonogenic properties.Citation47 From a functional perspective, a recently published article reported that autophagy is induced by all-trans retinoic acid treatment of acute promyelocytic leukemia (APL) and that autophagy plays an important role in the basal turnover, as well as therapy-induced proteolysis of the PML/RARA oncoprotein.Citation48
Genetic Alterations of Autophagy Genes in Human MDS/AML
Overall, characteristics found in cells from MDS and AML patients suggest a link between these diseases and autophagy/mitophagy; therefore, some abnormalities in autophagic gene expression could be expected to be revealed. However, as yet, studies of expression data from CD34+49 and CD15+50 cell populations, representing stem cell populations in MDS and AML myeloid progenitors respectively, have not highlighted autophagy genes as possible targets.
Indeed, a detailed review of expression data from two of the larger recent studies of MDS expression data revealed no significant drop in autophagy gene expression in BM mononuclear cellsCitation51 or in HSCs.Citation52 However, as noted by Lee et al. 2006,Citation50 each MDS karyotype shows different expression profiles, and it is likely that any given mechanism for MDS development applies to a specific subset of cases, which would be diluted in a general sampling of all MDS patients. Secondly, it is not yet clear whether autophagy is regulated at a transcriptional level and therefore whether a transcriptional analysis would reflect alterations in autophagic activity. Also, gene expression data would not necessarily reveal functionally deleterious point mutations in the coding regions of mitophagy/autophagy genes.
Similarly, multiple studies have examined genetic changes in myeloid neoplasm progression, including research into the role of mechanisms like acquired uniparental disomyCitation53 and methylation.Citation54 Such research has revealed alterations in a collection of tumor suppressor genes, such as Casitas B-cell lymphoma geneCitation53 and tet oncogene family member 2,Citation55 as prevalent in certain types of myeloproliferative neoplasms. However, these studies have, as yet, not found autophagy genes to be altered in these diseases.
In contrast, our review of recent studies of genetic abnormalities in AML patients has identified several key autophagy network genes, as recently defined by Behrends et al. (2010),Citation56 within the commonly deleted regions (CDRs) on 5q, 7q, 16q and 17p.Citation57–Citation59 Rucker et al. (2006) examined 60 AML patients with complex karyotypes and found losses of genetic material in these regions occurring at frequencies of 77% for 5q, 55% for 17p, 45% for 7q and 32% for 16q. This is significant because, as mentioned above, the chromosomal regions these CDRs encompass can include autophagy genes such as: ATG10 and ATG12 in the 5q CDR, ATG9B, PRKAG2 and KLHDC10 in the 7q CDR, GABARAPL2 and MAP1LC3B (both human ATG8 homologs) in the 16q CDR and GABARAP (another ATG8 homolog) in the 17p CDR.Citation57 Moreover, not only are these autophagy genes within CDRs, but Walter et al. (2009), found significantly reduced expression of both ATG12 and KLHDC10 mRNA in patients with the 5q deletion. Similarly, the autophagy gene GABARAPL1 is located in a small region of genetic loss identified by Akagi et al. (2010). In addition, a recent paper has identified ATG16L2 as hypermethylated in samples from both leukemias and lymphomas.Citation60 Taken together, these data suggest a possible role for autophagy in AML consistent with previous data on frameshift mutations in the autophagy genes ATG2, ATG5, ATG9B and ATG12 in gastric and colorectal cancers.Citation61 As mentioned above, a number of studies have shown functional autophagy to aid in cancer resistance. In particular, Beclin 1,Citation62 (ATG6), which is monoallelically deleted in almost half of human breast, ovarian and prostate cancers,Citation4 and UVRAGCitation63 have both been characterized as tumor suppressors. While deletions of at least one copy of autophagy genes is in keeping with the Knudson hypothesis for tumor suppressor genes, there is evidence to suggest that autophagy may not only overall prevent cancer but may also play a role in cancer cell survival,Citation64 such as mediating cell stress during BCR-Abl-driven leukemogenesis.Citation65 It is possible that autophagy and mitophagy are beneficial to primary oncogene-mediated leukemic transformation and for the survival of established tumors in hostile microenvironments, yet also have a protective role in genome stability in healthy cells, especially longer lived stem cells. Furthermore, heterozygous autophagy gene deletions followed by clonal selection first for deleterious mutations in the remaining allele then against them could explain this dual role,Citation6 but more research is required to assess this hypothesis. We suggest specifically this may be the case in MDS/AML.
