Abstract
A-type lamins are emerging as regulators of nuclear organization and function. Changes in their expression are associated with cancer and mutations are linked to degenerative diseases -laminopathies-. Although a correlation exists between alterations in lamins and genomic instability, the molecular mechanisms remain largely unknown. We previously found that loss of A-type lamins leads to degradation of 53BP1 protein and defective long-range non-homologous end-joining (NHEJ) of dysfunctional telomeres. Here, we determined how loss of A-type lamins affects the repair of short-range DNA double-strand breaks (DSBs) induced by ionizing radiation (IR). We find that lamins deficiency allows activation of the DNA damage response, but compromises the accumulation of 53BP1 at IR-induced foci (IRIF), hindering the fast phase of repair corresponding to classical-NHEJ. Importantly, reconstitution of 53BP1 is sufficient to rescue long-range and short-range NHEJ. Moreover, we demonstrate an unprecedented role for A-type lamins in the maintenance of homologous recombination (HR). Depletion of lamins compromises HR by a mechanism involving transcriptional downregulation of BRCA1 and RAD51 by the repressor complex formed by the Rb family member p130 and E2F4. In line with the DNA repair defects, lamins-deficient cells exhibit increased radiosensitivity. This study demonstrates that A-type lamins promote genomic stability by maintaining the levels of proteins with key roles in DNA DSBs repair by NHEJ and HR. Our results suggest that silencing of A-type lamins by DNA methylation in some cancers could contribute to the genomic instability that drives malignancy. In addition, lamins-deficient tumor cells could represent a good target for radiation therapy.
Introduction
A-type lamins—lamins A/C—are nuclear intermediate filaments which arise from differential splicing of the LMNA gene. Despite being established as mere structural components of the nucleus, recent studies reveal involvement of A-type lamins in many nuclear processes, ranging from positioning of nuclear pore complexes, to DNA replication and repair, as well as regulation of gene transcription.Citation1–Citation3 The fact that over 300 mutations in the LMNA gene are associated with a wide variety of disease phenotypes—muscular dysthrophies, lipodystrophies, neuropathies and premature aging syndromes such as Hutchinson Gilford Progeria Syndrome (HGPS)—is a testament to the varied functions of lamins A/C.Citation4
In contrast to laminopathies, which are associated with mutations in lamins or lamin-associated proteins, a number of human malignancies feature alterations in the expression of A-type lamins.Citation5,Citation6 Immunohistochemical data from more than 600 patients with colorectal cancer showed that 70% of the tumors overexpressed lamins A/C and these patients were almost twice more likely to die from their disease than similarly staged patients with low nuclear lamin expression.Citation7 On the other hand, studies in human lung carcinoma demonstrated that A-type lamins, though normally expressed in non-small cell lung cancer, are weakly expressed or completely absent in the great majority of small cell lung cancers.Citation5,Citation8 Furthermore, silencing of the LMNA gene by promoter hypermethylation in nodal diffuse large B-cell lymphoma was shown to be a significant predictor of overall survival.Citation9
To elucidate how alterations in lamins A/C contribute to laminopathies, researchers have investigated the relationship between mutations in the LMNA gene and genomic instability. Fibroblasts from HGPS patients and from a mouse model of progeria exhibit increased basal DNA damage, chromosomal aberrations, increased sensitivity to DNA-damaging agents,Citation10,Citation11 and faster telomere attrition.Citation12,Citation13 Furthermore, ectopic expression of mutant forms of lamin A in HeLa cells leads to deficient phosphorylation of H2AX (γH2AX) in response to cisplatin or UV irradiation and mislocalization of ATR.Citation14 Collectively, these studies correlate LMNA mutations with genomic instability, characterized by defects in telomere homeostasis and the DNA damage response (DDR) pathway. Alterations in telomere biology and DDR are among the leading causes of genomic instability, and are clear contributors to aging and cancer. As such, the molecular basis of their relationship with A-type lamins is of particular importance, yet remains largely unknown.
We recently showed that complete loss of A-type lamins leads to genomic instability in the absence of exogenous DNA damage. Lmna−/− mouse embryonic fibroblasts (MEFs) exhibited increased basal levels of γH2AX, aneuploidy, increased frequency of chromosome and chromatid breaks, and defects in telomere structure, length and function.Citation15,Citation16 Interestingly, Lmna−/− MEFs were also characterized by defects in NHEJ of dysfunctional telomeres, an example of long-range DSBs processing. The role of A-type lamins in NHEJ of deprotected telomeres and the increased frequency of chromosomal breaks in Lmna−/− MEFs suggest that lamins deficiency might also hinder the ability of cells to repair DNA DSBs that arise during normal metabolic processes—replication fork stalling and generation of reactive oxygen species—or due to exogenous genotoxic insult to the cells.
In mammalian cells, DNA DSBs repair occurs mainly by NHEJ and HR.Citation17–Citation20 NHEJ is the predominant repair pathway of IR-induced DNA DSBs. It is faster and more efficient than HR, but at the expense of low fidelity. Analysis of kinetics of repair of IR-induced DNA DSBs has revealed two types of NHEJ.Citation21 Classical-NHEJ represents the fast component of DSBs rejoining and relies on DNA-dependent protein kinase (DNA-PK) and the XRCC4/DNA Ligase IV complex.Citation22,Citation23 Recently, SIRT6 was shown to be required for mobilization of the DNA-PK catalytic subunit to chromatin in response to DNA damage.Citation24,Citation25 Although DNA-PK deficiency retards repair of DNA DSBs, damage is eventually repaired by a slower operating mechanism.Citation26 An alternative-NHEJ pathway has been described in vitro and in vivo as a backup to classical-NHEJ that involves DNA end-resection and ligation of ends with microhomology.Citation21,Citation27,Citation28 HR is an error-free repair pathway that is active primarily during late S and G2 phases, where sister chromatids are available for use as homology templates.Citation17,Citation29 HR requires nucleolytic degradation of DNA ends to generate 3′-ended ssDNA, a process mediated by the MRN (Mre11/Rad50/Nbs1) complex, CtIP and BRCA1 proteins. The ssDNA generated by end-resection is rapidly bound by Replication Protein A (RPA), which removes secondary structures in ssDNA and allows the formation of the RAD51 nucleoprotein filament that drives DNA strand invasion and exchanges during HR.Citation30–Citation34
We previously found that loss of A-type lamins induced destabilization of 53BP1.Citation16 A whole body of evidence indicates that 53BP1 participates in long-range DNA end-joining processes, such as class switch recombination,Citation35,Citation36 V(D)J recombination,Citation37 and chromosome end-to-end fusions.Citation38 The role of 53BP1 in short-range DSBs repair is not clear, as its depletion results in only a minor decrease in the repair of IR-induced DNA DSBs.Citation36 Recent findings suggest that rather than being an essential protein for NHEJ of short-range DSBs, 53BP1 functions as an inhibitor of alternative mechanisms of DNA DSBs repair.Citation39,Citation40 For example, in the context of BRCA1 deficiency, 53BP1 accumulation at DSBs prevents the 5′ to 3′ resection of DNA ends required for HR.Citation40 Thus, 53BP1 deficiency could be the mechanism linking loss of A-type lamins to decreased long-range NHEJ of dysfunctional telomeres. It is also possible that destabilization of 53BP1 impacts on the ability of A-type lamins-deficient cells to repair short-range DNA DSBs.
