Abstract
DNA strand break repair is essential for the prevention of multiple human diseases, particularly those which feature neuropathology. To further understand the pathogenesis of these syndromes, we recently developed animal models in which the DNA single-strand break repair (SSBR) components, XRCC1 and DNA Ligase III (LIG3), were inactivated in the developing nervous system. Although biochemical evidence suggests that inactivation of XRCC1 and LIG3 should share common biological defects, we found profound phenotypic differences between these two models, implying distinct biological roles for XRCC1 and LIG3 during DNA repair. Rather than a key role in nuclear DNA repair, we found LIG3 function was central to mitochondrial DNA maintenance. Instead, our data indicate that DNA Ligase 1 is the main DNA ligase for XRCC1-mediated DNA repair. These studies refine our understanding of DNA SSBR and the etiology of neurological disease.
Introduction
Normal cellular metabolic processes in mammals result in the production of reactive oxygen species (ROS). Although oxygen consumption is essential for life, the ROS byproduct is a potential cellular genotoxin, as ROS-mediated attack of DNA generates direct DNA single-strand breaks (SSBs), and, accordingly, DNA SSBs are one of the most common DNA lesions that occur within the cell.Citation1,Citation2 The substantial oxygen requirement for maintenance of the CNS renders neurons especially sensitive to DNA damage from the oxidative effects of metabolic ROS.Citation3–Citation5 Consequently, neurons require rapid and efficient DNA strand-break surveillance and repair mechanisms to deal with these types of lesions.
Repairing DNA Single-Strand Breaks
DNA SSBs can include a multitude of 3′- and 5′-end-modifications, which must be processed by specific DNA end-processing enzymes prior to gap-filling and resealing of the DNA backbone.Citation6–Citation8 DNA SSBs can occur directly to DNA via ROS-induced deoxyribose breakdown or abortive topoisomerase 1 (Top1)-DNA intermediates (Top1-3′-DNA) that form Top1-SSBs upon collision with RNA polymerase II or due to proximal secondary DNA damage.Citation9–Citation12 Indirect DNA SSBs can also occur as an intermediate of base excision repair (BER) due to enzymatic incision at an apurinic-apyrimidinic (AP) site by APE1 or DNA glycoslase.Citation9
DNA SSBs are repaired utilizing a dedicated repair pathway () that centrally involves XRCC1 and components of the BER pathway.Citation9,Citation13 Detection of direct SSBs is mediated by poly(ADP-ribose) polymerase (PARP), which acts to post-translationally modify the XRCC1 scaffolding protein for recruitment to the DNA break site. XRCC1 is a key factor for DNA SSBR, as it orchestrates a variety of enzymes involved in DNA end-processing (TDP1, APTX, APE1, PKNP), DNA Polβ-mediated short-patch gap-filling (single-nucleotide insertion) and DNA nick sealing (ligation), generally considered to be via XRCC1-bound DNA ligase IIIα (LIG3). However, in some instances, DNA nick gap-filling may continue for 2–12 nucleotides (long-patch polymerization) via Polβ/δ/ε, thereby requiring the Flap endonuclease 1 (FEN1) to remove the resultant displaced nucleotides followed by DNA ligation via PCNA-bound DNA ligase 1 (LIG1).Citation6,Citation14
Human syndromes that feature pronounced neuropathology can arise from defects in end-processing enzymes.Citation6,Citation15–Citation17 For example, spinocerebellar ataxia with axonal neuropathy (SCAN1) and ataxia with oculomotor apraxia 1 (AOA1) are syndromes that are associated with defective single-strand break repair (SSBR) and feature ataxia linked to cerebellar degeneration and neuropathy.Citation18,Citation19 SCAN1 results from disruption of tyrosyl-DNA phosphodiesterase 1 (TDP1), an enzyme that is required for the end-processing of 3′-phosphoglycolate and 3′-DNA-Top1 moieties.Citation20,Citation21 Lymphoblastoid cells derived from SCAN1 individuals are defective in their ability to repair DNA breaks after DNA SSB-inducing agents, such as peroxide (H2O2) or the Top1 poison camptothecin (CPT), while DNA double-strand break repair (DSBR) responses are normal.Citation20,Citation22 Mice deficient in Tdp1 (Tdp1-/-) show cerebellar hypoplasia and defective DNA SSBR responses to similar SSB-inducing agents.Citation21 In AOA1, mutations in aprataxin (APTX), a nucleotide hydrolase, causes defects in the removal of 5′-adenylate-DNA intermediates that arise due to abortive DNA ligation reactions.Citation23,Citation24 AOA1 lymphoblastoid cells and neural cells from Aptx-deficient mice (Aptx-/-) are defective in DNA short-patch repair.Citation23–Citation25 More recently, individuals with mutations in polynucleotide kinase 3′-phosphatase (PNKP) have been shown to develop microcephaly with early-onset seizures and developmental delay (a syndrome termed MCSZ).Citation26 Cell lines from MCSZ patients are defective in their ability to repair peroxide- or CPT-induced DNA damage. Similarly, human A549 cells deficient in PNKP display genomic instability and sensitivity to a variety of DNA damaging agents.Citation27 Collectively, these syndromes highlight the importance of SSB resolution in avoiding neuropathology. Nonetheless, the mechanistic contribution of SSBR during neurogenesis and the maintenance of neural homeostasis remain ill-defined.