In this review, we have built the case for a possible connection between autophagy defects, the death of erythrocyte precursors and the transformation of HSCs into leukemia initiating cells. However, it is important to note that the genetic alterations affecting autophagy genes in human MDS/AML may be bystander rather than driver mutations (i.e., may be a result of the genetic instability of transformed cells rather than a causative agent behind the tumorigenesis), or may at best contribute to the transformation process driven by other mutations. To date, it is the phenotype resulting from the ablation of autophagy in murine HSCs that has provided the strongest evidence for a link between autophagy and myeloid malignancy. There is an obvious need to investigate this connection in the human disease in order to establish a clear link between autophagy and leukemia development. This could shed light on the disease etiology and on how autophagy-modulating drugs may be beneficial in treating these diseases.
Abbreviations
| MDS | = | myelodysplastic syndrome |
| AML | = | acute myeloid leukemia |
| HSC | = | hematopoietic stem cells |
| ROS | = | reactive oxygen species |
Figures and Tables
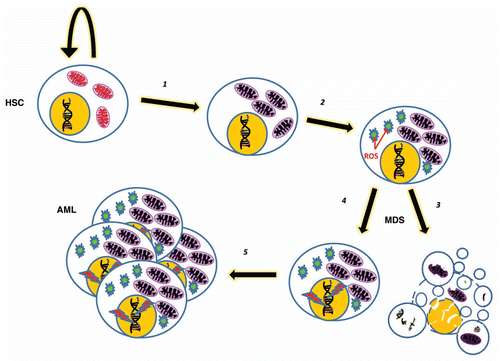
Figure 1 A simplified model where an HSC or early hematopoietic precursor develops a fault in autophagy/mitophagy (1) which leads to the build up of damaged mitochondria. These in turn cause an increase in metabolic by-products, including ROS (2) which result in cellular damage including genetic instability and more mitochondrial damage. A loss of quiescence results, along with both increased apoptosis (3) and proliferation (4) in an MDS phenotype. Oncogenic mutations may result in transformation and AML (5).
Acknowledgments
We would like to thank the National Sciences and Engineering Council of Canada and The Nuffield Department of Medicine for providing the funding of A.W.'s PhD. We also gratefully acknowledge the Det Frie Forskningsråd Sundhed og Sygdom (FSS) for a generous postdoctoral fellowship for M.M. The Wellcome Trust, the BBSRC and the NIHR have supported this research.
References
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859 - 864
- Levine B, Kroemer GCP. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42
- Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis 2011; In Press
- Rosenfeldt MT, Ryan KMC. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009; 11:36
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062 - 1075
- Mathew R, White ECP. Autophagy in tumorigenesis and energy metabolism: friend by day, foe by night. Curr Opin Genet Dev 2011; 21:113 - 119
- Farquhar MJ, Bowen DT. Oxidative stress and the myelodysplastic syndromes. Int J Hematol 2003; 77:342 - 350
- Steensma DP, Tefferi A. The myelodysplastic syndrome(s): a perspective and review highlighting current controversies. Leuk Res 2003; 27:95 - 120
- Cuijpers MLH, Raymakers RAP, MacKenzie MA, Witte TJM, Swinkels DW. Recent advances in the understanding of iron overload in sideroblastic myelodysplastic syndrome. Br J Haematol 2010; 149:322 - 333
- Buckstein R, Jang K, Friedlich J, Zhang L, Reis M, Chesney A, et al. Estimating the prevalence of myelodysplastic syndromes in patients with unexplained cytopenias: a retrospective study of 322 bone marrows. Leuk Res 2009; 33:1313 - 1318
- Estey E, Döhner H. Acute myeloid leukaemia. Lancet 2006; 368:1894 - 1907
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9 - 14
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi-Akha E, et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J Exp Med 2011; 208:455 - 467
- Kundu M, Lindsten T, Yang CY, Wu JM, Zhao FP, Zhang J, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493 - 1502
