Abstract
Upon exposure to genotoxic stress, skeletal muscle progenitors coordinate DNA repair and the activation of the differentiation program through the DNA damage-activated differentiation checkpoint, which holds the transcription of differentiation genes while the DNA is repaired. A conceptual hurdle intrinsic to this process relates to the coordination of DNA repair and muscle-specific gene transcription within specific cell cycle boundaries (cell cycle checkpoints) activated by different types of genotoxins. Here, we show that, in proliferating myoblasts, the inhibition of muscle gene transcription occurs by either a G1- or G2-specific differentiation checkpoint. In response to genotoxins that induce G1 arrest, MyoD binds target genes but is functionally inactivated by a c-Abl-dependent phosphorylation. In contrast, DNA damage-activated G2 checkpoint relies on the inability of MyoD to bind the chromatin at the G2 phase of the cell cycle. These results indicate an intimate relationship between DNA damage-activated cell cycle checkpoints and the control of tissue-specific gene expression to allow DNA repair in myoblasts prior to the activation of the differentiation program.
Introduction
The response to DNA damage has been extensively investigated in proliferating cells.Citation1–Citation3 In these cells, DNA damage can occur by endogenous sources (e.g., stalled replication forks during S phase and increased levels of oxygen reactive species) or as a consequence of the exposure to exogenous genotoxic stress, such as radiations or chemotherapic agents.Citation4,Citation5 DNA damage signaling activates a complex cellular response to temporally coordinate cell cycle progression and DNA repair.Citation6,Citation7 The transient arrest of proliferation in cells exposed to genotoxic cues restricts the repair process to discrete boundaries of the cell cycle: prior to and during the DNA synthesis (referred as to G1- and S phase-checkpoints, respectively) or before mitosis (the G2-checkpoint).Citation8–Citation15 These different DNA damage-activated cell cycle checkpoints permit the monitoring of genomic integrity in proliferating cells and avoid the propagation of unrepaired DNA lesions that often prelude to neoplastic transformation or cellular senescence.Citation16 In progenitors of terminally differentiated tissues, such as skeletal muscles and neurons, the DNA damage response is complicated by their commitment toward the differentiation programs, which includes the irreversible exit from the cell cycle.Citation17 Previous work identified a DNA damage-activated differentiation checkpoint in myoblasts that holds the differentiation program, while DNA lesions are repaired.Citation18 Two key effectors of this program are the DNA damage-activated cAbl tyrosine kinaseCitation19 and the muscle regulatory factor (MRF) MyoD. In the absence of DNA damage MyoD typically initiates the differentiation program in myoblasts upon the arrest of the cell cycle.Citation20 Upon DNA damage, MyoD becomes phosphorylated on tyrosine by c-Abl, leading to the inhibition of muscle gene transcription.Citation18 It is speculated that the differentiation checkpoint permits the temporal coordination between cell cycle progression, DNA repair and differentiation, thereby preventing the formation of terminally differentiated myotubes with unrepaired lesions.Citation17,Citation21 However, the precise relationship between DNA damage-activated cell cycle checkpoints, DNA repair and the mechanism underlying the inhibition of MyoD-dependent transcription has not been elucidated.
In the present work, we show that proliferating myoblasts exposed to different types of genotoxic agents activate distinct differentiation checkpoints at specific cell cycle boundaries. We found that the type of DNA damage and the related timing of DNA repair correlate with the length of latency of the differentiation program. Moreover, we show that the cell cycle phase at which myoblasts arrest in response to distinct genotoxic insults determines the mechanism of inhibition of muscle gene transcription.
Results
Cell cycle phase-specific differentiation checkpoints in proliferating myoblasts.