XRCC1-Dependent SSBR is Neuroprotective In Vivo
Understanding the roles of the SSBR/BER pathway in the mammalian nervous system requires the establishment of tractable models. However, germline inactivation of key SSBR factors in the mouse have not yielded insight, as they do not show overt neurodegeneration (as is the case in Aptx-/- and Tdp1-/- mice) or they result in early embryonic lethality (Xrcc1-/-, Ape1-/-, Polβ -/-, Lig1-/- and Lig3-/- mice).Citation28–Citation34 Therefore to understand the etiology of SSBR deficiency as it relates to neurodegenerative disease, we produced a conditional Xrcc1 allele using Nestin-cre to drive Xrcc1 deletion in the developing murine nervous system and generated Xrcc1Nes-cre mice.
Neuraxis-wide Xrcc1 deficiency led to pronounced neurological dysfunction that was characterized by the loss of cerebellar interneurons and hippocampal dysfunction associated with a progressive seizure-like phenotype.Citation35 In vivo and in vitro analyses of Xrcc1Nes-cre mice revealed that widespread DNA damage accumulated throughout the nervous system. Alkali comet analysis of Xrcc1Nes-cre neurons and astrocytes showed deficient DNA repair in response to a variety of DNA SSB-inducing agents, including ionizing radiation (IR), H2O2, CPT and methyl methanesulphonate (MMS, an alkylating agent). Xrcc1 deficiency sensitized Pax2-immunopositive cerebellar interneuron progenitors to undergo DNA damage-induced, p53-mediated cell cycle arrest, resulting in a failure to populate the cerebellar molecular layer with basket, stellate and Golgi interneurons.Citation35 Furthermore, elsewhere in the cerebellum, Xrcc1-deficiency resulted in progressive DNA damage-induced p53-mediated granule neuron apoptosis and disrupted hippocampal homeostasis, likely causing the observed seizures in these mice. The data from the Xrcc1Nes-cre mouse have augmented our understanding of the biology of SSBR/BER and are relevant to understanding the etiology and neuropathology associated with defective DNA SSBR responses.
XRCC1 and LIG3 Are Tightly Associated in the Cell Nucleus
Efficient DNA SSBR requires XRCC1-mediated assembly of the repair machinery, and the final step in DNA nick repair requires a DNA ligase to reseal the DNA backbone. Because XRCC1 and LIG3 are tightly associated, this ligase has been assumed to fulfill this requirement. XRCC1 and LIG3 can be co-immunoprecipitated from whole cell extracts, and recombinant XRCC1 and LIG3 form a strong interaction that is resistant to high salt concentrations.Citation36–Citation39 Furthermore, Xrcc1 and Lig3 germline knockout mice die at a similar early embryonic stage.Citation32,Citation33 LIG3 protein levels and activity have also been shown to be dependent on Xrcc1 protein expression, as Xrcc1Nes-cre CNS neural tissue and Xrcc1-deficient Chinese hamster ovary (CHO) cell lines (EM7, EM9, EM11-C11 and EM-C12) have markedly reduced LIG3 protein levels and defective DNA substrate ligation activity.Citation36,Citation39,Citation40 Given the close biochemical relationship between XRCC1 and LIG3, the substantial DNA repair deficiency after XRCC1 inactivation and the essential requirement for ligation as a final step during repair, LIG3 activity is potentially critical during XRCC1-mediated SSBR.