Here, we determine how loss of A-type lamins and the associated 53BP1 deficiency affect long-range and short-range DNA DSBs repair. We demonstrate that A-type lamins are required for the repair of IR-induced short-range DNA DSBs by both NHEJ and HR. Cells deficient in A-type lamins display decreased accumulation of 53BP1 at IRIF, as well as defects in the fast-phase of DNA DSBs repair, corresponding to NHEJ. In addition, loss of A-type lamins inhibits HR by a mechanism that involves transcriptional downregulation of BRCA1 and RAD51. This novel link between A-type lamins and transcriptional regulation of BRCA1/RAD51 is mediated by the Rb family member p130, which forms a transcriptionally repressive complex with E2F4. Consistent with deficient DNA DSBs repair, lamins A/C deficient cells are significantly more sensitive to IR. Our findings provide two distinct mechanisms by which A-type lamins maintain DNA repair and the stability of the genome.
Results
A-type lamins are essential for fast-phase repair of IR-induced DNA double-strand breaks.
Given the role of A-type lamins in the stabilization of 53BP1 and the processing of long-range NHEJ processes such as deprotected telomeres, we hypothesized that lamins deficiency might also hinder the ability of cells to repair short-range DNA DSBs, such those induced by IR. One of the earliest responses to IR-induced DNA DSBs is phosphorylation of H2AX (γH2AX) in the surrounding chromatinCitation41 followed by recruitment of 53BP1 to the demarcated site.Citation42 To test if loss of A-type lamins alters the cellular response to IR, we monitored the formation of γH2AX and 53BP1 IR-induced foci (IRIF) in Lmna+/+ and Lmna−/− MEFs treated with 0.5 Gy of IR. Consistent with the decreased global levels of 53BP1 protein, we observed a profound and consistent decreased intensity of fluorescence of 53BP1 IRIF in Lmna−/− MEFs with respect to Lmna+/+ controls at all times post-IR tested (). Striking differences were observed at 30 min, 1 h and 2 h after IR. By 24 h, 53BP1 protein was dispersed throughout the nucleus in Lmna+/+ MEFs, mirroring the localization of the protein in cells that were not irradiated. In contrast, Lmna−/− MEFs displayed decreased 53BP1 intensity and changes in the nuclear distribution of the protein, which was not dispersed throughout but rather accumulated in a few large foci. In addition, protein gel blots performed in MEFs showed that the levels of 53BP1 increase upon radiation in both lamins-proficient and -deficient cells, however the levels of 53BP1 in Lmna−/− cells remain much lower than in wild-type cells (Fig. S1A). In contrast, the intensity of labeling of γH2AX foci was indistinguishable between Lmna+/+ and Lmna−/− MEFs throughout the time course (Fig. S1B). Furhermore, ATM-dependent phosphorylation of p53 at Ser15, a central event in DDR signaling, was not affected by the loss of A-type lamins (Fig. S1C). These results indicate that the loss of A-type lamins does not hinder the activation of the DDR pathway. However, analysis of the kinetics of formation and resolution of γH2AX foci showed the presence of unrepaired DNA damage 24 h post-irradiation in Lmna−/− cells, suggesting defects in the repair of short-range IR-induced breaks in these cells ().
To determine if loss of A-type lamins and the associated decreased accumulation of 53BP1 at IRIF impacts on short-range DNA DSBs repair, we performed neutral comet assays. Lmna+/+ and Lmna−/− MEFs were treated with 8 Gy of IR. At different times post-IR, cells were collected and the extent of DNA DSBs repair evaluated by single-cell gel electrophoresis.Citation43 Olive moment, a quantification of the amount of DNA and its distribution in the comet tail, is a measure of unrepaired DNA DSBs. Repair of DSBs after IR follows bimodal kinetics with fast and slow repair phases.Citation21 There is substantial evidence implicating classical-NHEJ as the major mechanism during the fast phase of repair and alternative-NHEJ and to a lesser extent HR, in the slow phase.Citation26,Citation44 This bimodal form of repair is clearly observed in Lmna+/+ MEFs, such that the fast phase occurred within 60 min post-IR, followed by a relatively slow phase of DSBs repair onwards. However, Lmna−/− MEFs did not display the fast-phase of repair. Instead, a shift toward a slower mechanism of repair was observed in these cells (). This indicates that DNA DSBs repair is compromised by the loss of A-type lamins.
To determine if 53BP1 could rescue the defective fast phase of repair in Lmna−/− MEFs, cells were retrovirally transduced with 53BP1 or EV control and neutral comet assays were performed. While Lmna−/− MEFs transduced with EV still presented defects in fast-phase repair, reconstitution of 53BP1 restored the normal kinetics of DNA DSBs repair ( and B). Furthermore, overexpression of 53BP1 in Lmna+/+ MEFs did not have any effect on DNA repair kinetics. These results were intriguing, since previous reports indicated that 53BP1 is not essential for the repair of short-range DNA DSBs.Citation36 Accordingly, depletion of 53BP1 in wild-type MEFs did not inhibit the fast-phase of repair (Fig. S2). These results indicate that 53BP1 deficiency in lamins A/C-deficient cells is not solely responsible for the observed defects in the fast-phase of repair. We conclude that the combined deficiency of A-type lamins and 53BP1 is responsible for the shift in the kinetics for DNA DSBs repair toward a slower operating mechanism. This slower form of repair could represent lower efficiency of classical-NHEJ or activation of an alternative mechanism of repair. In either case, the fact that reconstitution of 53BP1 restores normal kinetics of repair of IR-induced DNA DSBs, supports a role for 53BP1 in promoting classical-NHEJ repair and inhibiting alternative mechanisms of DNA repair which become prominent upon loss of A-type lamins.