To investigate the relationship between cell cycle arrest in proliferating myoblasts, DNA repair and transcription of muscle genes, we exposed C2C7 skeletal myoblasts to distinct genotoxic agents while proliferating in growth medium (GM). These cells were then transferred in culture conditions permissive for differentiation (differentiation medium-DM), in the absence of genotoxins to activate the differentiation checkpoint (see Puri et al.Citation18 and scheme in ). The cell cycle profile of these cells was monitored by FACS (), and the kinetic of DNA damage repair was determined by alkaline comet assayCitation22 at different time points ( and C). All the genotoxic agents that we tested activated a DNA damage response, as determined in primary human skeletal myoblasts (HSMBs) by the re-distribution of Nbs1 into discrete nuclear foci (Sup. Fig. 1), and inhibited the formation of MyHC-positive myotubes ( and Sup. Fig. 1). Importantly, the same inhibition was observed in primary cultures of mouse satellite muscle cells (Sup. Fig. 2A), as was also previously shown with other myogenic cell lines.Citation18 However, while some genotoxic agents (methylmethane sulfonate-MMS-, hydrogen peroxide and cisplatin) did not alter significantly the cell cycle profle of differentiating myoblasts, which typically arrest in G1 upon culture in DM, others (Doxorubicin-Dox-, Etoposide-Eto-, bleomycin, mitomycin) caused a significant accumulation of myoblasts into the G2/M phase ( and ). These differences did not correlate with different kinetics of repair, since the DNA lesions caused by MMS and etoposide were repaired within the same time frame ( and C), although these agents induced G1 and G2/M arrest, respectively. By contrast, Dox-induced DNA damage was repaired within a more extended time frame ( and C), with some cells being unable to repair the DNA damage even after 48 h. Interestingly, the different kinetics of DNA repair after exposure to distinct agents correlated with differences in the temporal windows of inhibition of the differentiation program (). Indeed, we previously showed that myoblasts treated with MMS and etoposide showed a delay in the activation of the early markers of differentiation (e.g., myogenin) ranging between 12–18 h of DM culture as compared with control cells.Citation18 In Dox-treated myoblasts, the latency of the differentiation program extended over the first 30 h of incubation in DM (Sup. Fig. 2B), with a variable percentage of cells being permanently unable to resume the differentiation program. This is consistent with the complex nature of the DNA lesions induced by Dox, which can complicate the efficiency of the repair. Thus, distinct genotoxic agents elicit different types of differentiation checkpoints whose duration correlates with the specific kinetic of the DNA repair machinery, regardless of the cell cycle boundary at which cells arrest. This notion is consistent with the putative function of the differentiation checkpoint to coordinate the temporal sequence of DNA repair and activation of the differentiation program to avoid the accumulation of unrepaired lesions in terminally differentiated muscles.
To further analyze the relationship between cell cycle and the differentiation checkpoint, we performed a microarray in HSMBs induced to differentiate after being exposed to Dox. A comparison between the Dox-treated and control myoblasts revealed functional categories of Dox-modulated genes (≥ 2-fold changes). The complete list of genes upregulated and down-regualted is provided in Supplemental Table 1. Among the genes downregulated by Dox (, right panel), muscle-specific genes scored in the top categories and included genes involved in the activation of the myogenic program—myogenin—and contractile activity—myosin binding protein H (MYBPH), myosin light chain 4 (MYL4) and troponin T type 1 (TNNT1). In addition, cell cycle-related categories displayed high ranking in the GeneGO Process Networks due to the downregulation of genes such as cell division cycle protein 20 homolog (CDC20) and DNA topoisomerase 2-α (TOP2A). Among the upregulated genes (, left part), the top scoring categories were enriched in DNA remodeling process due to the upregulation of several histone proteins (HIST1H2AC, HIST1H2BC, HIST1H4C) and the DNA damage-response genes p21 (CDKN1A), xeroderma pigmentosum complementation group C protein (XPC), DNA damage-binding protein 2 (DDB2) and proliferating cell nuclear antigen (PCNA). Among Dox-upregulated genes were also annotated genes involved in the activin A signaling and containing, among the others, follistatin, p300 and the TGFβ receptor type 3. We validated the Dox-induced upregulation of the inhibitor of cyclin kinases p21 (Sup. Fig. 3A). p21 expression normally increases after regular differentiation and was further augmented by Dox treatment. Two other genes—XPC and DDB2—associated to the repair machinery activated by Dox treatment were validated (Sup. Fig. 3B and C). Among them, XPC is involved in the recognition of bulky DNA adducts in nucleotide excision repair,Citation23 and DDB2 is a small subunit of a heterodimeric protein complex that participates in nucleotide excision repair, mediating the ubiquitylation of histones H3 and H4, which facilitates the cellular response to DNA damage.Citation24
Collectively, data from the microarray analysis indicate a tight relationship between the inhibition of the myogenic program, the activation of the cell cycle arrest and the DNA repair in myoblasts exposed to DNA damage.
Different mechanism of MyoD inhibition by G1- vs. G2-associated differentiation checkpoint.
We next determined the molecular link between DNA repair and activation of cell cycle-specific differentiation checkpoints by monitoring MyoD recruitment to target genes in myoblasts exposed to distinct genotoxic agents.
Previous work demonstrated that in G1-arrested myoblasts, MyoD is phosphorylated at tyrosine 30 by the DNA damage-activated cAbl tyrosine kinase. The cAbl-mediated phosphorylation inactivates MyoD, thereby holding the activation of muscle gene expression.Citation18 We used chromatin immunoprecipitation (ChIP) to monitor MyoD binding to regulatory sequences at muscle-specific loci after treatment with MMS, which induces G1 arrest (). We found that MMS did not alter MyoD binding and local H3 acetylation on myogenin promoter and MCK enhancer (). However, MMS reduced H3K4 trimethylation (H3K43m) levels (see ), suggesting that DNA damage-mediated inactivation of MyoD in G1 phase occurs through selective inhibition of recruitment or activity of H3K4 methyltransferases. In contrast, in myoblasts arrested at the G2-phase upon Dox treatment, MyoD showed reduced occupancy on the regulatory elements of myogenin and MCK in association with decreased levels of H3 acetylation (). This is in agreement with a previous work indicating that during the G2/M phase, the chromatin is condensed and is not permissive for MyoD binding to its target DNA sequences.Citation25 Thus, MyoD inhibition upon treatment with drugs that induce G2 arrest is achieved by the intrinsic failure of MyoD to bind the chromatin of target genes and promote local hyperacetylation at the G2/M phase of the cell cycle.