LIG3 is Also Present in the Mitochondria
A mitochondrial isoform of LIG3 (mtLIG3) is also present in the cell. MtLIG3 is encoded from the single nuclear LIG3 gene via an alternate 5′-exon encoding an N-terminal mitochondrial targeting sequence.Citation41 Notably, mitochondrial extracts derived from Xrcc1-deficient EM9 CHO cells display intact ligase activity, while BER-dependent ligase activity is absent in the corresponding nuclear extracts.Citation42 Antisense-mediated reduction of mtLIG3 in HT1080 cells compromised mitochondrial DNA (mtDNA) integrity, resulting in reduced mtDNA levels and increased mtDNA nicks.Citation43 Thus, LIG3 can function in both the nucleus and mitochondria, but its stability is only dependent upon XRCC1 in the nucleus. This suggests that LIG3 functions in both nuclear DNA repair and in maintaining mitochondrial integrity.
LIG3 Inactivation Results in Neuropathology Distinct to XRCC1 Loss
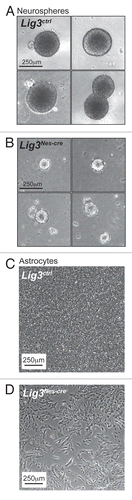
Given the DNA damage-associated neuropathology present in the Xrcc1Nes-cre mice, we reasoned that much of the damage was a failure to repair strand breaks due to Lig3 loss. To establish if this was the situation and to determine the DNA repair role of Lig3, we generated a conditional Lig3 allele and inactivated this gene throughout the developing nervous system using Nestin-cre.Citation44 We noted striking biological differences between Lig3Nes-cre and Xrcc1Nes-cre mice. Compared to Xrcc1Nes-cre mice that developed a seizure-like phenotype by three months of age, Lig3Nes-cre mice became profoundly ataxic and growth retarded by two weeks of age and failed to survive beyond postnatal day 20. Additionally, the Lig3-deficient cerebellum was substantially smaller than its Xrcc1-deficient counterpart and was characterized by pronounced developmental abnormalities associated with extensive granule cell loss. However, in contrast to Xrcc1 loss, the Lig3Nes-cre molecular layer displayed a normal complement of cerebellar interneurons. This indicates that the Xrcc1-mediated DNA repair pathway that prevents damage accumulation in the interneuron progenitors, leading to failed neurogenesis, occurs independently of Lig3. Furthermore, unlike Xrcc1Nes-cre mice, we found that cortical and cerebellar Lig3Nes-cre oligodendrocytes numbers were markedly reduced. Primary astrocytes or neurospheres derived from Lig3Nes-cre tissues display pronounced cell growth defects compared to wild-type (WT) and Xrcc1Nes-cre counterparts (). These data highlight the differing neuropathological bases for the observed phenotypes in these mice, and, despite the close biochemical relationship between XRCC1 and LIG3, the biological roles for these factors within the CNS differ greatly.
Mitochondrial Lig3 Preserves mtDNA in the Murine CNS
Because of the differences between inactivation of Xrcc1 and Lig3 in the mouse nervous system, we considered that disruption of mtLig3, rather than nuclear Lig3, underpinned the striking Lig3Nes-cre phenotype. This notion is consistent with the broad spectrum of phenotypes found in human syndromes associated with mtDNA defects.Citation45–Citation48 We also observed that media from Lig3Nes-cre primary astrocyte cultures quickly turned yellow, reflective of lactic acid accumulation in the media due to defective mitochondrial oxidative respiration. Subcellular analysis using mitotracker-Red and picogreen stains, combined with cellular oxygen consumption assays, confirmed that Lig3-deficient cells accumulated profound mitochondrial defects.Citation44 Analysis of Lig3Nes-cre tissues by electron microscopy (EM) and fluorescent immunostaining techniques also indicated substantial mitochondrial deficiency. These data demonstrated that, unlike Xrcc1Nes-cre mice, the pronounced neuropathology observed in Lig3Nes-cre mice was due to mitochondrial dysfunction. Furthermore, as mitochondrial Lig3 expression is independent of Xrcc1, the difference between the Lig3Nes-cre and Xrcc1Nes-cre mouse models highlights the mechanistic differences by which Lig3 and Xrcc1 promote and maintain neuronal homeostasis. As LIG3 is the only DNA ligase expressed in the mitochondria, its loss, especially in high oxygen-consumptive cells, such as neurons, would be dire, consistent with the neuropathology observed in Lig3Nes-cre mice.