In contrast to short-range DNA DSBs repair, the essential role of 53BP1 in long-range NHEJ has been clearly demonstrated.Citation35–Citation38 Thus, we hypothesized that the previously reported defects in the processing of dysfunctional telomeres upon loss of A-type laminsCitation15,Citation16 could be caused by the decrease in 53BP1 levels. To test this hypothesis, U2OS cells were retrovirally transduced with 53BP1 or an empty vector (EV) control followed by lentiviral transduction with a shRNA specific for depletion of A-type lamins (shLmna) or a shRNA control (shCtrl) (). Next, we retrovirally transduced cells with a dominant negative form of the telomere binding protein TRF2 (TRF2ΔBΔM) to induce telomere deprotection. To quantify the resulting end-to-end fusions, we scored metaphases based on four different categories of increasing chromosome fusions ranging from “less than five chromosomes fused” (category 1) to “more than half of the chromosomes fused” (category 4) (). In cells that express A-type lamins and have normal levels of endogenous 53BP1 (EV/shCtrl/TRF2ΔBΔM), 53% of metaphases were scored in category 1 and 31% in category 4 (). In contrast, lamins A/C-depleted cells (EV/shLmna/TRF2ΔBΔM) exhibited an overall decrease in the extent of end-to-end fusions (66% category 1 and only 14% category 4). Most importantly, reconstitution of 53BP1 into lamins A/C-depleted cells (53BP1/shLmna/TRF2ΔBΔM) resulted in a rescue of chromosome fusions (36% category 1 and 40% category 4). As a control, we monitored fusions in cells transduced with an empty vector instead of TRF2ΔBΔM (53BP1/shLmna/EV). As expected, 100% of metaphases belong to category 1, indicating that 53BP1 expression itself does not induce fusions (data not shown). We conclude that the effect of A-type lamins in NHEJ of dysfunctional telomeres is due to their ability to stabilize 53BP1 protein.
All together, our results indicate the loss of A-type lamins hinders the repair or short-range and long-range DNA DSBs (IR-induced breaks and dysfunctional telomeres, respectively) by destabilizing 53BP1 protein.
Loss of A-type lamins downregulates RAD51 and BRCA1, inhibiting homologous recombination.
Based on the theory of competition among DDR pathways and reports suggesting that 53BP1 is a potent HR inhibitor,Citation39,Citation40,Citation45,Citation46 we hypothesized that decreased 53BP1 in lamins A/C-depleted cells would lead to increased HR. To test this hypothesis we monitored HR proficiency by using a chromosomally integrated HR reporter substrate, DR-GFP, in MCF-7 cells.Citation47,Citation48 In this system, expression of GFP is the readout of successful HR during repair of I-SceI induced DNA DSBs. While depletion of A-type lamins in MCF-7 DR-GFP cells led to a substantial reduction in 53BP1 (), contrary to our expectations, we observed a 40% reduction in HR in these cells when compared with their shCtrl counterparts (). As a positive control, we found that inactivation of p53 by the viral oncogene E6 led to increased HR, as previously reported in reference Citation49 (Fig. S3A). Thus, the combined loss of A-type lamins and 53BP1 resulted in a phenotype that was inconsistent with only 53BP1 deficiency, suggesting that additional events during HR were affected.
To elucidate the molecular mechanisms behind HR defects, we monitored recruitment of RAD51, an essential HR protein, to IRIF. We treated lamins A/C proficient and deficient MCF-7 DR-GFP cells with 8 Gy of IR and performed immunofluorescence to detect RAD51 foci 6 h post-IR. We found a significant decrease in the formation of RAD51 foci, such that only 27% of shLmna cells scored positive for RAD51 IRIF, as opposed to 64% of the shCtrl cells ( and D). This decrease was consistent with a profound reduction in the global levels of the protein, as assessed by immunofluorescence () and by protein gel blot (). To ensure that our results were not cell type- or shRNA sequence-specific, we performed acute depletion of A-type lamins in wild-type MEFs, using a mouse-specific shRNA. Consistently, loss of A-type lamins led to a decrease in the levels of RAD51 protein and in the formation of RAD51 IRIF ( and G). These results demonstrate that A-type lamins are important regulators of RAD51 levels. Furthermore, neutral comet assays performed in MCF7 DR-GFP cells depleted of A-type lamins showed that these cells are also defective in NHEJ (Fig. S3B). Overall, our results indicate that A-type lamins are required for the maintenance of factors—53BP1 and RAD51—with key roles in the repair of DSBs by NHEJ and HR.
To determine if steps upstream from RAD51 recruitment to DSBs were affected by the loss of A-type lamins, we monitored the levels of two proteins with an essential role in HR-BRCA1 and RPA. As shown in , acute depletion of A-type lamins in MCF-7 DR-GFP cells led to a marked decrease in BRCA1 protein levels, but did not affect global RPA levels. However, the recruitment of RPA to DNA repair foci was inhibited in lamins-deficient cells. Furthermore, qRT-PCR on MCF-7 DR-GFP cells showed a significant decrease in transcripts of RAD51 and BRCA1 but not of RPA2 or 53BP1 (). A similar decrease in RAD51 transcript levels was also observed in MEFs depleted of A-type lamins (Fig. S3C). Altogether, these data demonstrate an unprecedented role for A-type lamins in the transcriptional regulation of two key factors in HR, BRCA1 and RAD51, in both mouse and human cells.
Downregulation of BRCA1 and RAD51 is mediated by p130/E2F4 repressor complexes.
Previous studies revealed that transcription of RAD51 and BRCA1 genes can be repressed by E2F4/p130 complexes that bind to E2F sites in their promoters.Citation50,Citation51 The co-regulation of BRCA1 and RAD51 observed upon loss of A-type lamins (), together with the fact that lamins deficiency induces profound decreases in pRb and p107 levels with only minor effects in p130,Citation16,Citation52 (Fig. S3D) led us to hypothesize that this E2F4/p130 repressive complex might participate in the repression of RAD51 and BRCA1 genes.
To test directly whether p130 was required for the transcriptional downregulation of RAD51 upon loss of A-type lamins, we depleted lamins in cells double null for pRb and p107 (DKO: Rb−/−;p107−/−) and in cells null for all 3 Rb family members (TKO: Rb−/−;p107−/−;p130−/−). Depletion of A-type lamins in DKO cells—proficient in p130—led to a marked decrease in RAD51 protein levels (). In contrast, depletion of A-type lamins in TKO cells did not result in downregulation of RAD51 levels, indicating that p130 is required for downregulation of RAD51 upon loss of A-type lamins. Consistent with a role for p130 in the regulation of RAD51 expression, TKO cells exhibit higher levels of RAD51 than wild-type or DKO MEFs.