The execution of the G1 and G2 differentiation checkpoints is dependent on the cell cycle boundary at which the cells arrest to repair DNA.
To gain further insight into the relationship between cell cycle arrest and the execution of the differentiation checkpoint, we used agents that bypass either the G1 or the G2 block of the cell cycle, and we monitored the ability of cells to perform the specific differentiation checkpoint.
Caffeine, which specifically resumes the cell cycle in G2/M-arrested cells,Citation26 did not significantly alter the cell cycle profle in myoblasts entering the differentiation program in normal conditions or after treatment with MMS (, top and middle panels) but dramatically reduced the percentage of myoblasts accumulated in the G2/M phase after Dox treatment (, bottom panel). In cells exposed to either treatment, caffeine prevented the phosphorylation of p53 on serine 15 in response to DNA damage (). The ability of caffeine to reverse the G2/M arrest correlated with its ability to bypass the G2-differentiation checkpoint and to allow the expression of muscle-specific genes in Dox-treated myoblasts but not the G1-differentiation checkpoint induced by MMS (). Consistently, caffeine treatment restored both MyoD chromatin-recruitment and hyperacetylation at the myogenin and MCK regulatory regions in Dox-treated, but not in MMS-treated, myo-blasts ( and B).
The same results were also replicated in HSMBs, in which caffeine specifically rescued the inhibition of myotube formation from the G2-arresting drugs, Etoposide and Dox (Sup. Fig. 4). It is presumed that the population of myoblasts that escaped the G2/M cell cycle arrest in response to Dox or Etoposide progresses through the cell cycle and eventually differentiates once arrested in the G1 phase. By contrast, caffeine did not rescue the inhibition of myotube formation in MMS-treated myoblasts that were already confined in the G1 phase of the cell cycle (Sup. Fig. 4). Caffeine is a non-specific inhibitor of the DNA damage-activated Ataxia Teleangectasia Mutant (ATM) kinase;Citation27 however, its ability to rescue the G2-mediated inhibition of muscle gene expression appears to rely on its effect on the cell cycle, because selective inhibition of ATM by shRNA or by a soluble inhibitor (KU-55933), which blocks ATM, but does not interfere with the G2 arrest, did not replicate the effect of caffeine (Simonatto MS and Puri PL, unpublished data).
We used insulin-like growth factor 1 (IGF-1) to bypass the G1 arrest induced by MMS damage. It has been shown that IGF1 extends the replicative life span of skeletal muscle satellite cells by promoting cell cycle progression into S phase.Citation28,Citation29 IGF1 bypassed the G1-associated differentiation checkpoint imposed by MMS treatment in HSMBs ( and B) and C2C12 myoblasts (data not shown) but did not interfere with the G2-differentiation checkpoint induced by Dox ( and B). ChIP analysis of the MCK enhancer in C2C12 myoblasts shows that IGF1 rescues muscle differentiation upon treatment with MMS, but not Dox, and this correlates with restoration of H3K4me3 (), a reliable indication of the transition from inactive to active chromatin conformation.Citation30
Overall, these data indicate that the inhibition of muscle gene expression induced by DNA damage occurs through a mechanism that is specific to the phase of the cell cycle at which myoblasts are arrested in response to different genotoxic drugs. This evidence supports the conclusion that distinct cell cycle-intrinsic mechanisms mediate two differentiation checkpoints induced by different genotoxic agents. The G1-associated differentiation checkpoint relies on the inactivation of MyoD-mediated transcription by cAbl-mediated phosphorylation of tyrosine 30 in the activation domain of MyoD, as previously reported, whereas the G2-associated differentiation checkpoint relies on the inability of MyoD to bind the chromatin at the G2/M phase of the cell cycle.Citation25
Discussion
The maintenance of the genomic integrity in post-mitotic tissues, such as skeletal muscles, is important to warrant their correct function during life span. The premature aging observed in mice deficient for key effectors of the DNA damage signalingCitation31,Citation32 supports the relationship between genomic instability and the functional decline of organs and tissues that is typically observed in aged organisms.Citation33 However, it is still unclear how organs and tissues safeguard their genome from the genotoxic insults present during development and throughout the adult life.
We have previously described a “differentiation checkpoint” that inhibits the activation of the myogenic program in undifferentiated myoblasts exposed to genotoxic stress by holding the transcription of muscle genes while the DNA is repaired.Citation18 In the present work, we have investigated the impact of different types of genotoxic agents on muscle gene expression and DNA repair in growing myoblasts.
