Abstract
Increasing evidence supports a connection between cancer and metabolism and emphasizes the need to understand how tumors respond to the metabolic microenvironment and how tumor cell metabolism is regulated. The insulin receptor (IR) and its close family member the insulin-like growth factor-1 receptor (IGF-1R) mediate the cellular response to insulin in normal cells and their function is tightly regulated to maintain metabolic homeostasis. These receptors are also expressed on tumor cells and their expression correlates with tumor progression and poor prognosis. Understanding how the IR/IGF-1R pathway functions in tumors is increasing in importance as the efficacy of drugs that target metabolic pathways, such as metformin, are investigated in prospective clinical trials. This review will focus on key signaling intermediates of the IR and IGF-1R, the Insulin Receptor Substrate (IRS) proteins, with an emphasis on IRS-2, and discuss how these adaptor proteins play a pivotal role at the intersection of metabolism and cancer.
Introduction
In recent years there has been a renewed interest in the connection between cancer and metabolism, both at the organismal and cellular level. Many studies have focused on the association between obesity and metabolic diseases such as type 2 diabetes and the risk of developing cancer, as well as their impact on cancer progression and recurrence (reviewed in ref. Citation1). The findings support that diabetes and cancer are co-diagnosed more frequently than would be expected from chance and that exogenous insulin is associated with an increased cancer risk.Citation1 The insulin receptor (IR) and its close family member the insulin-like growth factor-1 receptor (IGF-1R) mediate the cellular response to insulin, and are expressed on both normal and tumor cells.Citation2 Under normal conditions, fluctuations in glucose and insulin levels are tightly regulated through feedback mechanisms that inhibit IR/IGF-1R signaling and ensure that metabolic homeostasis is maintained.Citation3 However, numerous microenvironmental insults can inhibit the function of the IR/IGF-1R signaling pathways and cause insulin resistance, hyperglycemia and hyperinsulinemia. These include increased expression of inflammatory cytokines, free fatty acids and oxidative stress, all of which are associated with obesity and metabolic dysfunction.Citation4 Further evidence to support a connection between dysregulation of IR/IGF-1R signaling and cancer has come from studies using the most widely prescribed oral hypoglycemic agent, metformin. Treatment of transgenic MMTV-HER2/Neu mice with metformin decreases the incidence, latency and size of mammary adenocarcinomas.Citation5 Moreover, retrospective clinical trials suggest that metformin may reduce the risk of developing cancer.Citation1 The mechanism by which metformin acts to reduce cancer risk is thought to be through its ability to lower blood glucose and insulin levels and restore metabolic homeostasis.
In addition to its cancer prevention role, metformin may also have anti-tumor effects that are independent of its hypoglycemic actions. Specifically, metformin suppresses the mTORC1-signaling pathway that plays an important role in sensing the nutrient microenvironment and promoting tumor growth, and is itself an active target for cancer drug development.Citation6 Metformin mediates its inhibitory function by activating the tumor suppressor LKB1 and its downstream target AMP-activated protein kinase (AMPK) and by inhibiting the RagGTPases that are essential for mTORC1 activation by amino acids.Citation7,Citation8 The dual role of metformin in inhibiting both the activation of the IR/IGF-1R by lowering systemic insulin levels, and interfering with signaling downstream of these receptors by inhibiting mTORC1 signaling, highlights the IR/IGF-1R pathway as a key mediator of both the response of tumor cells to the metabolic microenvironment and the regulation of tumor cell metabolism. Understanding how this pathway functions in tumors is increasing in importance as the efficacy of drugs that target metabolic pathways, such as metformin, are investigated in prospective clinical trials. This review will focus on key signaling intermediates of the IR and IGF-1R, the Insulin Receptor Substrate (IRS) proteins, and discuss how these adaptor proteins play a pivotal role at the intersection of metabolism and cancer.