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931 - 937
- Kent G, Minick OT, Volini FI, Orfei E. AUTOPHAGIC VACUOLES IN HUMAN RED CELLS. Am J Pathol 1966; 48:831 - 857
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, et al. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232 - 235
- Zhang J, Randall MS, Loyd MR, Dorsey FC, Kundu M, Cleveland JL, et al. Mitochondrial clearance is regulated by Atg7-dependent and -independent mechanisms during reticulocyte maturation. Blood 2009; 114:157 - 164
- Mortensen M, Ferguson DJP, Simon AK. Mitochondrial clearance by autophagy in developing erythrocytes Clearly important, but just how much so?. Cell Cycle 2010; 9:1901 - 1906
- Koury MJ, Koury ST, Kopsombut P, Bondurant MC. In vitro maturation of nascent reticulocytes to erythrocytes. Blood 2005; 105:2168 - 2174
- Pua HH, Guo J, Komatsu M, He YW. Autophagy Is Essential for Mitochondrial Clearance in Mature T Lymphocytes. J Immunol 2009; 182:4046 - 4055
- Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy 2009; 5:625 - 635
- Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 2010; 107:832 - 837
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010; 116:4806 - 4814
- van de Loosdrecht AA, Brada SJL, Blom NR, Hendriks DW, Smit JW, et al. Mitochondrial disruption and limited apoptosis of erythroblasts are associated with high risk myelodysplasia. An ultrastructural analysis. Leuk Res 2001; 25:385 - 393
- Bader-Meunier B, Mielot F, Breton-Gorius J, Cramer E, Guichard J, Landrieu P, et al. Hematologic involvement in mitochondrial cytopathies in childhood: A retrospective study of bone marrow smears. Pediatr Res 1999; 46:158 - 162
- Lin CW, Manshouri T, Jilani I, Neuberg D, Patel K, Kantarjian H, et al. Proliferation and apoptosis in acute and chronic leukemias and myelodysplastic syndrome. Leuk Res 2002; 26:551 - 559
- Economopoulou C, Pappa V, Papageorgiou S, Kontsioti F, Economopoulou P, Charitidou E, et al. Cell cycle and apoptosis regulatory gene expression in the bone marrow of patients with de novo myelodysplastic syndromes (MDS). Ann Hematol 2010; 89:349 - 358
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants and aging. Cell 2005; 120:483 - 495
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009; 417:1 - 13
- Feig DI, Reid TM, Loeb LA. Reactive oxygen species in tumorigenesis. Cancer Res 1994; 54:1890 - 1894
- Kirches E. Mitochondrial and nuclear genes of mitochondrial components in cancer. Curr Genomics 2009; 10:281 - 293
- Reddy PL, Shetty VT, Dutt D, York A, Dar S, Mundle SD, et al. Increased incidence of mitochondrial cytochrome c-oxidase gene mutations in patients with myelodysplastic syndromes. Br J Haematol 2002; 116:564 - 575
- Linnartz B, Anglmayer R, Zanssen S. Comprehensive scanning of somatic mitochondrial DNA alterations in acute leukemia developing from myelodysplastic syndromes. Cancer Res 2004; 64:1966 - 1971
- Shin MG, Kajigaya S, Levin BC, Young NS. Mitochondrial DNA mutations in patients with myelodysplastic syndromes. Blood 2003; 101:3118 - 3125
- Wulfert M, Kupper AC, Tapprich C, Bottomley SS, Bowen D, Germing U, et al. Analysis of mitochondrial DNA in 104 patients with myelodysplastic syndromes. Exp Hematol 2008; 36:577 - 586
- Cohen AM, Alexandrova S, Bessler H, Mittelman M, Cycowitz Z, Djaldetti M. Ultrastructural observations on bone marrow cells of 26 patients with myelodysplastic syndromes. Leuk Lymphoma 1997; 27:165 - 172
- Szumiel I. Autophagy, reactive oxygen species and the fate of mammalian cells. Free Radic Res 2011; 45:253 - 265
- Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J 2003; 22:5501 - 5510
- Downes CP, Walker S, McConnachie G, Lindsay Y, Batty IH, Leslie NR. Acute regulation of the tumour suppressor phosphatase, PTEN, by anionic lipids and reactive oxygen species. Biochem Soc Transact 2004; 32:338 - 342
