Abstract
Cancer is thought to be a disease associated with aging. Interestingly, normal aging is driven by the production of ROS and mitochondrial oxidative stress, resulting in the cumulative accumulation of DNA damage. Here, we discuss how ROS signaling, NFκB- and HIF1-activation in the tumor microenvironment induces a form of “accelerated aging,” which leads to stromal inflammation and changes in cancer cell metabolism. Thus, we present a unified model where aging (ROS), inflammation (NFκB) and cancer metabolism (HIF1), act as co-conspirators to drive autophagy (“self-eating”) in the tumor stroma. Then, autophagy in the tumor stroma provides high-energy “fuel” and the necessary chemical building blocks, for accelerated tumor growth and metastasis. Stromal ROS production acts as a “mutagenic motor” and allows cancer cells to buffer—at a distance—exactly how much of a mutagenic stimulus they receive, further driving tumor cell selection and evolution. Surviving cancer cells would be selected for the ability to induce ROS more effectively in stromal fibroblasts, so they could extract more nutrients from the stroma via autophagy. If lethal cancer is a disease of “accelerated host aging” in the tumor stroma, then cancer patients may benefit from therapy with powerful antioxidants. Antioxidant therapy should block the resulting DNA damage, and halt autophagy in the tumor stroma, effectively “cutting off the fuel supply” for cancer cells. These findings have important new implications for personalized cancer medicine, as they link aging, inflammation and cancer metabolism with novel strategies for more effective cancer diagnostics and therapeutics.
One way to think about cancer is as a disease associated with normal aging.Citation1,Citation2 During aging, mitochondrial ROS production results in DNA damage and mitochondrial dys-function.Citation3–Citation5 Interestingly, we see that cancer cells can also induce a form of “accelerated aging” in normal adjacent fibroblasts, via ROS production and oxidative stress.Citation6–Citation15
Oxidative stress in cancer-associated fibroblasts then results in the activation of two major transcription factors, namely NFκB and HIF1.Citation10 NFκB is the master regulator of the innate immune response (inflammation), while HIF1 drives metabolic re-programming with the onset of aerobic glycolysis (the “Warburg effect”).Citation16,Citation17 Both NFκB and HIF1 are also key activators of autophagy and mitophagy.Citation12 Thus, “accelerated aging” via ROS production drives inflammation, cancer metabolism and autophagy (). This, in turn, results in DNA damage in cancer cells (driving genomic instability) and fuels oxidative mitochondrial metabolism in cancer cells (via the stromal production of high-energy metabolites, such as lactate, ketones and glutamine).Citation10,Citation12,Citation13 Ketones and lactate, in turn, stimulate tumor growth and metastasis.Citation18 A loss of stromal caveolin-1 (Cav-1) provides a robust biomarker for this “accelerated aging” phenotype in the tumor stroma, and is functionally associated with oxidative stress, inflammation, aerobic glycolysis and autophagy.Citation15
Four recent papers published in the journal Cell Cycle further validate this model of cancer as “accelerated host aging.”
The first paper establishes that stromal fibroblasts and cancer cells are metabolically coupled, via a lactate shuttle that transfers lactate from glycolytic cancer-associated fibroblasts to oxidative cancer cells, via the compartmentalization of MCT transporters.Citation19 This is accomplished, in part, by the upregulation of MCT4 expression in cancer-associated fibroblasts, via oxidative stress.Citation19 Thus, aggressive cancer cells use lactate as a fuel source, via oxidative mitochondrial metabolism.
The second paper shows that oxidative stress in cancer-associated fibroblasts results in a “cytokine storm,” via NFκB-activation.Citation20 Individually, each of these inflammatory cytokines was also sufficient to induce autophagy in stromal fibroblasts.Citation20 This observation explains how inflammation literally “fuels” tumor growth, via stromal autophagy.Citation20
The third paper analyzes the transcriptional profiles of laser-captured tumor stroma, associated with a lethal tumor microenvironment in human breast cancer patient samples.Citation21 In this paper, a Cav-1-deficient tumor microenvironment shows the same patterns of gene expression as those normally associated with aging, Alzheimer disease, DNA damage, oxidative stress, inflammation, hypoxia, HIF1- and NFκB-activation ().Citation21 In fact, the Alzheimer disease brain signature can identify which breast cancer patients will develop metastasis.Citation7,Citation8
Finally, the fourth paper shows that when breast cancer cells (MCF7) use lactate as a “fuel supply,” they undergo mitochondrial biogenesisCitation10,Citation12 and transcriptionally they appear similar to “stem cells.”Citation22 These lactate-induced gene signatures predict recurrence, metastasis, and poor overall survival in breast cancer patients, directly linking oxidative mitochondrial metabolism with clinical outcome.Citation22 These observations provide a new starting point for personalized cancer medicine, linked to cancer metabolism, termed “metabolo-genomics.”Citation22 Interestingly, administration of lactate to a human breast cancer xenograft model (MDA-MB-231 cells) increases the rate of lung metastasis >10-fold.Citation18
In accordance with this “accelerated host aging” model, a loss of Cav-1 in the tumor stroma is a marker of oxidative stress and inflammation in the tumor microenvironment,Citation7,Citation8,Citation10,Citation23,Citation24 and is associated with metastasis and poor prognosis in breast and prostate cancer patients.Citation24–Citation28 Similarly, in mouse animal models, a loss of Cav-1 is associated with accelerated aging,Citation29 and an increased susceptibility towards various oncogenic stimuli,Citation30–Citation34 as well as oxidative stress, DNA damage and autophagy.Citation7,Citation10 Thus, “accelerated host aging” provides a “fertile soil” for tumor growth and metastasis.