To further investigate the role of E2F4/p130 in RAD51 repression in lamins A/C deficient cells, we determined if depletion of A-type lamins would promote formation of the p130/E2F4 repressive complex by performing co-immunoprecipitation studies. Total cell lysates from wild-type MEFs were subjected to immunoprecipitation with E2F4 antibody, and the presence of p130 monitored by protein gel blot. While depletion of A-type lamins did not affect the global levels of E2F4 ( and inputs), it promoted the formation of p130/E2F4 complexes, as shown by the increase in p130 co-immunoprecipitated with the E2F4 antibody ( and IPs). The same results were obtained upon depletion of A-type lamins in DKO cells, proficient in p130. Thus, we conclude that depletion of A-type lamins promotes the ability of p130 to form repressor complexes with E2F4, which in turn leads to downregulation of RAD51 and BRCA1, and results in defective repair of DNA DSBs by HR.
The p130/E2F4 repressor complex is active primarily during G0/G1 phases of the cell cycle. In addition, the levels of RAD51 and BRCA1 have been shown to fluctuate during the cell cycle, being reduced during G1 and upregulated at late G1, S, and G2 phases of the cycle. Here, we determined if depletion of A-type lamins induces a growth arrest in G1, which in turn would increase p130/E2F4 repressor complex activity and repression of RAD51 and BRCA1 genes. Monitoring the cell cycle profile, we found that depletion of A-type lamins does not lead to an accumulation of cells in G1 phase of the cell cycle, as assessed by FACS analysis (Fig. S4). These results indicate that the effect of depletion of A-type lamins on components of HR-mediated repair is not linked to cell cycle arrest.
Loss of A-type lamins increases radiosensitivity.
Radiation therapy is a common modality in the treatment of cancer. IR preferentially kills repair-compromised cells, which are unable to deal with the extensive DNA damage generated. To determine if loss of A-type lamins and the associated deficiency in 53BP1 and RAD51/BRCA1 affect sensitivity to IR, we performed colony formation assays. Lmna+/+ and Lmna−/− MEFs were treated with increasing doses of IR up to 6 Gy, and their clonogenic capability assessed after 10 d in culture. The cell survival curves shown in describe the relationship between the radiation dose and the proportion of cells that retain reproductive integrity. In line with severely compromised DNA repair, Lmna−/− MEFs were significantly more sensitive to IR than wild-type controls. Overall, our studies demonstrate that loss of A-type lamins hinders the repair of IR-induced DNA DSBs by NHEJ and HR promoting radiosensitivity. A-type lamins affect NHEJ and HR via distinct mechanisms. NHEJ is maintained through post-translational stabilization of the 53BP1 protein while HR is regulated through p130-mediated transcriptional regulation of RAD51 and BRCA1 ().
Discussion
Given the well established contribution of genomic instability to aging and cancer, identifying the molecular mechanisms involved in maintaining genomic integrity is of utmost importance. DNA DSBs repair, which occurs mainly by NHEJ and HR, is critical for maintaining genomic stability.Citation17,Citation19,Citation53–Citation57 Here, we demonstrate that the structural nuclear proteins A-type lamins preserve the integrity of the genome in part by maintaining the ability of cells to repair DNA DSBs by NHEJ and HR. We provide evidence that the mechanism which allows A-type lamins to promote long-range (dysfunctional telomeres) and short-range (IR-induced DNA DSBs) classical-NHEJ is the stabilization of 53BP1. In addition, we show that loss of A-type lamins leads to a severe compromise in HR. The mechanism behind this finding is the unprecedented role for A-type lamins in the transcriptional co-regulation of two key factors in HR, RAD51 and BRCA1, by means of formation of p130/E2F4 repressor complexes. Finally, we demonstrate that compromised DSBs repair in lamins A/C-deficient cells leads to increased sensitivity to IR. Overall, our study indicates that the main function of A-type lamins in DNA repair relies on their ability to stabilize 53BP1 and maintain transcription of BRCA1/RAD51 ().
The function of A-type lamins in the localization and stabilization of different proteins, including Rb family members, ING1 and ATR was previously established in references Citation14, Citation52 and Citation58. In the case of Rb family members and 53BP1, loss of A-type lamins is associated with increased degradation by the proteasome.Citation16,Citation52 We also found that loss of A-type lamins promotes degradation of 53BP1 by lysosomal proteases of the Cathepsin family (unpublished results). It is tempting to speculate that A-type lamins serve as a platform for retention of nuclear proteins 53BP1, pRb and p107, preventing their proteasomal or lysosomal degradation. Alternatively or concomitantly, A-type lamins could play an active role in maintaining the stability of these proteins. For instance, 53BP1, pRb and p107 undergo a number of posttranslational modifications, which could potentially regulate the stability of the proteins and in turn be modulated by A-type lamins.
Previously, we provided evidence for the importance of A-type lamins in NHEJ of dysfunctional telomeres.Citation16 Recent studies have shown that 53BP1 plays an essential role in the end-to-end fusion of dysfunctional telomeres resulting from depletion of the shelterin complex component TRF2.Citation38,Citation59 This process is mediated by the classical-NHEJ mechanism of DNA repair. In contrast, 53BP1 was dispensable for the chromosome end-to-end fusions that result from removal of Tpp1-Pot1a/b, which are mediated by alternative-NHEJ.Citation59 Our reconstitution experiments clearly demonstrate that the destabilization of 53BP1 upon loss of A-type lamins is responsible for the defects in the processing of dysfunctional telomeres by classical-NHEJ. Importantly, in addition to the role of A-type lamins in long-range NHEJ, our studies demonstrate their participation in short-range DSBs repair, as exemplified by the defects in the fast-phase of repair of IR-induced DSBs, corresponding to classical-NHEJ. Interestingly, these defects were also rescued by overexpression of 53BP1, suggesting that 53BP1 deficiency contributes to this phenotype. However, the fact that depletion of 53BP1 did not alter the kinetics of DSBs repair in wild-type cells (Fig. S2) indicates that the combined deficiency in A-type lamins and 53BP1 is responsible for the defects in short-range DSBs repair reported here. Altogether, our data suggest that the role of 53BP1 in wild-type cells is masked by a functional redundancy that is lost in A-type lamins-deficient cells. A-type lamins or mechanisms regulated by them overcome the necessity for 53BP1 in short-range DNA DSBs repair. We speculate that loss of A-type lamins allows enacting compensatory mechanisms that repair DSBs with slower kinetics. Restoration of 53BP1 in this context would then alter the balance between different DSBs repair pathways; tilting it in favor of the fast-phase classical-NHEJ.Citation39 Future studies will address whether loss of A-type lamins is associated with upregulation of other repair pathways, such as alternative-NHEJ.