In proliferating myoblasts, the DNA damage is resolved by the repair of the lesions; hence, the block of the differentiation program is transient, with the expression of muscle genes being resumed in coincidence with the resolution of the DNA lesionsCitation18 (see also and Sup. Fig. 1). Our data indicate an intimate relationship between the cell cycle boundary at which the cells arrest to repair the DNA lesion and the mechanism that inhibits the activation of the myogenic program. The arrest at the G2/M checkpoint precludes MyoD binding to the chromatin of target genes,Citation25 thereby providing a cell cycle-intrinsic inhibition of muscle gene transcription that can be reversed by agents such as caffeine, which bypass the G2 checkpoint. In contrast, myoblasts arrested at the G1 checkpoint are permissive for the recruitment of MyoD to the chromatin of target genes, but transcription is inhibited by cAbl-mediated tyrosine phosphorylation.Citation18 The G1 checkpoint is reversed by IGF-1, which bypasses the DNA damage-induced G1 arrest in myoblasts.Citation29 Thus, the activation of differentiation checkpoints that are superimposed at cell cycle checkpoints appears an “economic” strategy for the cells to coordinate multiple tasks, such as DNA repair and gene expression, within restricted windows along the cell cycle progression.
During development, muscle progenitors are exposed to intrinsic sources of DNA damage (e.g., oxidative stress, S-phase intrinsic DNA damage) generated by their high mitotic activity, and MyoD plays a unique role among the muscle bHLH proteins as a target of DNA damage-activated signaling.Citation34 In this context, the coordination between cell cycle, DNA repair and activation of the differentiation program is essential to prevent the formation of genetically unstable myofibers carrying unrepaired DNA lesions, which could not otherwise be repaired in terminally differentiated nuclei.Citation35 We therefore speculate that the failure to execute this program might lead to an accumulation of unrepaired DNA lesions. The resulting genomic instability could predispose to “accelerated” aging phenotypes of skeletal muscle and might reveal a previously unappreciated developmental origin of sarcopenia. Likewise, during post-natal life, the differentiation checkpoint triggered in activated satellite cells can control the genomic integrity of myofibers during muscle repair or physiological myonuclear turnover and can therefore be implicated in the maintenance of muscle homeostasis along the life span.
Materials and Methods
Cell culture and treatments.
The murine C2C12 and C2C7 skeletal muscle cell lines were cultured in growth medium (GM; Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM glutamine). Normal Human Skeletal Muscle Myoblasts (HSMBs) was purchased from Clonetics® and cultured in Basal Medium with SingleQuots® as described in the data sheet (Lonza). Muscle differentiation was induced, exposing cells to differentiation medium (DM; Dulbecco's modifed Eagle's medium supplemented with 2% horse serum).
Single muscle fibers with associated satellite cells were isolated as described in reference Citation36. Briefly, the hind limb muscles were digested with collagenase, and single myofibers were plated on matrigel (Sigma, 1 mg/ml ECM gel) coated dishes in GM1 (DMEM supplemented with 10% horse serum (Gibco), 0.5% chick embryo extract (MP biomedicals) and penicillin-streptomycin (Gibco) at 37°C). Three days later the medium was replaced with proliferation medium (GM2-20% FBS, 10% horse serum, 1% chick embryo extract in DMEM) to promote proliferation of detached cells (delaminated satellite cells). After 4–5 d, the cells were allowed to differentiate, replacing the medium with differentiation medium (DM-2% HS and 0.5% chick embryo extract in DMEM).
Genotoxic treatments were carried by incubating cells in GM for 12–16 h to the following DNA damaging agents: 0.4 µM Doxorubicin, 0.5 µM Etoposide, 75 µM MMS; when indicated, cells were pretreated 30 min with 5 mM caffeine or 1 µg/ml IGF-1 before drug exposure. The acute treatments were carried for 1 h with the following dosage: 3 µM Doxorubicin, 10 µM Etoposide, 250 µM MMS. After drug exposure, cells were incubated in DM for 24 up to 72 h.
Alkaline comet analysis.
C2C12 cells were treated with 250 µM MMS, 10 µM Etoposide and 3 µM Doxorubicin for 1 h then shifted in differentiation medium and harvested at different time points. DNA breaks and repair kinetics were measured as previously described in reference Citation37 with minor modifications. Single cells were analyzed with “TriTek CometScore version 1.5” software. The tail moment was used as measure of DNA damage. One hundred cells for each experimental point were scored.
Isolation of total RNA and microarray analysis.
HSMBs were cultured in Basal Medium with SingleQuots,® as described in the data sheet (Lonza), and then treated for 18 h with Doxorubicin 0.4 µM prior exposure to differentiation medium (DM) for 24 h. Total RNA was extracted with Trizol,® as described in the data sheet (Invitrogen).