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther 2010; 10:537 - 542
- Houwerzijl EJ, Pol HW, Blom NR, van der Want JJ, de Wolf JT, Vellenga E. Erythroid precursors from patients with low-risk myelodysplasia demonstrate ultrastructural features of enhanced autophagy of mitochondria. Leukemia 2009; 23:886 - 891
- Boultwood J, Fidler C, Mills KI, Frodsham PM, Kusec R, Gaiger A, et al. Amplification of mitochondrial DNA in acute myeloid leukaemia. Br J Haematol 1996; 95:426 - 431
- Essick EE, Sam FCP. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid Med Cell Longev 2010; 3:168 - 177
- Martelli AM, Evangelisti C, Chiarini F, Grimaldi C, Manzoli L, McCubrey JA. Targeting the PI3K/AKT/mTOR signaling network in acute myelogenous leukemia. Expert Opin Investig Drugs 2009; 18:1333 - 1349
- Horn S, Bergholz U, Jucker M, McCubrey JA, Trumper L, Stocking C, et al. Mutations in the catalytic subunit of class IA PI3K confer leukemogenic potential to hematopoietic cells. Oncogene 2008; 27:4096 - 4106
- Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, et al. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005; 105:2527 - 2534
- Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010; 116:2324 - 2331
- Hofmann WK, de Vos S, Komor M, Hoelzer D, Wachsman W, Koeffler HP. Characterization of gene expression of CD34(+) cells from normal and myelodysplastic bone marrow. Blood 2002; 100:3553 - 3560
- Lee S, Chen JJ, Zhou GL, Shi RZ, Bouffard GG, Kocherginsky M, et al. Gene expression profiles in acute myeloid leukemia with common translocations using SAGE. Proc Natl Acad Sci USA 2006; 103:1030 - 1035
- Mills KI, Kohlmann A, Williams PM, Wieczorek L, Liu WM, Li R, et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 2009; 114:1063 - 1072
- Pellagatti A, Cazzola M, Giagounidis A, Perry J, Malcovati L, Della Porta MG, et al. Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells. Leukemia 2010; 24:756 - 764
- Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 2009; 460:904 - 908
- Jiang Y, Dunbar A, Gondek LP, Mohan S, Rataul M, O'Keefe C, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood 2009; 113:1315 - 1325
- Bacher U, Haferlach C, Schnittger S, Kohlmann A, Kern W, Haferlach T. Mutations of the TET2 and CBL genes: novel molecular markers in myeloid malignancies. Ann Hematol 2010; 89:643 - 652
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68 - 76
- Walter MJ, Payton JE, Ries RE, Shannon WD, Deshmukh H, Zhao Y, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA 2009; 106:12950 - 12955
- Akagi T, Ogawa S, Dugas M, Kawamata N, Yamamoto G, Nannya Y, et al. Frequent genomic abnormalities in acute myeloid leukemia/myelodysplastic syndrome with normal karyotype. Haematologica 2009; 94:213 - 223
- Rucker FG, Bullinger L, Schwaenen C, Lipka DB, Wessendorf S, Frohling S, et al. Disclosure of candidate genes in acute myeloid leukemia with complex karyotypes using microarray-based molecular characterization. J Clin Oncol 2006; 24:3887 - 3894
- Dunwell T, Hesson L, Rauch TA, Wang LH, Clark RE, Dallol A, et al. A Genome-wide screen identifies frequently methylated genes in haematological and epithelial cancers. Mol Cancer 2010; 9:44
- Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Kim SS, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol 2009; 217:702 - 706
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 676
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8:688 - 698
- White E, Karp C, Strohecker AM, Guo Y, Mathew RC. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol 2010; 22:212 - 217
- Altman BJ, Jacobs SR, Mason EF, Michalek RD, MacIntyre AN, Coloff JL, et al. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011; 30:1855 - 1867