Decreased levels of RAD51 and BRCA1 are behind HR deficiency upon loss of A-type lamins. Since BRCA1 associates with the MRN complex,Citation33 which displays nucleolytic activity, it is possible that HR-dependent end-resection itself is afflicted in these cells. This notion is consistent with the defective recruitment of RPA to DNA DSBs upon loss of A-type lamins in the absence of detectable changes in the global levels of the protein. BRCA1 also interacts with phosphorylated CtIP, a protein that is involved in DNA end-resection and known to function in both HR and alternative-NHEJ.Citation32,Citation33 Disrupting the interaction of BRCA1 with CtIP is detrimental to HR, but does not affect alternative-NHEJ.Citation32 Thus, consistent with our discussion of classic- vs. alternative-NHEJ in lamins A/C deficient cells, it is possible that loss of BRCA1 and RAD51 inhibits HR, but does not affect the ability of cells to enact alternative-NHEJ.
Our findings are important in the context of cancer, as alterations in the expression of A-type lamins have significant effects on genome stability. Several lines of evidence indicate that not only are A-type lamins either silenced or upregulated in a variety of human cancers, but that these changes significantly impact patient outcome.Citation6,Citation9 This study provides a link between A-type lamins depletion and defects in mechanisms of DNA repair, suggesting that the silencing of the LMNA gene observed in some tumors contributes to the genomic instability that drives malignancy. Increased genomic instability has also been reported in diseases caused by expression of mutant forms of lamins A/C. The best example is the genomic instability observed in progeria patients and mouse models.Citation10,Citation60,Citation61 In particular, progeria cells are defective in RAD51 recruitment to IRIF, although they do not exhibit lowered protein levels. Interestingly, progeria cells were not deficient in NHEJ. Rather, an increase in NHEJ was reported inn reference Citation10, and associated with increased accumulation of XPA at DSBs.Citation62,Citation63 These differences are reminders of the fact that depletion of A-type lamins can cause significantly different effects from specific mutations in A-type lamins. Unravelling which mutations in A-type lamins destabilize 53BP1 and Rb family members and/or transcriptionally regulate RAD51/BRCA1 will allow us to predict which lamins-related diseases present with defects in specific mechanisms of DNA repair.
In summary, this study shows that A-type lamins play a role in the two main pathways of repair of IR-induced DNA DSBs: NHEJ and HR. Our finding that depletion of A-type lamins impairs DNA repair and induces radiosensitivity, along with clinical data indicating that lamins expression can affect prognosis in certain malignancies, introduces the possibility of using these proteins as targets for cancer therapeutics. In addition, identification of 53BP1, RAD51 and BRCA1 as molecular targets of A-type lamins provides new tools to screen disease-associated mutations in the LMNA gene for defects in DNA repair and genomic instability, which could contribute to their pathophysiology. Understanding the molecular mechanisms regulating the levels of these proteins could bring about new therapeutic strategies for laminopathies as well as to enhance the killing of tumor cells by ionizing radiation.
Materials and Methods
Cell culture.
Lmna+/+ and Lmna−/− MEFs were generated in the laboratory of Colin L. Stewart as described in reference Citation64. DKO (Rb−/−;p107−/−) and TKO (Rb−/−;p107−/−;p130−/−) MEFs were provided by Julien Sage. MCF-7 and U2OS cells were obtained from ATCC (Manassas, VA). The MCF-7 DR-GFP cell line was previously described.Citation65 All cells were maintained in DMEM-Glutamax (Invitrogen) supplemented with 10% BGS, antibiotics and antimycotics.
Viral transduction.
Retro- and lenti-viral transductions were performed as described in reference Citation16. Briefly, 293T cells were transfected with viral packaging (pUMVC3 or pHR'8.2ΔR) and envelope plasmids (p-CMV-VSV-G)Citation66 along with the appropriate vector containing the cDNA or shRNA of interest. After 48 h, virus-containing media was harvested to infect target cells (MEFs, U2OS or MCF-7 cells). Retroviral transductions were performed as two 4–6 h infections on sequential days and lentiviral as one 4 h infection. Cells were allowed to recover for 48 h, and selected with the appropriate drugs. Viral envelope and packaging plasmids were gifts from Sheila Stewart (Washington University). shRNAs were obtained from Sigma-Aldrich (St. Louis, MO), 53BP1 expression plasmid from Addgene.
Immunofluorescence analysis.
Immunofluorescence was performed with antibodies: γH2AX (1:600, Upstate Biotechnology), 53BP1 (1:600, Novus Biologicals NB100-304) or RAD51 (1:100, Santa Cruz sc-8349). Cells were grown on coverslips until 70–80% confluent and irradiated with a dose of either 0.5 Gy (53BP1 and γH2AX) or 8 Gy (RAD51). At different times post-IR, cells were fixed with 3.7% formaldehyde and 0.2% Triton-X100 for 10 min at RT. Cells were blocked for 1 h at 37°C with 1% goat serum or BSA in PBS and incubated with primary antibodies for 1 h at 37°C. Secondary antibody incubations were performed for 1 h at 37°C using Alexa- and Cy3-labeled antibodies. Slides were counterstained using DAPI in Vectashield (Vector). Fluorescent images were taken using a Nikon 90i upright microscope or a confocal microscope Zeiss L510.
Protein gel blotting.
Cells were lysed in RIPA buffer (0.15 M NaCl, 0.05 M TRIS-HCl pH 7.2, 1% Triton-X100, 1% DOC, 0.1% SDS) and subjected to gel electrophoresis. Proteins were detected using the following antibodies: Lamins A/C (Santa Cruz-20681), RPA2 (Calbiochem, NA18), actin (Clone C4, MPB), β-tubulin (Sigma), γH2AX (Upstate, 07-164) and BRCA1 (Santa Cruz-6954).
Radiation treatment.
Irradiation was performed using a PANTAK pmc1000 X-ray machine with a 0.1 Cu+ 2.5 AL filter at a dose rate of 1.1 Gy/min. For immunofluorescence studies cells were irradiated with 0.5 Gy or 8 Gy, and for comet assays cells were given 8 Gy.
Comet assays.
Neutral comet assays were performed using CometSlide assay kits (Trevigen). Cells were irradiated with 8 Gy and incubated at 37°C for different periods of time (0, 30, 60, 90, 120 and 150 min) to allow for DNA damage repair. Cells were embedded in agarose, lysed and subjected to neutral electrophoresis. Before image analysis, cells were stained with ethidium bromide and visualized under a fluorescence microscope. Single-cell electrophoresis results in a comet-shaped distribution of DNA. The comet head contains high molecular weight and intact DNA, and the tail contains the leading ends of migrating fragments. Olive comet moment was calculated by multiplying the percentage of DNA in the tail by the displacement between the means of the head and tail distributions, as described in reference Citation67. We utilized the program CometScore™ Version 1.5 (TriTek) to calculate Olive Comet Moment. A total of 25 to 30 comets were analyzed per sample in each experiment.
Fluorescence in situ hybridization.