RNA (500 ng) was reverse-transcribed by M-MLV reverse transcriptase. The transcripts were labeled with biotin using an RNA amplification kit (Ambion). The cDNA samples were mixed with a Hyb E1 hybridization buffer containing 37.5% (w/w) formamide. The hybridization mix was dispensed on the Sentrix Human Ref-8 BeadChip (Illumina) containing 24,000 transcripts of the 22,000 genes represented in the consensus Reference Sequence (RefSeq) human genome database. Hybridization was performed for 18 h at 55°C. Array chips were then washed with an E1BC solution, then with 100% ethanol and, lastly, with the E1BC solution again. The chips were blocked with an E1 blocking buffer followed by staining with streptavidin-Cy3, washing with the E1BC solution and drying. Array chips were scanned using a BeadArray Reader (Illumina). The resulting images were analyzed using the BeadStudio image processing software (Illumina). The chip contained 30 to 40 beads with the attached oligonucleotide DNA corresponding to an individual gene of the RefSeq database. Differentially expressed transcripts resulting from the comparison between the Doxorubicin-treated sample and the control sample (≥ 2-fold) were divided into upregulated and downregulated. Both subsets of genes were then analyzed for the presence of overrepresented GO categories (Biological Process), GeneGo process Networks and GeneGo Pathway Maps using MetaCore™ software from GeneGo Inc. Gene expression of XPC DDB2 and p21 was validated by RT-PCR using the following primers.
Human p21:
FWD: TGT CAC TGT CTT GTA CCC TTG
REV: GGC GTT TGG AGT GGT AGA A
Human XPC:
FWD: GTC TCT ACA GCC AAT TCC TCT G
REV: CCT TTG CTG GTC TTT GGT TTG
Human DDB2:
FWD: GGC TGC AAG ACT TTA AAG GC
REV: ACA TCC AGG CTA CAA AAC CAG.
Protein gel blot and immunofuorescence.
C2C12 mouse cell line were treated with 0.4 µM Doxorubicin and MMS 75 µM for 16 h; when indicated, cells were pretreated with 5 mM caffeine and IGF-1 before genotoxic agents exposure. After drug treatments, cells were shifted in DM for 48 h. Proteins were extracted with Ripa buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 1% Np40, 1 mM EDTA), separated on polyacrilamyde gel and transferred to nitrocellulose filters. The following primary antibodies were used to detect endogenous protein level: MF20 mouse monoclonal antibody to detect Myosin Heavy Chain (MyHC), monoclonal antibodies against Myogenin (F5D), MyoD antibody (M-318 rabbit polyclonal Santa Cruz), phospho-p53 Ser15 (Cell Signaling), Tubulin (Ab4 from NeoMarkers). Primary antibodies were visualized with the ECL (Amersham) chemioluminescent kit following the manufacture's instruction.
For immunostaining, HSMBs and satellite cells were plated on glass coverslips; when indicated, cells were treated with genotoxic agents as described above. After DNA damage exposure, cells were shifted in DM. After 4 days, cells were fixed with 3.7% formaldehyde and permeabilized 10 min with PBS supplemented with 0.2% Triton. Single or double fluorescence were performed with the following primary antibodies (Ab): MoAb MF20 for Myosin Heavy Chain (MyHC), rabbit polyclonal anti total NbsI (Novus), rabbit polyclonal anti phospho 139 H2AX (Upstate). We used rodamine-conjugated goat anti-mouse IgG and fluoresceine-conjugated goat anti-rabbit IgG secondary antibodies (Jackson Immunoreserch) to detect the primary Ab, according to manufacture's instructions. Nuclei were visualized by 4′,6′-diamino-2-phenylindole (DAPI).
RNA extraction and RT-PCR.
Total RNA was extracted with Trizol (Invitrogen) according manufacturer instructions. 0.5–1 µg of RNA was retrotranscribed using the Taqman reverse transcription kit (Applied Biosystems). Real-time quantitative PCR was performed to analyze relative gene expression levels using SYBR Green Master mix (Applied Biosystems) and following manufacturer indications. Primers sequences are as follows:
Human mck:
Fwd: GGC ACA ATG ACA ACA AGA GC
Rev: GAA AAG AAG AGG ACC CTG CC
Human myogenin:
Fwd: GCC ACA GAT GCC ACT ACT TC
Rev: CAA CTT CAG CAC AGG AGA CC
GAPDH:
Fwd: CAC CAT CTT CCA GGA GCG AG
Rev: CCT TCT CCA TGG TGG TGA AGA C.
Chromatin inmunoprecipitation (ChIP).
ChIP assay on C2C12 was performed using the following antibodies: anti-acetylated histone 3 (Upstate), MyoD (Santa Cruz SC-760), H3-K4 tri-methylation (Millipore). Normal rabbit IgG (Santa Cruz, SC-2027) antibody was used as a control. Real-time PCR was performed on input samples and equivalent amounts of inmunoprecipitated material using the SYBR Green Master Mix (Applied Biosystems). Relative recruitment is calculated as the amount of amplified DNA normalized to input and relative to values obtained after normal rabbit IgG inmunoprecipitation, which were set as the background (one unit).