The IRS Proteins and Normal Metabolism
To understand the role of the IRS proteins in cancer metabolism, it is necessary to first discuss their role in normal metabolic regulation. The IRS proteins are a family of cytoplasmic adaptor proteins that transmit signals from the insulin and IGF-1 receptors to elicit a cellular response. IRS-1, the first member of the family to be identified, was initially characterized as a 185 kD phosphoprotein in response to insulin stimulation.Citation9,Citation10 IRS-2 was discovered as an alternative insulin receptor substrate, initially named 4PS, in insulin-stimulated cells derived from Irs-1−/− mice.Citation11,Citation12 IRS-1 and IRS-2 are ubiquitously expressed and are the primary mediators of insulin-dependent mitogenesis and regulation of glucose metabolism in most cell types (reviewed in ref. Citation3). Humans express one additional family member, IRS-4, which is more restricted in its expression pattern and is found primarily in brain, kidney, thymus and liver.Citation13 Although they share significant homology, the phenotypes of knockout mice provide strong evidence that the IRS proteins have non-redundant normal functions. Irs-1−/− mice are born ∼70% the size of WT mice and remain small throughout their lives, implicating a role for this IRS protein in organismal growth regulation.Citation11,Citation14 In contrast, Irs-2−/− mice are normal in size but have tissue-specific defects.Citation15 Specifically, Irs2−/− mice have small brains, due to a 50% decrease in neuronal proliferation, have reduced numbers of photoreceptor cells and females are infertile, due to small, anovulatory ovaries and pituitary defects.Citation16–Citation18 Both Irs1−/− and Irs2−/− mice develop peripheral insulin resistance, but only Irs-2−/− mice develop early-onset diabetes due to a loss of β-cell function.Citation14,Citation15,Citation19 Irs-4−/− mice are phenotypically normal, with only mild growth, reproductive and insulin sensitivity defects.Citation20
The IRS proteins contain no intrinsic enzymatic activity and they mediate IR/IGF-1R signaling through their function as protein scaffolds to organize signaling complexes.Citation10 They are recruited to activated upstream receptors through PH and PTB domains located in their N-termini.Citation21 They are subsequently phosphorylated by receptor kinases on tyrosine residues in their C-termini, generating binding sites to recruit downstream effectors.Citation22 IRS-2 contains an additional domain, the KRLB domain that interacts with the tyrosine kinase domain of the IR, and may function to limit IRS-2 tyrosine phosphorylation.Citation23 This inhibitory interaction of the KRLB domain in IRS-2 with the IR provides a potential mechanism for distinguishing the functions of IRS-1 and IRS-2 and may contribute to their distinct knockout phenotypes. Interestingly, the KRLB domain doesn't inhibit phosphorylation of IRS-2 by the IGF-1R, suggesting that this receptor, or potentially hybrid IR/IGF-1Rs, may play a more active role than the IR in signaling through IRS-2.Citation23 Downstream effectors that have been characterized to bind to the IRS proteins in response to insulin or other physiological stimuli include the p85 regulatory subunit of PI3K, Grb-2, SHP-2, Fyn, c-Crk, CrkII and Nck.Citation3 However, numerous studies support that a dominant role for the IRS proteins in IR/IGF-1R-mediated metabolic regulation is the amplification of PI3K signaling to activate the serine threonine kinase AKT.Citation24–Citation26 The IRS proteins each contain multiple consensus PI3K binding motifs (YXXM) that recruit and activate PI3K through the SH2-domains in the p85 regulatory subunit.Citation3 Increased PIP3 generated by PI3K recruits AKT to the plasma membrane where it is activated by phosphorylation on T308 by PDK1.Citation27 Additional phosphorylation on S473 by TORC2 enhances AKT activity and expands the substrate targets of the kinase.Citation28,Citation29 The involvement of AKT in metabolic regulation is multifold, with several downstream substrates playing key roles in the response of cells to IR/IGF-1R signaling including Akt Substrate of 160 kD (AS160), the FOXO transcription factors and mTORC1.Citation30
The IRS proteins have been implicated as essential signaling intermediates in insulin-regulated glucose homeostasis through the promotion of glucose uptake and the regulation of genes essential for the utilization of glucose for energy production and for the biosynthesis of macromolecules including proteins, lipids and nucleic acids that are required for cell growth and proliferation.Citation31 Glucose uptake is controlled by a family of facilitative glucose transporter proteins, the GLUT proteins, that transport glucose, and in some cases fructose, across the cell membrane.Citation32 The major insulin-stimulated glucose transporter is GLUT4.Citation33 Both IRS-1 and IRS-2 have been implicated in regulating GLUT4-dependent glucose uptake in response to insulin. IRS-dependent activation of AKT stimulates phosphorylation of the Rab-GAP