This new model of cancer as “accelerated host aging” suggests that we should be treating cancer patients with powerful antioxidants. Interestingly, “catalase therapy” in pre-clinical models efficiently blocks both tumor recurrence and metastasis.Citation35–Citation42 Genetic reductions in mitochondrial oxidative stress (via transgenic overexpression of catalase) reduces metastatic tumor burden in MMTV-PyMT mice by ∼13-fold.Citation43,Citation44 Similarly, treatment of breast cancer patients with antioxidants (Vitamins C and E), significantly reduces both mortality and recurrence, and this protective effect is nearly doubled if the patients did not undergo radiation therapy.Citation45 Radiation therapy is known to increase oxidative stress and DNA damage, thereby negating the positive effects of antioxidants.Citation45
Successful antioxidant therapy should prevent DNA damage, stopping cancer cell evolution and halting autophagy in the tumor stroma, effectively “starving” cancer cells. Thus, we should re-consider using antioxidants (N-acetyl-cysteine and Vitamins C/E), mitochondrial poisons (metformin) and autophagy inhibitors (chloroquine and hydroxy-chloroquine), as potential anti-cancer agents.
Unfortunately, most current cancer therapies are toxic and work by increasing oxidative stress and DNA damage, thereby “accelerating host aging.” In fact, most current therapies (chemo-therapeutics and radiation) also increase a patient's risk for secondary malignancies, such as leukemia or lymphoma.
As a consequence, we should carefully re-think how to more effectively treat cancer patients, without increasing oxidative stress and “accelerated aging.”
Aging studies in flies and mice also point towards a causative role for inflammation in reduced life span.Citation46,Citation47 For example, flies selected for increased longevity show the specific downregulation of gene transcripts normally associated with inflammation and the immune response.Citation47 Similar results were obtained in mice, where they observed that gene transcripts associated with cytokine production, the immune response and inflammation were specifically upregulated during aging.Citation46 Interestingly, caloric restriction specifically reduced the expression of these aging-associated inflammation genes, but not other aging-associated gene transcripts.Citation46 Thus, there appears to be a direct metabolic link between aging and inflammation. We believe this metabolic link is that inflammation induces systemic increases in autophagy/mitophagy and aerobic glycolysis.Citation20 Thus, “cutting off the fuel supply” via caloric restriction decreases inflammation, which is promoting aging and cancer via increased ROS production and the ensuing DNA damage.
In further support of this notion, N-acetyl-cysteine (NAC), which functions as a powerful antioxidant and NFκB-inhibitor, increases lifespan in flies and in mice by >25%.Citation48–Citation50 NAC is also protective against tumor formation.Citation51,Citation52 Thus, exploiting the dietary use of NAC and other antioxidants (metformin) or autophagy inhibitors (hydroxychloroquine) may be helpful in preventing “accelerated host aging” in the tumor microenvironment.Citation10,Citation11
Figures and Tables
Figure 1 Accelerated aging drives DNA damage, inflammation, aerobic glycolysis and autophagy in the tumor microenvironment, fueling tumor progression and metastasis. See text for further details. (A) Cancer cells induce ROS production (accelerated aging) in adjacent normal fibroblasts, which results in NFκB-activation (inflammation) and HIF1-stabilization (cancer metabolism). Their combined effect drives autophagy in the tumor stroma. Stromal autophagy, via the production of high-energy nutrients (lactate, ketones and glutamine), stimulates oxidative mitochondrial metabolism in cancer cells. (B) Accelerated aging in fibroblasts produces ROS that induces DNA damage in fibroblasts and adjacent cancer cells (via a Bystander effect), fostering tumor-stroma co-evolution.