FISH on metaphase spreads was performed as described in reference Citation68. In brief, cells were arrested in mitosis by treating with colcemid for 4 h and prepared for FISH by hypotonic swelling in 0.56% KCl, followed by fixation in 3:1 methanol:acetic acid. Cell suspensions were dropped onto slides and FISH was performed using a Cy3-telomeric PNA probe and DNA counterstained using DAPI. Images were taken using a Nikon 90i upright microscope.
Homologous recombination assays.
Proficiency of HR is monitored by using a chromosomally integrated HR reporter substrate, DR-GFP, in MCF-7 cells.Citation65 The DR-GFP substrate consists of two tandem GFP sequences that have been mutated to abrogate expression of GFP and an I-SceI recognition site in one sequence. Transient expression of the I-SceI produces a DSB at the recognition site. Repair of this break by intragenic HR with the downstream GFP sequence as the homology substrate results in restoration of a functional GFP gene. Thus, expression of GFP is a readout of successful HR.Citation65 MCF-7 DR-GFP cells were transfected with an I-SceI expressing plasmid. After 48 h, flow cytometry was used to determine the percent of cells expressing GFP as an indication of successful HR.
Colony formation assays.
Clonogenic analysis was performed as described in reference Citation69. Briefly, cells were plated in p60 culture dishes to facilitate formation of 30–40 colonies per plate and allowed to become adherent by incubating at 37°C for 2–3 h. Cells were immediately treated with 0, 2, 4 or 6 Gy of ionizing radiation and allowed to grow undisturbed for 7–10 d. Colonies were then counted and the surviving fractions calculated. Colony formation experiments were done three times, with triplicate samples within each experiment.
Quantitative real time PCR.
qRT-PCR was performed using the 7900HT Fast Real-Time PCR system (Applied Biosystems) with the Taqman® Universal PCR Master Mix (Applied Biosystems, California). Generation of cDNA was performed by reverse transcription of 1 µg total RNA using the GeneAmp® RNA PCR kit, also from Applied Biosystems. RAD51, BRCA1 and RPA2 transcripts were detected by TaqMan® Gene Expression Assays (Hs00153418_m1, Hs01556193_m1 and Hs00358315_m1 respectively). All PCR reactions were done in triplicate (within experiments) to amplify endogenous target genes, with 18S controls in the same plate. Data was analyzed by relative quantitation.
Figures and Tables
Figure 1 Spatio-temporal formation and resolution of DNA repair foci induced by ionizing radiation. (A) Lmna+/+ and Lmna−/− MEFs were irradiated with 0.5 Gy or left untreated. Cells were fixed at different times post-IR (from 30 min to 24 h) and subjected to immunofluorescence (IF) with an antibody recognizing 53BP1. Pictures of fields selected randomly were taken under the same conditions of exposure. Representative images of four cells per condition are shown in each part. (B) Lmna+/+ and Lmna−/− MEFs were irradiated and at different times post-IR cells were fixed and subjected to IF with an antibody recognizing γH2AX. Cells presenting more than 5 γH2AX foci were scored as positive for responding to IR. A total of 200–300 cells were scored per time point and per experiment. The average ± standard deviation of three independent experiments is shown.
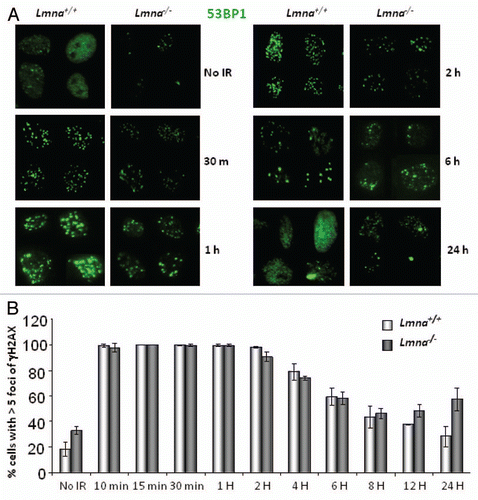
Figure 2 Defective repair of IR-induced DNA DSBs upon loss of A-type lamins. (A) Asynchronously growing cells were irradiated with 8 Gy. At different times post-irradiation (0, 30, 60, 90, 120 and 150 min) cells were collected and neutral comet assays were performed. Kinetics of repair of IR-induced DSBs was assessed by Olive Comet Moment, a measure of unrepaired DNA. A total of 25 to 30 cells were analyzed per sample and per time-point. (B) Protein gel blot to monitor the levels of 53BP1 protein in Lmna+/+ and Lmna−/− MEFs retrovirally transduced with EV control or a 53BP1-expressing vector. Short and long exposures of 53BP1 are shown. Actin levels are used as loading control.
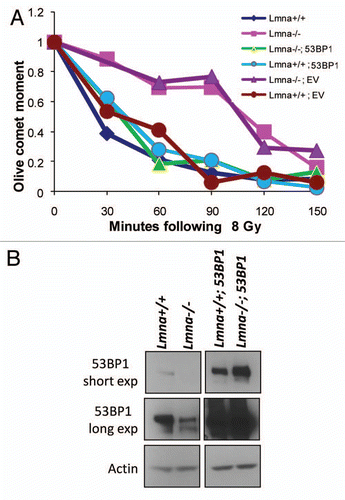
Figure 3 Defects in NHEJ of dysfunctional telomeres are rescued by reconstitution of 53BP1. (A) Levels of 53BP1 and A-type lamins in U2OS cells after ectopic expression of 53BP1 (EV, empty vector control), followed by lentiviral transduction with shLmna or shCtrl. β-tubulin was used as loading control. Note how ectopic expression of 53BP1 prevents the decrease in protein levels upon depletion of A-type lamins. (B) Key of the different categories of metaphases based on the extent of chromosome end-to-end fusions induced by expression of TRF2ΔBΔM. Representative images of the different categories are shown. (C) Histograms showing the percentage of metaphases belonging to each category from the different cell lines. Top part: cells transduced with EV, shCtrl and TRF2ΔBΔM. Middle part: cells transduced with EV, shLmna and TRF2ΔBΔM. Bottom part: cells transduced with 53BP1, shLmna and TRF2ΔBΔM. 79–88 metaphases were analyzed per condition.