While a detailed understanding of mtLIG3 function is unavailable, LIG3 is likely to have critical roles in both mtDNA replication and repair. It is generally accepted that XRCC1-independent BER mediates repair of mtDNA nicks.Citation49–Citation54 In addition to mtLIG3 as the only mitochondrial DNA ligase, DNA polymerase γ (Polγ) is the sole mitochondrial DNA polymerase and performs critical gap-filling functions that are normally attributed to DNA Pol β/δ/ε in nuclear DNA replication and SSBR.Citation55–Citation57 Defective Polγ activity resulting from inherited hypomorphic mutations results in a broad spectrum of human diseases.Citation45 More recently, BER pathway components, including APTX, TDP1 and FEN1, have been identified in mitochondria,Citation58–Citation60 further supporting the active requirement for BER in maintaining the integrity of mtDNA.Citation49–Citation54
Normal DNA SSBR activity in Lig3-deficient neural cells. While Lig3 loss clearly impacted mitochondrial homeostasis, its contribution to nuclear DNA repair was uncertain. To assess this aspect of Lig3 function, we performed DNA repair assays using primary cortical astrocyte cultures derived from Lig3Nes-cre mice. These assays were done in parallel with primary cell lines derived from Xrcc1Nes-cre, Aptx-/- and Tdp1-/- mice, lines previously shown to be defective in SSBR/BER.Citation21,Citation23–Citation25 Although Lig3 loss resulted in mitochondrial dysfunction and compromised cell growth, we were able to establish Lig3 primary astrocytes at early passage and grew these to quiescence for DNA repair studies. Surprisingly, Lig3-deficient cells displayed similar DNA repair activity to WT cells after a variety of genotoxins that induce different classes of DNA damage, including IR, H2O2, CPT, MMS. One clear exception was found after ultraviolet radiation (UV)-induced damage, which displayed a repair deficiency when compared to WT cells, although this repair defect was substantially less than after Xrcc1 loss.
These data were somewhat paradoxical, as Lig3 is considered important during SSBR repair.Citation9,Citation61,Citation62 It is likely that the reported studies linking Lig3 inactivation to repair deficiency may, in some cases, actually reflect Xrcc1 inactivation to promote Lig3 destabilization, as direct inactivation of LIG3 would compromise cellular viability because of mitochondrial dysfunction. Consistent with our data, normal repair is observed after oxidative damage when the XRCC1-LIG3 interaction is disabledCitation63 and knockdown of LIG3 via RNA interference results in repair deficiency after UV damage.Citation64 Furthermore, a companion study to our own used nuclear Lig3-deficient ES cells complemented with a mitochondrial version of Lig3 to maintain cellular viability and showed survival of these ES cells was similar to controls after an assortment of genotoxic agents.Citation65 These data strengthen the case that nuclear LIG3 is dispensable for survival after DNA damaging agents that require repair via the SSBR/BER pathway.