Primers used were as follows:
Mouse myogenin promoter:
Fwd: TGG CTA TAT TTA TCT CTG GGT TCA TG
Rev: GCT CCC GCA GCC CCT
Mouse mck enhancer:
Fwd: AGG GAT GAG AGC AGC CAC TA
Rev: CAG CCA CAT GTC TGG GTT AAT
Human myogenin promoter:
Fwd: GCC ATG CGG GAG AAA GAA G
Rev: AGC CAA CGC CAC AGA AAC C
Human mck enhancer:
Fwd: CCT TGC CCT GAG TTT GAA TCT C
Rev: GGC AGT CTA ACC CCA GAA ACC.
Cytofuorimetric analysis.
For cell cycle analysis, C2C7 skeletal muscle cells were treated 16 h with different DNA damaging agents and then shifted in DM for 24 h. Cells were collected and than stained for 30 min at 37°C with a solution containing propidium iodide at 100 mg/ml, RNase at 200 mg/ml and 0.2% Triton X-100 and analyzed with an EPICS XL cytofluorimeter (Coulter).
Figures and Tables
Figure 1 Diverse genotoxic agents induce cell cycle arrest at different boundaries and display different kinetics of DNA repair. C2C7 mouse myoblasts were exposed to Methylmethanesulfonate (MMS) 75 µM, Doxorubicin (Dox) 0.4 µM and Etoposide (Eto) 0.5 µM before incubation in differentiation medium (DM) for 24 h. See scheme on top. (A) The cell cycle profle was analyzed by cytofuorimetric analysis in the conditions described above or in control (untreated) cells cultured in GM or shifted in DM for 18 h. (B) The kinetic of repair of the DNA lesions caused by the different genotoxins was monitored by comet assay at different time points (T) after shifting the cells in DM. Representative fields of no-treated cells (NT) MMS-, Eto- and Dox-treated cells at different time points. (C) Quantification of the repair in the same experiment shown in (B) by plotting the average of the tail moment (calculated using Tritek CometScore™) of at least 100 cells per experimental point. This experiment is representative of multiple, independent experiments, which consistently show the same pattern of repair for each genotoxic agent, despite of the variability of the values that is intrinsic to each comet assay. (D) Human myoblasts were treated as described above and the expression levels of myogenin at different time points after DNA damage were assessed by real time PCR.
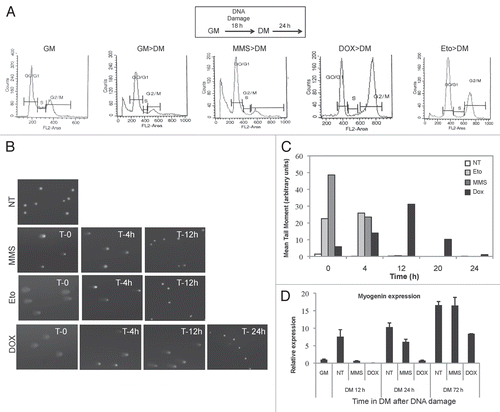
Figure 2 Transcription profle of myoblasts exposed to Doxorubicin treatment. Primary Human Skeletal Muscle Myoblasts (HSMBs) were exposed for 18 h to Doxorubicin 0.4 µM while growing in GM and then shifted in DM for additional 24 h. Total RNA was extracted with Trizol® as described in the data sheet (Invitrogen). A microarray was performed using Sentrix Human Ref-8 BeadChip (Illumina). In the panel is shown the analysis of the differentially expressed transcripts divided in upregulated (left) and downregulated (right). Bar charts represent the functional categories statistically more significant (≥ 2-fold change) in our groups of genes that include GO Biological Process, GeneGo Process Network and GeneGo Pathway Maps. Analyzed using MetaCore™ software from GeneGo Inc.

Figure 3 Different mechanisms are responsible for the differentiation checkpoint induced by genotoxic agents that activate the G1 or G2 checkpoints. C2C12 cells were untreated or treated with Doxorubicin and MMS, as shown in . When indicated, 5 mM caffeine was added 30 min before drug exposure. After genotoxic treatments cells, were shifted to differentiation medium for an additional 24 h. (A) ChIP analysis was performed to monitor MyoD binding and the H3K9/14 acetylation status (AcH3) to the chromatin of myogenin promoter or muscle creatin kinase (MCK) enhancer after MMS treatment. (B) ChIP analysis was performed as in (A) after Dox treatment. (C) The effect of caffeine treatment on the cell cycle was monitored by cytofluorimetric analysis. (D) The phosphorylation of p53 on Serine15 was assessed by protein gel blot on cells collected after genotoxins exposure. Tubulin was used as a loading control. (E) Expression levels of muscle-specific proteins (Myogenin and Myosin Heavy Chain-MyHC) were monitored by protein gel blot. Tubulin was used as a loading control.