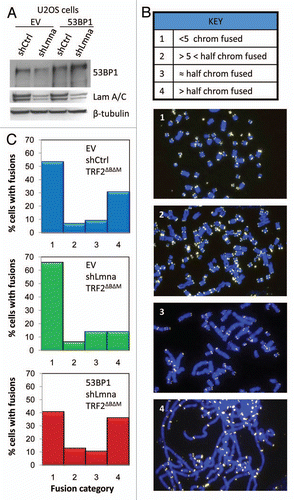
Figure 4 A-type lamins promote HR by maintaining RAD51 levels and recruitment to DSBs. (A) Protein gel blots showing decreased 53BP1 protein upon depletion of A-type lamins in MCF-7 cells carrying an HR reporter construct (DR-GFP). (B) Percent of GFP-positive MCF7-DR-GFP cells resulting from HR of I-SceI-induced DSBs. Depletion of A-type lamins leads to a 40% reduction in HR. (C) Percentage of MCF7-DR-GFP cells positive for RAD51 foci (more than 10 foci throughout the nucleus) 6 h after treatment with 8 Gy. A total of 200 cells per condition were analyzed per experiment. The average ± standard deviation of three independent experiments is shown. (D) Representative images of RAD51 foci. Blue images show DAPI stained nuclei, green shows RAD51 IF. (E) Protein gel blots showing the decrease in global levels of RAD51 upon depletion of A-type lamins in MCF-7 DR-GFP cells. β-tubulin was used as loading control. (F) Protein gel blots showing lower RAD51 upon depletion of A-type lamins in MEFs. (G) Percentage of MEFs positive for RAD51 foci 6 h after treatment with 8 Gy. A total of 290-10 cells per condition were analyzed.
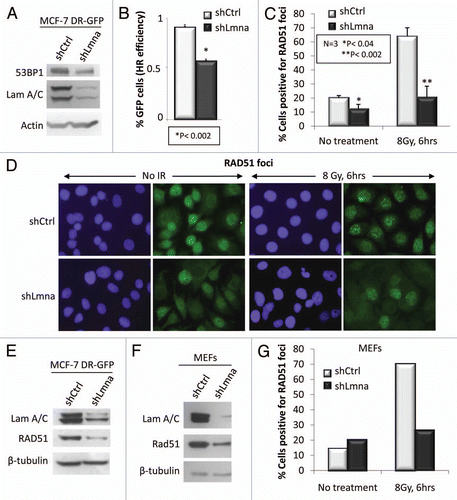
Figure 5 Loss of A-type lamins significantly affects transcriptional regulation of RAD51 and BRCA1. (A) Protein gel blots showing lower levels of BRCA1, but not RPA2 in cells depleted of A-type lamins. β-tubulin was used as loading control. (B) Percent of cells positive for formation of RPA2 foci 6 h after treatment with 8 Gy IR. (C) qRT-PCR to determine transcript levels of RAD51, BRCA1 and RPA2. Shown are the averages and standard deviations from three independent experiments.
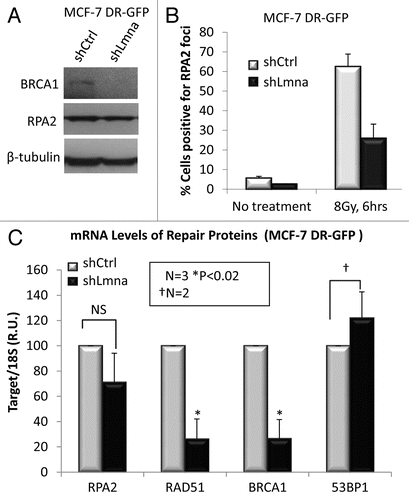
Figure 6 Decreased RAD51 in lamins A/C-deficient cells requires p130, a member of the pocket family of proteins. (A) Protein gel blot showing depletion of A-type lamins from MEFs that are null for two (DKO: Rb−/−; p107−/−) or all three (TKO: Rb−/−; p107−/−; p130−/−) of the pocket family proteins. Note how loss of lamins leads to a dramatic decrease of RAD51 only in the cells that are p130 proficient (DKO). (B) Co-immunoprecipitation of p130 with E2F4. Lystates from lamins A/C-proficient or -deficient cells were subjected to immunoprecipitation with an E2F4 antibody. Protein gel blots (left) shows successful immunoprecipitation of E2F4 and co-precipitation of p130. Inputs are shown on the right part.
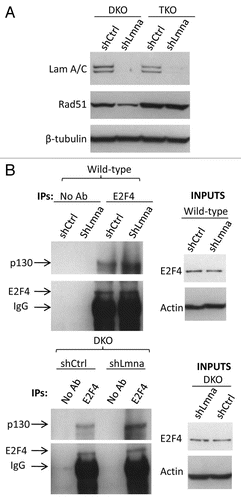
Figure 7 Radiosensitivity and model of the dual role of A-type lamins in DNA DSBs repair. (A) Clonogenic survival in Lmna+/+ and Lmna−/− MEFs in response to increasing doses of radiation (0 to 6 Gy). Shown are the surviving fractions and standard deviation of the mean from three independent experiments. (B) A-type lamins play a role in the stabilization of the pocket family proteins pRb and p107, as well as 53BP1, in part by preventing their degradation by the proteasome. By stabilizing 53BP1, A-type lamins promote classical-NHEJ. In addition, A-type lamins regulate transcriptionally two key factors in HR, RAD51 and BRCA1. Loss of A-type lamins leads to increased formation of p130/E2F4 complexes, which in turn can bind the RAD51 and BRCA1 gene promoters and inhibit their transcription. Loss of A-type lamins leads to defects in the two major mechanisms of DNA DSBs repair (NHEJ and HR), increased genomic instability and radiation sensitivity.
Additional material
Download Zip (1 MB)Acknowledgments
A.B.R. performed many of the experiments and contributed to the conception and design of the project as well as the writing of the manuscript. S.M.P. and R.P.V. performed some IF studies and comet assays. Z.F., W.S. and J.Z. provided the DR-GFP system and performed the HR experiments in collaboration with A.B.R. J.Z. also provided advice and criticisms throughout the development of the project. The rest of the authors contributed to different aspects of the project.
We are grateful to A. Laszlo for sharing his technique for immunofluorescence to monitor IRIF with 53BP1 and γH2AX antibodies and for helpful discussions. We thank T. de Lange for providing TRF2ΔBΔM retroviral vector, D. Hodzig for providing shLmna vector for depletion of A-type lamins in mouse cells and B. Sleckman for providing the shRNA for depletion of 53BP1. We thank C. Franz and R. Waller for their contribution to this work during their laboratory rotation and M. Neumann for wonderful technical assistance. We are in debt with Z. You for suggestions and critical reading of the manuscript. Research in the laboratory of S.G. was supported by an Institutional Research Grant from American Cancer Society IRG-58-010-50, an Alzheimer Disease Research Center Pilot Grant (ADRC-WUSM), a Grant from NIH (1RO1GM094513-01) and a Siteman Cancer Center Research Development Award.