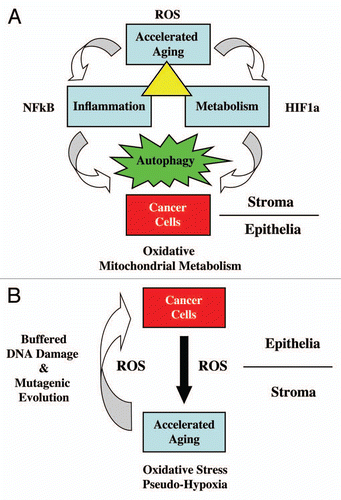
Figure 2 A loss of Cav-1 in the tumor stroma of human breast cancer patients is associated with aging, DNA damage, inflammation, cancer metabolism and autophagy. The transcriptional profiles of Cav-1-positive (+) tumor stroma (n = 4) versus Cav-1-negative tumor stroma (n = 7) were compared, via laser-capture microdissection. Note that Cav-1-deficient stroma shows the upregulation of aging (73 transcripts), the DNA damage response (67 transcripts), NFκB signaling (11 transcripts), the immune response (31 transcripts), HIF1/hypoxia target genes (65 transcripts), glycolysis/pyruvate metabolism (15 transcripts) and autophagy (22 transcripts). Modified and reproduced with permission from reference Citation21. In human breast cancer patients, a loss of stromal Cav-1 is associated with early tumor recurrence, lymph-node metastasis, drug-resistance and overall poor survival, conferring a “lethal” tumor microenvironment.Citation24–Citation28

Acknowledgments
F.S. and her laboratory were supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA), and a Research Scholar Grant from the American Cancer Society (ACS). M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. A.K.W. was supported by a Young Investigator Award from the Breast Cancer Alliance, Inc., and a Susan G. Komen Career Catalyst Grant. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
References
- Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle 2010; 9:4788 - 4794
- Blagosklonny MV, Campisi J, Sinclair DA. Aging: Past, present and future. Aging (Albany NY) 2009; 1:1 - 5
- Giem P, Beeson WL, Fraser GE. The incidence of dementia and intake of animal products: preliminary findings from the Adventist Health Study. Neuroepidemiology 1993; 12:28 - 36
- Singh PN, Sabate J, Fraser GE. Does low meat consumption increase life expectancy in humans?. Am J Clin Nutr 2003; 78:526 - 532
- Fraser GE. Vegetarian diets: What do we know of their effects on common chronic diseases?. Am J Clin Nutr 2009; 89:1607 - 1612
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of Stromal Caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010; 9:2201 - 2219
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “reverse Warburg effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer disease and “neuron-glia metabolic coupling”. Aging (Albany NY) 2010; 2:185 - 199
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: Aerobic glycolysis in cancer-associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer-associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer-associated fibroblast phenotype via Caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer-associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533
- Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010; 9:4297 - 4306
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther 2010; 10:537 - 542
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromalepithelial metabolic coupling in cancer: Integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 2011; 43:1045 - 1051
- Blagosklonny MV. Hypoxia-inducible factor: Achilles' heel of antiangiogenic cancer therapy. Int J Oncol 2001; 19:257 - 262
- Blagosklonny MV. Antiangiogenic therapy and tumor progression. Cancer Cell 2004; 5:13 - 17
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 3514
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 1783
- Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: Role of stromal caveolin-1 as a key regulator. Cell Cycle 2011; 10:1784 - 1793
- Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Pavlides S, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle 2011; 10:1794 - 1809
- Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011; 10:1271 - 1286
- Trimmer C, Sotgia F, Whitaker-Menezes D, Balliet R, Eaton G, Martinez-Outschoorn UE, et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal micro-environment: A new genetically tractable model for human cancer-associated fibroblasts. Cancer Biol Ther 2011; 11:383 - 394
- Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8:1167 - 1175
- Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of Stromal Caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther 2010; 10:135 - 143
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of Stromal Caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 2034
- Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of Stromal Caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8:2420 - 2424