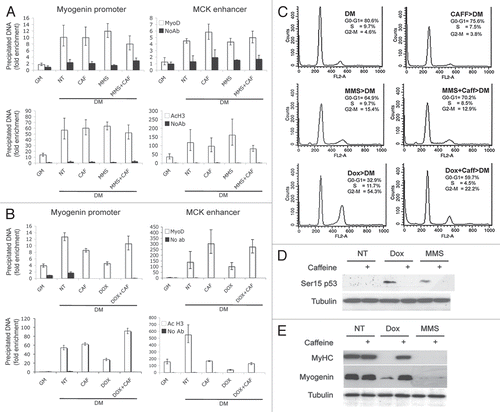
Figure 4 IGF-1 bypass the G1-differentiation checkpoint imposed by MMS treatment. Human Skeletal Muscle Myoblasts (HSMBs) were exposed to Dox and MMS in the presence or in the absence of 5 mM caffeine and 1ug/ml IGF-1 then shifted to DM. Cells were immunostained for MyHC and DAPI (A).The expression of myogenin was assessed by protein gel blot. Tubulin was used as a loading control (B). (C) C2C12 cells were treated as in (B), and ChIP analysis was performed to monitor the H3 tri-methylation in lysine 4 (MetK4) to the chromatin of muscle creatin kinase (MCK) enhancer.
Table 1 Analysis of cell cycle and inhibition of myogenic differentiation by DNA damaging agents
Additional material
Download Zip (4.1 MB)Acknowledgments
P.L.P. is Associate Telethon Scientist of the Dulbecco Telethon Institute (DTI). This work was partly supported by grants from Sanford Children's Health Research Center, AIRC and R01AR052779 from NIH/NIAMS. F.M. was supported by a AFM Ph.D. fellowship We thank Carmen D'Angelo, Silvia Di Cesare and Maria Teresa Cencioni for FACS analysis, and the Sanford-Burnham Medical Research Institute microarray facility.
References
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739 - 745; PMID: 18082599; http://dx.doi.org/10.1016/j.molcel.2007.11.015
- Hartwell LH, Kastan MB. Cell cycle control and cancer. Science 1994; 266:1821 - 1828; PMID: 7997877; http://dx.doi.org/10.1126/science.7997877
- Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature 2004; 432:316 - 323; PMID: 15549093; http://dx.doi.org/10.1038/nature03097
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 2004; 5:792 - 804; PMID: 15459660; http://dx.doi.org/10.1038/nrm1493
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 1078; PMID: 19847258; http://dx.doi.org/10.1038/nature08467
- Rouse J, Jackson SP. Interfaces between the detection, signaling and repair of DNA damage. Science 2002; 297:547 - 551; PMID: 12142523; http://dx.doi.org/10.1126/science.1074740
- Bartek J, Lukas J. DNA damage checkpoints: from initiation to recovery or adaptation. Curr Opin Cell Biol 2007; 19:238 - 245; PMID: 17303408; http://dx.doi.org/10.1016/j.ceb.2007.02.009
- Li L, Zou L. Sensing, signaling and responding to DNA damage: organization of the checkpoint pathways in mammalian cells. J Cell Biochem 2005; 94:298 - 306; PMID: 15578575; http://dx.doi.org/10.1002/jcb.20355
- Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 2002; 297:599 - 602; PMID: 12142537; http://dx.doi.org/10.1126/science.1074023
- Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 2010; 11:208 - 219; PMID: 20177396; http://dx.doi.org/10.1038/nrm2852
- Niculescu AB 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol 1998; 18:629 - 643; PMID: 9418909
- Harrington EA, Bruce JL, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 1998; 95:11945 - 11950; PMID: 9751770; http://dx.doi.org/10.1073/pnas.95.20.11945
- Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev 2005; 19:1040 - 1052; PMID: 15833913; http://dx.doi.org/10.1101/gad.1301205
- Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Stewart AF, et al. Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol 2005; 347:509 - 521; PMID: 15755447; http://dx.doi.org/10.1016/j.jmb.2005.01.041
- Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell 2006; 124:943 - 955; PMID: 16530042; http://dx.doi.org/10.1016/j.cell.2005.12.041
- Campisi J, Yaswen P. Aging and cancer cell biology, 2009. Aging Cell 2009; 8:221 - 225; PMID: 19627264; http://dx.doi.org/10.1111/j.1474-9726.2009.00475.x
- Simonatto M, Latella L, Puri PL. DNA damage and cellular differentiation: more questions than responses. J Cell Physiol 2007; 213:642 - 648; PMID: 17894406; http://dx.doi.org/10.1002/jcp.21275
- Puri PL, Bhakta K, Wood LD, Costanzo A, Zhu J, Wang JY. A myogenic differentiation checkpoint activated by genotoxic stress. Nat Genet 2002; 32:585 - 593; PMID: 12415271; http://dx.doi.org/10.1038/ng1023
- Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene 2000; 19:5643 - 5650; PMID: 11114745; http://dx.doi.org/10.1038/sj.onc.1203878