AS160, inhibiting its GAP activity.Citation34 As a result, Rab proteins remain in their active GTP-bound form and promote trafficking of GLUT4 from a perinuclear compartment to the cell surface to transport glucose into the cell. AKT regulates glucose utilization through both the FOXO transcription factors and mTORC1, which control the expression of metabolic pathway genes. FOXO factors promote gluconeogenesis and negatively regulate the expression of genes that promote glucose utilization including those involved in glycolysis, the pentose-phosphate shunt pathway and lipogenesis.Citation35 Phosphorylation by AKT inhibits FOXO function by preventing the translocation of these factors into the nucleus where they can suppress gene expression.Citation36,Citation37 mTORC1 regulates the expression of transcription factors such as HIF-1α and SREBP1c that regulate genes important for glucose utilization, and contributes additionally to gene expression through enhancing protein translation.Citation38 mTORC1 activity is dually regulated by AKT phosphorylation. AKT-mediated phosphorylation of TSC2 inhibits its GAP activity, allowing GTP-bound RHEB to stimulate TORC1 activity.Citation39,Citation40 Additionally, AKT phosphorylates PRAS40, which relieves its direct inhibition of mTORC1 catalytic activity.Citation41
The importance of the IRS proteins in regulating metabolic homeostasis is emphasized by the role that these proteins play in the feedback regulation of IR/IGF-1R signaling. Serine phosphorylation of the IRS proteins, mediated by many of their direct downstream effectors, interferes with their function by targeting these adaptor proteins for inactivation and/or proteasomal degradation (reviewed in ref. Citation4). For example, phosphorylation on serines 302 and 307 in IRS-1 disrupt function by inhibiting the interaction of IRS-1 with the IR. Phosphorylation of serine residues within the PI3K-binding region inhibit interactions between the IRS proteins and PI3K, selectively inhibiting the activation of this essential downstream signaling pathway. Serine phosphorylation of IRS-1 and IRS-2 can also target these adaptor proteins for ubiquitination and degradation via the 26S proteasome. This downregulation is mediated by an mTORC1-dependent negative feedback loop that also involves p70S6-kinase. The net result of these feedback events is that the magnitude and duration of the insulin signaling response is limited and insulin sensitivity and glucose homeostasis is maintained. Dysregulation of this feedback mechanism can lead to insulin resistance and diabetes.Citation4 For example, the inflammatory cytokine tumor necrosis factor-α (TNF-α) inactivates IRS-1 through a JNK-mediated phosphorylation of S307, which causes insulin resistance. Elevated free fatty acids and oxidative stress have also been shown to promote the negative serine phosphorylation of the IRS proteins and interfere with IR/IGF-1R signaling.
The IRS Proteins and Cancer Metabolism
The IR and IGF-1R are commonly expressed in human cancer and their expression has been shown to be associated with poor prognosis.Citation2 IRS-1 and IRS-2 are also ubiquitously expressed in many types of cancer.Citation42 Examination of IRS function in human tumor cell lines and in transgenic and knockout mouse tumor models has provided important clues regarding IRS function in cancer, and in the regulation of tumor cell metabolism. The majority of this research has been done in breast cancer and will be the primary focus of this review. Overexpression of either IRS-1 or IRS-2 in the mouse mammary gland results in mammary hyperplasia and tumorigenesis, which correlates with constitutive tyrosine phosphorylation of the IRS proteins, activation of Akt and Erk1/2.Citation43 Similar to the results from the transgenic models, tumor onset and growth are equivalent in the absence of either Irs-1 or Irs-2 using the PyV-MT mouse model of mammary tumor progression.Citation44,Citation45 However, mammary tumor metastasis is significantly diminished in the absence of Irs-2 and Irs-1 cannot compensate for this loss.Citation44 Moreover, Irs-2 activation is enhanced in Irs1−/− tumors that are highly metastatic.Citation45 These results correlate well with observations of IRS-1 and IRS-2 function in human breast carcinoma cells. That is, IRS-1 is expressed and signals predominantly in estrogen receptor positive (ER+), well-differentiated breast carcinoma cell lines, whereas IRS-2 is predominantly expressed in ER−, poorly-differentiated metastatic breast carcinoma cells.Citation46,Citation47 A general theme that emerges from these studies is that IRS-1 and IRS-2 may play redundant roles in tumor initiation and primary tumor growth, but have distinct roles in tumor progression. Specifically, IRS-2 promotes aggressive tumor behavior, while IRS-1 may negatively regulate tumor progression. Recent mechanistic studies suggest that IRS-2 plays this dominant role in tumor progression in large part through its ability to respond to the metabolic microenvironment and regulate tumor cell metabolism ().