LIG1 is Important for DNA SSBR
As discussed above, DNA repair assays of Lig3-deficient primary neural cells suggest that Lig3 is generally dispensable for Xrcc1-mediated repair. Three mammalian DNA ligases have been identified: LIG1, LIG3 and LIG4.Citation62 While LIG3 has generally been considered to be the primary SSBR ligase because it associates with XRCC1, a proportion of DNA SSBs undergo repair that utilizes a variation on SSBR/BER termed long-patch repair, wherein PCNA-bound LIG1 performs the final DNA nick resealing event.Citation66,Citation67 LIG1 also functions during DNA replication in ligating Okazaki fragments within the lagging strand at DNA replication forks.Citation68 LIG4 functions in concert with XRCC4 as a key factor during non-homologous end-joining of DNA DSBsCitation69 and is not considered important for SSBR.Citation66 Thus, a central question arising from our studies on Lig3 inactivation is what ligase is critical for SSBR/BER? To address this, we used lentiviral-delivered shRNAs to knockdown Lig1, Lig3 and Lig4, both individually and in combination, for DNA repair studies in quiescent primary mouse embryonic fibroblasts. We achieved greater than 90% target knockdown of each ligase and measured DNA repair in these cells after a variety of DNA damage-inducing agents. Our analysis showed that like Lig3Nes-cre cells, shLig3 (Lig3-deficient) cells had DNA repair rates similar to control cells. Lig4 loss had minimal impact on SSBR activity, as previously reported.Citation66 However, our analyses showed that Lig1 was the key functional SSBR ligase in vivo, as shLig1 cells accumulated/retained the highest proportion of DNA breaks compared to control, shLig3 and shLig4 cells. Nevertheless, compared to the repair deficiency of Tdp1-/- quiescent cells after IR, H2O2 and MMS treatment, Lig1 inactivation did not result in the same magnitude of DNA repair deficiency. However, cells deficient for both Lig1 and Lig3 (shLig1/shLig3) accumulated/retained more DNA breaks than shLig1 cells alone and at levels comparable to Tdp1-deficient cells, thereby indicating that Lig1 and Lig3 coordinate the full breadth of DNA end-ligation events in SSBR. Thus, this approach provided the identification of a functional contribution for Lig3 during nuclear DNA SSBR.
Biological Roles for LIG1 and LIG3 in DNA SSB Repair?
The data outlined above serves to focus on the relative in vivo contribution for the mammalian ligases during specific types of DNA repair. However, we still do not know the specific biological roles of nuclear LIG3 in the context of DNA repair and, from a disease perspective, its role in neural homeostasis. As the Lig3Nes-cre mice succumb to mitochondrial defects around two weeks of age, this mouse model is not suited to address this question. There is a great deal of speculation suggesting that inefficient SSBR leads to age-related neurological decline;Citation14,Citation16 however, the lack of tractable biological models has prevented any sort of definitive study addressing this question. One potentially useful model to address nuclear Lig3 function would involve generating a mouse that exclusively expresses mitochondrial Lig3. This would overcome the deleterious effects of mtLig3-deficiency attributed to the Lig3Nes-cre mouse and facilitate analysis of the consequences of nuclear Lig3 deficiency.
Another related question is whether conditional loss of Lig1 alone or together with nuclear Lig3 phenocopies the neurological defects observed in Xrcc1Nes-cre mice. A similar approach in studying Lig1 in the context of the CNS in vivo would be useful (i.e., generation of Lig1Nes-cre mice), as Lig1 is required for cellular viabilityCitation70 and Lig1-deficient mice develop hematopoietic defects and die during embryonic development.Citation28,Citation34
Should Lig1Nes-cre mice share common neuropathology with Xrcc1Nes-cre mice, it would support a direct biochemical relationship between XRCC1 and LIG1 through protein-protein interaction or via PCNA or DNA Polβ.Citation67 Whatever the situation, generation of a conditional Lig1 allele will facilitate understanding the relative roles during tissue homeostasis and DNA repair of the three mammalian ligases.
Figures and Tables
Figure 1 DNA single-strand break repair. Resolution of DNA single-strand breaks involves specialized factors that process and repair specific types of breaks. Direct DNA breaks (which may arise from radiation, oxidation or abortive Top1-DNA intermediates) undergo detection by PARP, which signals the breaksite through poly-ADP ribosylation of XRCC1 and DNA end-processing factors. The formation of ADP-ribose polymers on these proteins serves to augment affinity and facilitate protein complex recruitment to the DNA breaksite. Damaged 5′- and 3′-end DNA termini undergo end-modification by DNA processing factors to generate 5′- end phosphate and 3′-end hydroxyl termini, which allows DNA polymerase to replace the missing nucleotide(s) and DNA ligase to restore integrity of the DNA strand backbone. DNA short-patch repair involves integration of a single nucleotide via DNA polymerase β into the damaged strand and DNA ligation by XRCC1-bound LIG3. Indirect DNA breaks arise from enzymatic excision of apurinic-apyrimidinic (AP) bases (which can occur from base alkylation) via APE1 or DNA glycosylases. Although this substrate can undergo XRCC1-mediated end-processing, followed by DNA short-patch repair, certain classes of damage undergo long-patch DNA repair, whereby DNA polymerase δ and ε generate an extended nucleotide polymer (2–12 nucleotides) to replace DNA bases at and proximal to the DNA break site. Flap-endonuclease 1 (FEN1) cleaves the extraneous DNA flap, and the intact DNA backbone is ligated via PCNA-bound LIG1. The shaded box represents the aspect of SSBR involving LIG3, which is now less clear based on new dataCitation44,Citation65 discussed in this paper.