- Puri PL, Sartorelli V. Regulation of muscle regulatory factors by DNA-binding, interacting proteins and post-transcriptional modifications. J Cell Physiol 2000; 185:155 - 173; PMID: 11025438; http://dx.doi.org/10.1002/1097-4652(200011)185:2<155::AID-JCP1>3.0.CO;2-Z
- Polesskaya A, Rudnicki MA. A MyoD-dependent differentiation checkpoint: ensuring genome integrity. Dev Cell 2002; 3:757 - 758; PMID: 12479798; http://dx.doi.org/10.1016/S1534-5807(02)00372-6
- Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res 1990; 122:86 - 94; PMID: 2320728; http://dx.doi.org/10.2307/3577587
- Yokoi M, Masutani C, Maekawa T, Sugasawa K, Ohkuma Y, Hanaoka F. The xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem 2000; 275:9870 - 9875; PMID: 10734143; http://dx.doi.org/10.1074/jbc.275.13.9870
- Takedachi A, Saijo M, Tanaka K. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol 2010; 30:2708 - 2723; PMID: 20368362; http://dx.doi.org/10.1128/MCB.01460-09
- Batonnet-Pichon S, Tintignac LJ, Castro A, Sirri V, Leibovitch MP, Lorca T, et al. MyoD undergoes a distinct G2/M-specific regulation in muscle cells. Exp Cell Res 2006; 312:3999 - 4010; PMID: 17014844; http://dx.doi.org/10.1016/j.yexcr.2006.09.001
- Minemoto Y, Gannon J, Masutani M, Nakagama H, Sasagawa T, Inoue M, et al. Characterization of adria-mycin-induced G2 arrest and its abrogation by caffeine in FL-amnion cells with or without p53. Exp Cell Res 2001; 262:37 - 48; PMID: 11120603; http://dx.doi.org/10.1006/excr.2000.5072
- You Z, Bailis JM, Johnson SA, Dilworth SM, Hunter T. Rapid activation of ATM on DNA flanking doublestrand breaks. Nat Cell Biol 2007; 9:1311 - 1318; PMID: 17952060; http://dx.doi.org/10.1038/ncb1651
- Chakravarthy MV, Abraha TW, Schwartz RJ, Fiorotto ML, Booth FW. Insulin-like growth factor-I extends in vitro replicative life span of skeletal muscle satellite cells by enhancing G1/S cell cycle progression via the activation of phosphatidylinositol-3′-kinase/Akt signaling pathway. J Biol Chem 2000; 275:35942 - 35952; PMID: 10962000; http://dx.doi.org/10.1074/jbc.M005832200
- Zhang L, Kim M, Choi YH, Goemans B, Yeung C, Hu Z, et al. Diminished G1 checkpoint after gamma-irradiation and altered cell cycle regulation by insulin-like growth factor II overexpression. J Biol Chem 1999; 274:13118 - 13126; PMID: 10224065; http://dx.doi.org/10.1074/jbc.274.19.13118
- Seenundun S, Rampalli S, Liu QC, Aziz A, Palii C, Hong S, et al. UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogen-esis. EMBO J 2010; 29:1401 - 1411; PMID: 20300060; http://dx.doi.org/10.1038/emboj.2010.37
- Niedernhofer LJ. Tissue-specific accelerated aging in nucleotide excision repair deficiency. Mech Ageing Dev 2008; 129:408 - 415; PMID: 18538374; http://dx.doi.org/10.1016/j.mad.2008.04.010
- Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH. DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol 2008; 10:1241 - 1247; PMID: 18978832; http://dx.doi.org/10.1038/ncb1108-241
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair, genome stability and aging. Cell 2005; 120:497 - 512; PMID: 15734682; http://dx.doi.org/10.1016/j.cell.2005.01.028
- Innocenzi A, Latella L, Messina G, Simonatto M, Marullo F, Berghella L, et al. An evolutionarily acquired genotoxic response discriminates MyoD from Myf5, and differentially regulates hypaxial and epaxial myogenesis. EMBO Rep 2011; 12:164 - 171; PMID: 21212806; http://dx.doi.org/10.1038/embor.2010.195
- Narciso L, Fortini P, Pajalunga D, Franchitto A, Liu P, Degan P, et al. Terminally differentiated muscle cells are defective in base excision DNA repair and hypersensitive to oxygen injury. Proc Natl Acad Sci USA 2007; 104:17010 - 17015; PMID: 17940040; http://dx.doi.org/10.1073/pnas.0701743104
- Rosenblatt JD, Parry DJ, Partridge TA. Phenotype of adult mouse muscle myoblasts reflects their fiber type of origin. Differentiation 1996; 60:39 - 45; PMID: 8935927; http://dx.doi.org/10.1046/j.1432-0436.1996.6010039.x
- Fortini P, Pascucci B, Belisario F, Dogliotti E. DNA polymerase beta is required for efficient DNA strand break repair induced by methyl methanesulfonate but not by hydrogen peroxide. Nucleic Acids Res 2000; 28:3040 - 3046; PMID: 10931918; http://dx.doi.org/10.1093/nar/28.16.3040