A key finding that initially connected IRS-2 with tumor metabolism was the observation that Akt and mTorc1 activation are positively correlated with Irs-2 expression in PyV-MT mammary tumors.Citation45 Specifically, activity of these signaling pathways is elevated in Irs1−/− null tumors that express increased Irs-2 expression and activity (tyrosine phosphorylation), and decreased in Irs2−/− tumors. Subsequent analysis revealed that aerobic glycolysis, as assessed by glucose uptake and lactic acid production, is diminished significantly in Irs-2−/− cells compared to WT and Irs-1−/− cells, and restoration of Irs-2 expression in Irs-2−/− cells rescues the glycolytic rate to that observed in WT cells.Citation48 Glucose metabolism in cancer cells differs significantly from that of normal cells as observed initially by Otto Warburg. Specifically, cancer cells depend more on glycolysis than oxidative phosphorylation to generate ATP, even in high oxygen tensions, a phenomenon that has become known as the ‘Warburg’ effect.Citation49 Studies have affirmed the importance of aerobic glycolysis in tumor progression and have shown that it provides tumor cells with a selective advantage in their ability to progress towards invasive and metastatic disease.Citation50,Citation51 Of note, metastatic human breast carcinoma cells have enhanced aerobic glycolysis when compared with more well differentiated, non-metastatic cells.Citation50 There are several reasons why the ability to sustain aerobic glycolysis is advantageous for tumors to metastasize including the ability to survive fluctuations in oxygen tension that would be toxic to cells that depend on oxidative phosphorylation. Moreover, the acids (lactic and bicarbonic) that are generated by aerobic glycolysis can facilitate tumor invasion by degrading the extracellular matrix.Citation50
The uptake of glucose is considered to be the rate-limiting step in glycolysis.Citation52 In contrast to normal glucose regulation, which, as mentioned previously, relies primarily on GLUT4, GLUT1 (erythrocyte glucose transporter) has been implicated in controlling glucose uptake in most tumors. GLUT1 is largely undetectable in normal epithelial tissues but it is overexpressed in many carcinomas, the result of increased protein and mRNA expression stimulated by oncogenes or environmental stimuli, such as hypoxia.Citation53–Citation55 Expression of GLUT1 is higher in more poorly differentiated tumors than in low-grade tumors and high GLUT1 expression correlates with increased invasion and metastasis and poor prognosis.Citation56,Citation57 The mechanism by which IRS-2 enhances glucose transport is by increasing GLUT1 levels on the cell surface, similar to its role in stimulating GLUT4 trafficking in normal cells.Citation48 Irs-2-dependent regulation of Glut1 surface expression is rapamycin-sensitive, implicating the Akt/mTorc1 pathway in this selective regulation. Importantly, suppression of Glut1 expression inhibits Irs-2-dependent invasion, which links the enhancement of glycolysis with the ability of Irs-2 to promote metastasis.Citation48 A novel conclusion from these studies is that increased expression of GLUT1 alone may not be sufficient to confer enhanced glycolysis in human tumors because factors such as IRS-2 may be required for GLUT1 to localize to the cell surface where it can facilitate glucose uptake.