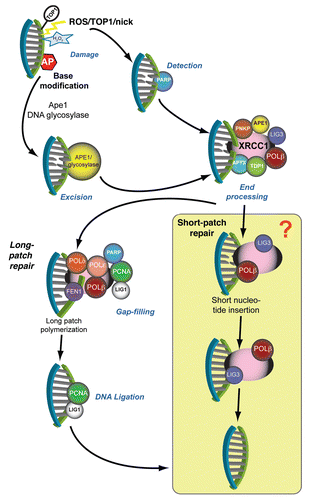
Figure 2 Primary Lig3Nes-cre neural cell lines have growth defects. Photomicrographs of primary neurospheres derived from (A) Lig3ctrl and (B) Lig3Nes-cre neural stem cells isolated from E14.5 mouse embryos. Lig3Nes-cre neurospheres are significantly smaller and display reduced cellularity compared to WT (Lig3ctrl) counterparts. (C) Primary astrocytes isolated from WT P3 mice form a tight confluent monolayer five days post-isolation. (D) In contrast, Lig3Nes-cre astrocytes cultures are growth delayed.
Acknowledgments
We would like to thank Dr. Youngsoo Lee for helpful discussions in preparing this manuscript. P.J.M. is supported by the National Institutes of Health (NS-37956, CA-21765), a Cancer Center Support Grant (P30 CA21765) and the American Lebanese and Syrian Associated Charities of St. Jude Children's Research Hospital. S.K. is a Neoma Boadway AP Endowed Fellow.
Related Research Data
References
- Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair (Amst) 2003; 2:955 - 969
- Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709 - 715
- Lombard DB, Chua KF, Mostoslavsky R, Franco S, Gostissa M, Alt FW. DNA repair genome stability and aging. Cell 2005; 120:497 - 512
- Barzilai A. The contribution of the DNA damage response to neuronal viability. Antioxid Redox Signal 2007; 9:211 - 218
- Chen L, Lee HM, Greeley GH Jr, Englander EW. Accumulation of oxidatively generated DNA damage in the brain: a mechanism of neurotoxicity. Free Radic Biol Med 2007; 42:385 - 393
- McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 2007; 8:37 - 55
- Wilson DM 3rd. Processing of nonconventional DNA strand break ends. Environ Mol Mutagen 2007; 48:772 - 782
- Wilson DM 3rd, Mattson MP. Neurodegeneration: nicked to death. Curr Biol 2007; 17:55 - 58
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet 2008; 9:619 - 631
- Kretzschmar M, Meisterernst M, Roeder RG. Identification of human DNA topoisomerase I as a cofactor for activator-dependent transcription by RNA polymerase II. Proc Natl Acad Sci USA 1993; 90:11508 - 11512
- Merino A, Madden KR, Lane WS, Champoux JJ, Reinberg D. DNA topoisomerase I is involved in both repression and activation of transcription. Nature 1993; 365:227 - 232
- Wu HY, Liu LF. DNA looping alters local DNA conformation during transcription. J Mol Biol 1991; 219:615 - 622
- Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair (Amst) 2007; 6:695 - 711
- Wilson DM 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst) 2007; 6:544 - 559
- Katyal S, McKinnon PJ. DNA repair deficiency and neurodegeneration. Cell Cycle 2007; 6:2360 - 2365
- Katyal S, McKinnon PJ. DNA strand breaks, neurodegeneration and aging in the brain. Mech Ageing Dev 2008; 129:483 - 491
- McKinnon PJ. DNA repair deficiency and neurological disease. Nat Rev Neurosci 2009; 10:100 - 112
- Takashima H, Boerkoel CF, John J, Saifi GM, Salih MA, Armstrong D, et al. Mutation of TDP1, encoding a topoisomerase I-dependent DNA damage repair enzyme, in spinocerebellar ataxia with axonal neuropathy. Nat Genet 2002; 32:267 - 272
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, et al. The gene mutated in ataxia-ocular apraxia 1 encodes the new HIT/Zn-finger protein aprataxin. Nat Genet 2001; 29:189 - 193
- El-Khamisy SF, Saifi GM, Weinfeld M, Johansson F, Helleday T, Lupski JR, et al. Defective DNA single-strand break repair in spinocerebellar ataxia with axonal neuropathy-1. Nature 2005; 434:108 - 113
- Katyal S, el-Khamisy SF, Russell HR, Li Y, Ju L, Caldecott KW, et al. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J 2007; 26:4720 - 4731