- Sloan EK, Ciocca D, Pouliot N, Natoli A, Restall C, Henderson M, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol 2009; 174:2035 - 2043
- Park DS, Cohen AW, Frank PG, Razani B, Lee H, Williams TM, et al. Caveolin-1 null (-/-) mice show dramatic reductions in life span. Biochemistry 2003; 42:15124 - 15131
- Capozza F, Williams TM, Schubert W, McClain S, Bouzahzah B, Sotgia F, et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am J Pathol 2003; 162:2029 - 2039
- Williams TM, Cheung MW, Park DS, Razani B, Cohen AW, Muller WJ, et al. Loss of caveolin-1 gene expression accelerates the development of dysplastic mammary lesions in tumor-prone transgenic mice. Mol Biol Cell 2003; 14:1027 - 1042
- Williams TM, Lee H, Cheung MW, Cohen AW, Razani B, Iyengar P, et al. Combined loss of INK4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis. J Biol Chem 2004;
- Williams TM, Medina F, Badano I, Hazan RB, Hutchinson J, Muller WJ, et al. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo: Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem 2004; 279:51630 - 51646
- Williams TM, Sotgia F, Lee H, Hassan G, Di Vizio D, Bonuccelli G, et al. Stromal and Epithelial Caveolin-1 Both Confer a Protective Effect Against Mammary Hyperplasia and Tumorigenesis: Caveolin-1 Antagonizes Cyclin D1 Function in Mammary Epithelial Cells. Am J Pathol 2006; 169:1784 - 1801
- Hyoudou K, Nishikawa M, Ikemura M, Kobayashi Y, Mendelsohn A, Miyazaki N, et al. Prevention of pulmonary metastasis from subcutaneous tumors by binary system-based sustained delivery of catalase. J Control Release 2009; 137:110 - 115
- Hyoudou K, Nishikawa M, Kobayashi Y, Umeyama Y, Yamashita F, Hashida M. PEGylated catalase prevents metastatic tumor growth aggravated by tumor removal. Free Radic Biol Med 2006; 41:1449 - 1458
- Nishikawa M. Reactive oxygen species in tumor metastasis. Cancer Lett 2008; 266:53 - 59
- Nishikawa M, Hashida M. Inhibition of tumour metastasis by targeted delivery of antioxidant enzymes. Expert Opin Drug Deliv 2006; 3:355 - 369
- Nishikawa M, Hashida M, Takakura Y. Catalase delivery for inhibiting ROS-mediated tissue injury and tumor metastasis. Adv Drug Deliv Rev 2009; 61:319 - 326
- Nishikawa M, Tamada A, Hyoudou K, Umeyama Y, Takahashi Y, Kobayashi Y, et al. Inhibition of experimental hepatic metastasis by targeted delivery of catalase in mice. Clin Exp Metastasis 2004; 21:213 - 221
- Nishikawa M, Tamada A, Kumai H, Yamashita F, Hashida M. Inhibition of experimental pulmonary metastasis by controlling biodistribution of catalase in mice. Int J Cancer 2002; 99:474 - 479
- Oberley LW. Mechanism of the tumor suppressive effect of MnSOD overexpression. Biomed Pharmacother 2005; 59:143 - 148
- Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C, et al. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011; 11:191
- Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: Should we use antioxidants as a key component of cancer treatment and prevention?. BMC Medicine 2011; 9:62
- Nechuta S, Lu W, Chen Z, Zheng Y, Gu K, Cai H, et al. Vitamin supplement use during breast cancer treatment and survival: a prospective cohort study. Cancer Epidemiol Biomarkers Prev 2011; 20:262 - 271
- Swindell WR. Genes and gene expression modules associated with caloric restriction and aging in the laboratory mouse. BMC Genomics 2009; 10:585
- Sarup P, Sorensen P, Loeschcke V. Flies selected for longevity retain a young gene expression profile. Age (Dordr) 2011; 33:69 - 80
- Brack C, Bechter-Thuring E, Labuhn M. N-acetylcysteine slows down ageing and increases the life span of Drosophila melanogaster. Cell Mol Life Sci 1997; 53:960 - 966
- De la Fuente M. Murine models of premature ageing for the study of diet-induced immune changes: improvement of leucocyte functions in two strains of old prematurely ageing mice by dietary supplementation with sulphur-containing antioxidants. Proc Nutr Soc 2010; 69:651 - 659
- Flurkey K, Astle CM, Harrison DE. Life extension by diet restriction and N-acetyl-L-cysteine in genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2010; 65:1275 - 1284
- Zhang XF, Tan X, Zeng G, Misse A, Singh S, Kim Y, et al. Conditional beta-catenin loss in mice promotes chemical hepatocarcinogenesis: role of oxidative stress and platelet-derived growth factor receptor alpha/phosphoinositide-3-kinase signaling. Hepatology 2011; 52:954 - 965
- Reliene R, Schiestl RH. Antioxidant N-acetyl cysteine reduces incidence and multiplicity of lymphoma in Atm deficient mice. DNA Repair (Amst) 2006; 5:852 - 859