Tumor cells that can develop a metabolic self-sufficiency through aerobic or anaerobic glycolysis can survive in stressful environments that lack oxygen and other essential nutrients for energy production, and continue to proliferate within the primary tumor.Citation58 Rapidly growing tumors develop areas of low oxygen (hypoxia) and nutrient content when their growth outpaces the development of new blood vessels. Exposure of tumor cells to hypoxia creates a selection for cells that can maintain their metabolic capacity and as a result develop a more aggressive, invasive behavior. As an example, antiangiogenic inhibitors that create a hypoxic environment by inhibiting blood vessels can elicit “evasive resistance” that results in increased tumor invasion and distant metastasis.Citation59,Citation60 Gene expression is tightly regulated by hypoxia as a means to preserve energy in oxygen and nutrient deficient environments. In general, the genes that are actively transcribed in response to hypoxia are thought to be essential for tumor cells to sustain their growth and survival.Citation61 For example, pro-angiogenic factors, such as VEGF, are upregulated by hypoxia to expand the tumor vasculature and restore oxygen concentration and nutrient flow.Citation62 GLUT1 is positively regulated by hypoxia to increase the uptake of glucose to support anaerobic glycolysis and sustain energy production in low oxygen environments.Citation32 We recently identified IRS-2 as a novel hypoxia-responsive gene, which strengthens the evidence that this adaptor protein plays an important role in regulating tumor cell metabolism.Citation63 The induction of IRS-2 expression in hypoxic conditions sustains active AKT signaling and promotes tumor cell survival and invasion. Significantly, IRS-1 expression decreases in response to prolonged exposure to hypoxic conditions, providing a mechanism by which the expression levels of IRS-1 and IRS-2 can be modulated by the tumor microenvironment to promote tumor progression.Citation63
Hypoxia represents one mechanism by which the relative balance of IRS-1 and IRS-2 expression can be modulated by the tumor microenvironment to impact tumor cell metabolism and function. Serine phosphorylation of the IRS proteins may be an additional mechanism by which the stromal microenvironment influences tumor behavior in an IRS-dependent manner.Citation64 In metastatic PyV-MT mouse mammary tumors, Irs-2 phosphorylation and association with p85 are increased, whereas Irs-1 is phosphorylated on serine residues in the PI3K binding region and the overall tyrosine phosphorylation and association with p85 is decreased.Citation45 One conclusion from these studies is that tumors suppress IRS-1 function to allow IRS-2-dependent signaling to dominate. The majority of studies to evaluate the negative feedback regulation of the IRS proteins have focused on IRS-1 and there is relatively little data on how IRS-2 may be effected by the same stimuli. The distinct functions of IRS-1 and IRS-2 in tumor progression may reflect a differential sensitivity of IRS-1 and IRS-2 to the effects of negative feedback regulation, which could alter the longevity and intensity of signals initiated through each individual adaptor protein.Citation64 In an extreme example of negative feedback, tumors with constitutive activation of mTORC1, such as those with mutations in the TSC-1 or TSC-2 genes, are benign and rarely progress to a more malignant state because both IRS-1 and IRS-2 are phosphorylated by p70-S6kinase and degraded and cannot sufficiently activate survival signals.Citation65–Citation67 Interestingly, a recent study that compared the effects of metformin and rapamycin on breast carcinoma cell growth showed that metformin suppressed growth to a greater extent than rapamycin, even though both of these drugs inhibit a similar target, mTORC1.Citation68 In MCF-7 breast carcinoma cells, both drugs relieve the mTORC1-dependent negative feedback loop that allows IRS-1 to activate PI3K/AKT signaling. However, metformin also stimulates the AMPK-dependent phosphorylation of IRS-1 on S794, which suppresses PI3K/AKT activation.Citation68 Given that MCF-7 cells express little IRS-2 and signal predominantly through IRS-1, an important question that arises from these studies is whether IRS-2 is also inhibited by AMPK-mediated phosphorylation. If IRS-2 continues to signal to AKT in the presence of metformin, it could promote additional resistance to this drug because AKT can inhibit AMPK by interfering with its activation by LKB1.Citation69 Future studies are needed to compare the negative feedback regulation of IRS-1 and IRS-2 to determine if the expression of these adaptor proteins would differentially impact how a tumor cell responds to the metabolic microenvironment and to specific targeted therapies that interfere with metabolic pathways.