- El-Khamisy SF, Hartsuiker E, Caldecott KW. TDP1 facilitates repair of ionizing radiation-induced DNA single-strand breaks. DNA Repair (Amst) 2007; 6:1485 - 1495
- El-Khamisy SF, Katyal S, Patel P, Ju L, McKinnon PJ, Caldecott KW. Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin. DNA Repair (Amst) 2009; 8:760 - 766
- Ahel I, Rass U, El-Khamisy SF, Katyal S, Clements PM, McKinnon PJ, et al. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006; 443:713 - 716
- Reynolds JJ, El-Khamisy SF, Katyal S, Clements P, McKinnon PJ, Caldecott KW. Defective DNA ligation during short-patch single-strand break repair in ataxia oculomotor apraxia 1. Mol Cell Biol 2009; 29:1354 - 1362
- Shen J, Gilmore EC, Marshall CA, Haddadin M, Reynolds JJ, Eyaid W, et al. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat Genet 2010; 42:245 - 249
- Rasouli-Nia A, Karimi-Busheri F, Weinfeld M. Stable downregulation of human polynucleotide kinase enhances spontaneous mutation frequency and sensitizes cells to genotoxic agents. Proc Natl Acad Sci USA 2004; 101:6905 - 6910
- Bentley D, Selfridge J, Millar JK, Samuel K, Hole N, Ansell JD, et al. DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability. Nat Genet 1996; 13:489 - 491
- Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase beta. EMBO J 2000; 19:1397 - 1404
- Ludwig DL, MacInnes MA, Takiguchi Y, Purtymun PE, Henrie M, Flannery M, et al. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat Res 1998; 409:17 - 29
- Xanthoudakis S, Smeyne RJ, Wallace JD, Curran T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc Natl Acad Sci USA 1996; 93:8919 - 8923
- Puebla-Osorio N, Lacey DB, Alt FW, Zhu C. Early embryonic lethality due to targeted inactivation of DNA ligase III. Mol Cell Biol 2006; 26:3935 - 3941
- Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, et al. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol 1999; 208:513 - 529
- Bentley DJ, Harrison C, Ketchen AM, Redhead NJ, Samuel K, Waterfall M, et al. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J Cell Sci 2002; 115:1551 - 1561
- Lee Y, Katyal S, Li Y, El-Khamisy SF, Russell HR, Caldecott KW, et al. The genesis of cerebellar interneurons and the prevention of neural DNA damage require XRCC1. Nat Neurosci 2009; 12:973 - 980
- Caldecott KW, Tucker JD, Stanker LH, Thompson LH. Characterization of the XRCC1-DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res 1995; 23:4836 - 4843
- Nash RA, Caldecott KW, Barnes DE, Lindahl T. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry 1997; 36:5207 - 5211
- Whitehouse CJ, Taylor RM, Thistlethwaite A, Zhang H, Karimi-Busheri F, Lasko DD, et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001; 104:107 - 117
- Caldecott KW, McKeown CK, Tucker JD, Ljungquist S, Thompson LH. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol Cell Biol 1994; 14:68 - 76
- Shen MR, Zdzienicka MZ, Mohrenweiser H, Thompson LH, Thelen MP. Mutations in hamster single-strand break repair gene XRCC1 causing defective DNA repair. Nucleic Acids Res 1998; 26:1032 - 1037
- Lakshmipathy U, Campbell C. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol Cell Biol 1999; 19:3869 - 3876
- Lakshmipathy U, Campbell C. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic Acids Res 2000; 28:3880 - 3886
- Lakshmipathy U, Campbell C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res 2001; 29:668 - 676
- Gao Y, Katyal S, Lee Y, Zhao J, Rehg JE, Russell HR, et al. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011; 471:240 - 244
- Chan SS, Copeland WC. DNA polymerase gamma and mitochondrial disease: understanding the consequence of POLG mutations. Biochim Biophys Acta 2009; 1787:312 - 319