The ability of metformin to stimulate AMPK activation could have a differential impact on IRS-1 and IRS-2 signaling that is independent of its potential to phosphorylate these adaptor proteins on serine residues. In addition to being post-translationally modified by phosphorylation, the IRS proteins can also be acetylated, which influences their ability to be phosphorylated on tyrosine by upstream receptors and to mediate downstream signaling. Limited data indicate that acetylation positively regulates IRS-1 function and negatively regulates IRS-2 function.Citation70,Citation71 Although the acetylases responsible for these modifications are unknown, IRS-1 is deactylated by HDAC2 and IRS-2 is deacetylated by the NAD+-dependent deacetylase SIRT1. Although SIRT1 interacts with both IRS-1 and IRS-2, it only deacetylates IRS-2, which selectively enhances IRS-2 tyrosine phosphorylation by the IR and amplifies its downstream signals.Citation71 Of note, SIRT1 and IRS-2 knockout mice have similar reproductive abnormalities, which supports that this regulatory pathway may be important for IRS-2 function.Citation72 With regard to metformin, AMPK activates SIRT1, and by doing so it has the potential to preferentially stimulate IRS-2-dependent signaling.Citation73
As discussed, most of the evidence to date supports that metformin is an antitumor agent. However, data from a few studies raise caution that metformin may promote tumor progression in some contexts. For example, systemic administration of metformin increased the orthotopic growth of the poorly differentiated MDA-MB-435 breast carcinoma cell line by enhancing the expression of VEGF and promoting angiogenesis.Citation74 In a separate study, metformin inhibited tumor growth but did not decrease mammary tumor lung metastasis in mice that were fed a high fat diet.Citation75 In normal cells, AMPK is activated when energy production is low causing increased AMP/ATP ratios.Citation76 In general, AMPK phosphorylates substrates that turn on catabolic processes, such as autophagy, and shut down anabolic processes to conserve energy. In addition, it phosphorylates targets that increase glucose uptake and mitochondrial biogenesis to increase energy production and restore balance.Citation76 These functions of AMPK would be expected to positively allow tumor cells to sustain viability in nutrient and oxygen poor microenvironments and could promote metastasis. Although it is anecdotal evidence, MDA-MB-435 cells express predominantly IRS-2 and may respond favorably to metformin because of a positive impact of AMPK on IRS-2 signaling. This is one more example of how the balance of IRS activity may be shifted in response to the metabolic microenvironment to favor IRS-2 over IRS-1 signaling to promote tumor progression.
Conclusions
The increasing interest in the connection between metabolism and cancer and the development of drugs that target metabolic pathways highlights the importance of understanding how the signaling pathways that sense and respond to the metabolic microenvironment are regulated. The IRS proteins play a central role in mediating the signals from the IR/IGF-1R that control tumor cell metabolism. The differential function of the IRS proteins in tumors highlights the importance of understanding how expression of IRS-1 and IRS-2 might impact drug response and the selection of patients for specific targeted therapies.
Abbreviations
| AMPK | = | AMP-activated protein kinase |
| AS160 | = | Akt substrate of 160 kD |
| ER | = | estrogen receptor |
| IGF-1R | = | insulin-like growth factor-1 receptor |
| IR | = | insulin receptor |
| IRS | = | insulin receptor substrate |
| KRLB | = | kinase regulatory loop binding |
| mTORC1 | = | target of rapamycin complex 1 |
| mTORC2 | = | target of rapamycin complex 2 |
| PI3K | = | phosphatidylinositol kinase-3 |
| TNF | = | tumor necrosis factor |
| VEGF | = | vascular endothelial growth factor |
Figures and Tables
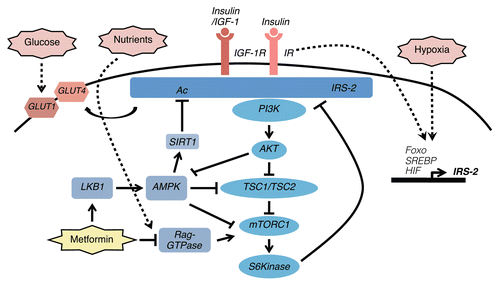
Figure 1 Schematic of IRS-2 regulation and signaling and the intersection with tumor metabolism. IRS-2 is recruited to the activated IR and IGF-1R in response to ligand stimulation and it is phosphorylated on tyrosine residues that mediate the recruitment and amplification of PI3K signaling. Shown are the stimulatory and inhibitory events that regulate the signaling molecules that are activated through IRS-2 and that impact upon tumor metabolism. Stimuli from the metabolic microenvironment that regulate IRS-2 expression are also indicated. The mechanism by which drugs that interfere with metabolism, such as metformin, intersect with IRS-2 signaling are shown.