- DiMauro S, Schon EA. Mitochondrial disorders in the nervous system. Annu Rev Neurosci 2008; 31:91 - 123
- Wallace DC, Fan W. The pathophysiology of mitochondrial disease as modeled in the mouse. Genes Dev 2009; 23:1714 - 1736
- Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet 2005; 6:389 - 402
- Gredilla R, Bohr VA, Stevnsner T. Mitochondrial DNA repair and association with aging—an update. Exp Gerontol 2010; 45:478 - 488
- LeDoux SP, Druzhyna NM, Hollensworth SB, Harrison JF, Wilson GL. Mitochondrial DNA repair: a critical player in the response of cells of the CNS to genotoxic insults. Neuroscience 2007; 145:1249 - 1259
- LeDoux SP, Wilson GL, Beecham EJ, Stevnsner T, Wassermann K, Bohr VA. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis 1992; 13:1967 - 1973
- Liu P, Demple B. DNA repair in mammalian mitochondria: Much more than we thought?. Environ Mol Mutagen 2010; 51:417 - 426
- Pinz KG, Bogenhagen DF. Efficient repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell Biol 1998; 18:1257 - 1265
- Weissman L, de Souza-Pinto NC, Stevnsner T, Bohr VA. DNA repair, mitochondria and neurodegeneration. Neuroscience 2007; 145:1318 - 1329
- Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol Biol Cell 2003; 14:1583 - 1596
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429:417 - 423
- Pinz KG, Shibutani S, Bogenhagen DF. Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J Biol Chem 1995; 270:9202 - 9206
- Liu P, Qian L, Sung JS, de Souza-Pinto NC, Zheng L, Bogenhagen DF, et al. Removal of oxidative DNA damage via FEN1-dependent long-patch base excision repair in human cell mitochondria. Mol Cell Biol 2008; 28:4975 - 4987
- Sykora P, Croteau DL, Bohr VA, Wilson DM 3rd. Aprataxin localizes to mitochondria and preserves mitochondrial function. Proc Natl Acad Sci USA 2011; 108:7437 - 7442
- Das BB, Dexheimer TS, Maddali K, Pommier Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc Natl Acad Sci USA 2010; 107:19790 - 19795
- Sleeth KM, Robson RL, Dianov GL. Exchangeability of mammalian DNA ligases between base excision repair pathways. Biochemistry 2004; 43:12924 - 12930
- Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: structural and functional insights. Annu Rev Biochem 2008; 77:313 - 338
- Breslin C, Caldecott KW. DNA 3′-phosphatase activity is critical for rapid global rates of single-strand break repair following oxidative stress. Mol Cell Biol 2009; 29:4653 - 4662
- Moser J, Kool H, Giakzidis I, Caldecott K, Mullenders LH, Fousteri MI. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell cycle-specific manner. Mol Cell 2007; 27:311 - 323
- Simsek D, Furda A, Gao Y, Artus J, Brunet E, Hadjantonakis AK, et al. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011; 471:245 - 248
- Mortusewicz O, Rothbauer U, Cardoso MC, Leonhardt H. Differential recruitment of DNA Ligase I and III to DNA repair sites. Nucleic Acids Res 2006; 34:3523 - 3532
- Tomkinson AE, Chen L, Dong Z, Leppard JB, Levin DS, Mackey ZB, et al. Completion of base excision repair by mammalian DNA ligases. Prog Nucleic Acid Res Mol Biol 2001; 68:151 - 164
- Montecucco A, Rossi R, Levin DS, Gary R, Park MS, Motycka TA, et al. DNA ligase I is recruited to sites of DNA replication by an interaction with proliferating cell nuclear antigen: identification of a common targeting mechanism for the assembly of replication factories. EMBO J 1998; 17:3786 - 3795
- Lee Y, McKinnon PJ. Responding to DNA double strand breaks in the nervous system. Neuroscience 2007; 145:1365 - 1374
- Petrini JH, Xiao Y, Weaver DT. DNA ligase I mediates essential functions in mammalian cells. Mol Cell Biol 1995; 15:4303 - 4308