Acknowledgments
This work was supported by National Institute of Health (NIH) grants CA090583 and CA142782. L.M.S. is a member of the University of Massachusetts Diabetes and Endocrinology Research Center (DERC) (DK32520) and the University of Massachusetts Memorial Cancer Center of Excellence.
References
- Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, et al. Diabetes and cancer: a consensus report. CA Cancer J Clin 2010; 60:207 - 221
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer 2008; 8:915 - 928
- White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab 2002; 283:413 - 422
- Gual P, Le Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005; 87:99 - 109
- Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, et al. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol 2005; 40:685 - 693
- Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol 2009; 10:307 - 318
- Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005; 310:1642 - 1646
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab 2010; 11:390 - 401
- White MF, Maron R, Kahn CR. I nsulin rapidly stimulates tyrosine phosphorylation of a Mr-185,000 protein in intact cells. Nature 1985; 318:183 - 186
- Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, et al. Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 1991; 352:73 - 77
- Araki E, Lipes MA, Patti ME, Bruning JC, Haag B 3rd, Johnson RS, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature 1994; 372:186 - 190
- Sun XJ, Wang LM, Zhang Y, Yenush L, Myers M Jr, Glasheen E, et al. Role of IRS-2 in insulin and cytokine signalling. Nature 1995; 377:173 - 177
- Lavan BE, Fantin VR, Chang ET, Lane WS, Keller SR, Lienhard GE. A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem 1997; 272:21403 - 21407
- Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature 1994; 372:182 - 186
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature 1998; 391:900 - 904
- Burks DJ, de Mora JF, Schubert M, Withers DJ, Myers MG, Towery HH, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature 2000; 407:377 - 382
- Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci 2003; 23:7084 - 7092
- Yi X, Schubert M, Peachey NS, Suzuma K, Burks DJ, Kushner JA, et al. Insulin receptor substrate 2 is essential for maturation and survival of photoreceptor cells. J Neurosci 2005; 25:1240 - 1248
- Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet 1999; 23:32 - 40
- Fantin VR, Wang Q, Lienhard GE, Keller SR. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction and glucose homeostasis. Am J Physiol Endocrinol Metab 2000; 278:127 - 133
- Voliovitch H, Schindler DG, Hadari YR, Taylor SI, Accili D, Zick Y. Tyrosine phosphorylation of insulin receptor substrate-1 in vivo depends upon the presence of its pleckstrin homology region. J Biol Chem 1995; 270:18083 - 18087
- Sun XJ, Crimmins DL, Myers M Jr, Miralpeix M, White MF. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol Cell Biol 1993; 13:7418 - 7428
- Wu J, Tseng YD, Xu CF, Neubert TA, White MF, Hubbard SR. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat Struct Mol Biol 2008; 15:251 - 258
- Okada T, Kawano Y, Sakakibara T, Hazeki O, Ui M. Essential role of phosphatidylinositol-3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J Biol Chem 1994; 269:3568 - 3573
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 1995; 376:599 - 602
- Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol 2009; 19:25 - 31
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7:261 - 269
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 307:1098 - 1101
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006; 127:125 - 137
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261 - 1274
- Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest 2006; 116:101 - 114
- Macheda ML, Rogers S, Best JD. Molecular and cellular regulation of glucose transporter (GLUT) proteins in cancer. J Cell Physiol 2005; 202:654 - 662
- Zaid H, Antonescu CN, Randhawa VK, Klip A. Insulin action on glucose transporters through molecular switches, tracks and tethers. Biochem J 2008; 413:201 - 215
- Kane S, Sano H, Liu SC, Asara JM, Lane WS, Garner CC, et al. A method to identify serine kinase substrates. Akt phosphorylates a novel adipocyte protein with a Rab GTPase-activating protein (GAP) domain. J Biol Chem 2002; 277:22115 - 22118
- Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. FoxO1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic and lipogenic gene expression. J Biol Chem 2006; 281:10105 - 10117
- Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem 1999; 274:17179 - 17183
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96:857 - 868
- Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 2010; 39:171 - 183
- Potter CJ, Pedraza LG, Xu T. Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 2002; 4:658 - 665
- Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002; 4:648 - 657
- Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 2007; 25:903 - 915
- Mardilovich K, Pankratz SL, Shaw LM. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal 2009; 7:14
- Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, Zhang X, et al. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol 2006; 26:9302 - 9314
- Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol 2004; 24:9726 - 9735
- Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, Shaw LM. Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol Cell Biol 2006; 26:9338 - 9351
- Jackson JG, White MF, Yee D. Insulin receptor substrate-1 is the predominant signaling molecule activated by insulin-like growth factor-I, insulin and interleukin-4 in estrogen receptor-positive human breast cancer cells. J Biol Chem 1998; 273:9994 - 10003
- Shaw LM. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide-3-OH kinase and promotion of invasion. Mol Cell Biol 2001; 21:5082 - 5093
- Pankratz SL, Tan EY, Fine Y, Mercurio AM, Shaw LM. Insulin receptor substrate-2 regulates aerobic glycolysis in mouse mammary tumor cells via glucose transporter 1. J Biol Chem 2009; 284:2031 - 2037
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 314
- Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis?. Nat Rev Cancer 2004; 4:891 - 899
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 2008; 7:11 - 20
- Hatanaka M. Transport of sugars in tumor cell membranes. Biochim Biophys Acta 1974; 355:77 - 104
- Hiraki Y, Rosen OM, Birnbaum MJ. Growth factors rapidly induce expression of the glucose transporter gene. J Biol Chem 1988; 263:13655 - 13662
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, et al. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem 2000; 275:21797 - 21800
- Chen C, Pore N, Behrooz A, Ismail-Beigi F, Maity A. Regulation of glut1 mRNA by hypoxia-inducible factor-1. Interaction between H-ras and hypoxia. J Biol Chem 2001; 276:9519 - 9525
- Younes M, Brown RW, Mody DR, Fernandez L, Laucirica R. GLUT1 expression in human breast carcinoma: correlation with known prognostic markers. Anticancer Res 1995; 15:2895 - 2898
- Kang SS, Chun YK, Hur MH, Lee HK, Kim YJ, Hong SR, et al. Clinical significance of glucose transporter 1 (GLUT1) expression in human breast carcinoma. Jpn J Cancer Res 2002; 93:1123 - 1128
- Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006; 441:437 - 443
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15:232 - 239
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220 - 231
- Seagroves TN, Ryan HE, Lu H, Wouters BG, Knapp M, Thibault P, et al. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol 2001; 21:3436 - 3444
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 1996; 16:4604 - 4613
- Mardilovich K, Shaw LM. Hypoxia regulates insulin receptor substrate-2 expression to promote breast carcinoma cell survival and invasion. Cancer Res 2009; 69:8894 - 8901
- Gibson SL, Ma Z, Shaw LM. Divergent roles for IRS-1 and IRS-2 in breast cancer metastasis. Cell Cycle 2007; 6:631 - 637
- Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 2004; 166:213 - 223
- Shah OJ, Wang Z, Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance and cell survival deficiencies. Curr Biol 2004; 14:1650 - 1656
- Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev 2005; 19:1773 - 1778
- Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat 2010; 123:271 - 279
- Horman S, Vertommen D, Heath R, Neumann D, Mouton V, Woods A, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem 2006; 281:5335 - 5340
- Kaiser C, James SR. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol 2004; 2:23
- Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem 2007; 282:34356 - 34364
- McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, et al. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol 2003; 23:38 - 54
- Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009; 458:1056 - 1060
- Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat 2009; 113:101 - 111
- Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat 2010; 123:333 - 344
- Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem Soc Trans 2011; 39:1 - 13