References
- Andrés V, Gonzalez JM. Role of A-type lamins in signaling, transcription and chromatin organization. J Cell Biol 2009; 187:945 - 957; PMID: 20038676; http://dx.doi.org/10.1083/jcb.200904124
- Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma?. Nat Cell Biol 2004; 6:1062 - 1067; PMID: 15517000; http://dx.doi.org/10.1038/ncb1104-062
- Mattout A, Dechat T, Adam SA, Goldman RD, Gruenbaum Y. Nuclear lamins, diseases and aging. Curr Opin Cell Biol 2006; 18:335 - 341; PMID: 16632339; http://dx.doi.org/10.1016/j.ceb.2006.03.007
- Worman HJ, Ostlund C, Wang Y. Diseases of the nuclear envelope. Cold Spring Harb Perspect Biol 2010; 2:760; PMID: 20182615; http://dx.doi.org/10.1101/cshperspect.a000760
- Broers JL, Raymond Y, Rot MK, Kuijpers H, Wagenaar SS, Ramaekers FC. Nuclear A-type lamins are differentially expressed in human lung cancer subtypes. Am J Pathol 1993; 143:211 - 220; PMID: 8391215
- Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar D, Barkan R, et al. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 2009; 13:1059 - 1085
- Willis ND, Wilson RG, Hutchison CJ. Lamin A: a putative colonic epithelial stem cell biomarker which identifies colorectal tumours with a more aggressive phenotype. Biochem Soc Trans 2008; 36:1350 - 1353; PMID: 19021554; http://dx.doi.org/10.1042/BST0361350
- Kaufmann SH, Mabry M, Jasti R, Shaper JH. Differential expression of nuclear envelope lamins A and C in human lung cancer cell lines. Cancer Res 1991; 51:581 - 586; PMID: 1985776
- Agrelo R, Setien F, Espada J, Artiga MJ, Rodriguez M, Perez-Rosado A, et al. Inactivation of the lamin A/C gene by CpG island promoter hypermethylation in hematologic malignancies, and its association with poor survival in nodal diffuse large B-cell lymphoma. J Clin Oncol 2005; 23:3940 - 3947; PMID: 15867203; http://dx.doi.org/10.1200/JCO.2005.11.650
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780 - 785; PMID: 15980864; http://dx.doi.org/10.1038/nm1266
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059 - 1063; PMID: 16645051; http://dx.doi.org/10.1126/science.1127168
- Allsopp RC, Vaziri H, Patterson C, Goldstein S, Younglai EV, Futcher AB, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA 1992; 89:10114 - 10118; PMID: 1438199; http://dx.doi.org/10.1073/pnas.89.21.10114
- Decker ML, Chavez E, Vulto I, Lansdorp PM. Telomere length in Hutchinson-Gilford progeria syndrome. Mech Ageing Dev 2009; 130:377 - 383; PMID: 19428457; http://dx.doi.org/10.1016/j.mad.2009.03.001
- Manju K, Muralikrishna B, Parnaik VK. Expression of disease-causing lamin A mutants impairs the formation of DNA repair foci. J Cell Sci 2006; 119:2704 - 2714; PMID: 16772334; http://dx.doi.org/10.1242/jcs.03009
- Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle 2009; 8:3860 - 3865; PMID: 19901537; http://dx.doi.org/10.4161/cc.8.23.10092
- Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J 2009; 28:2414 - 2427; PMID: 19629036; http://dx.doi.org/10.1038/emboj.2009.196
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 2009; 423:157 - 168; PMID: 19772495; http://dx.doi.org/10.1042/BJ20090942
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 1078; PMID: 19847258; http://dx.doi.org/10.1038/nature08467
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008; 7:1765 - 1771; PMID: 18675941; http://dx.doi.org/10.1016/j.dnarep.2008.06.018
- Adams BR, Hawkins AJ, Povirk LF, Valerie K. ATM-independent, high-fidelity nonhomologous end joining predominates in human embryonic stem cells. Aging (Albany NY) 2010; 2:582 - 596
- Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res 2004; 104:14 - 20; PMID: 15162010; http://dx.doi.org/10.1159/000077461
- Chen L, Trujillo K, Sung P, Tomkinson AE. Interactions of the DNA ligase IV-XRCC4 complex with DNA ends and the DNA-dependent protein kinase. J Biol Chem 2000; 275:26196 - 26205; PMID: 10854421; http://dx.doi.org/10.1074/jbc.M000491200
- Wang H, Zeng ZC, Perrault AR, Cheng X, Qin W, Iliakis G. Genetic evidence for the involvement of DNA ligase IV in the DNA-PK-dependent pathway of non-homologous end joining in mammalian cells. Nucleic Acids Res 2001; 29:1653 - 1660; PMID: 11292837; http://dx.doi.org/10.1093/nar/29.8.1653
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009; 1:109 - 121; PMID: 20157594
- Lombard DB. Sirtuins at the breaking point: SIRT6 in DNA repair. Aging (Albany NY) 2009; 1:12 - 16; PMID: 20157593
- Nevaldine B, Longo JA, Hahn PJ. The scid defect results in much slower repair of DNA double-strand breaks but not high levels of residual breaks. Radiat Res 1997; 147:535 - 540; PMID: 9146698; http://dx.doi.org/10.2307/3579619
- Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 2004; 279:55117 - 55126; PMID: 15498778; http://dx.doi.org/10.1074/jbc.M404524200
- Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 2006; 34:6170 - 6182; PMID: 17088286; http://dx.doi.org/10.1093/nar/gkl840
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706 - 5715; PMID: 12897142; http://dx.doi.org/10.1128/MCB.23.16.5706-15.2003
- Assenmacher N, Hopfner KP. MRE11/RAD50/NBS1: complex activities. Chromosoma 2004; 113:157 - 166; PMID: 15309560; http://dx.doi.org/10.1007/s00412-004-0306-4
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, et al. Human CtIP promotes DNA end resection. Nature 2007; 450:509 - 514; PMID: 17965729; http://dx.doi.org/10.1038/nature06337
- Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009; 459:460 - 463; PMID: 19357644; http://dx.doi.org/10.1038/nature07955
- Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 2008; 283:7713 - 7720; PMID: 18171670; http://dx.doi.org/10.1074/jbc.M710245200
- Schlegel BP, Jodelka FM, Nunez R. BRCA1 promotes induction of ssDNA by ionizing radiation. Cancer Res 2006; 66:5181 - 5189; PMID: 16707442; http://dx.doi.org/10.1158/0008-5472.CAN-05-3209
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat Immunol 2004; 5:481 - 487; PMID: 15077110; http://dx.doi.org/10.1038/ni1067
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, et al. 53BP1 is required for class switch recombination. J Cell Biol 2004; 165:459 - 464; PMID: 15159415; http://dx.doi.org/10.1083/jcb.200403021
- Difilippantonio S, Gapud E, Wong N, Huang CY, Mahowald G, Chen HT, et al. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature 2008; 456:529 - 533; PMID: 18931658; http://dx.doi.org/10.1038/nature07476
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 2008; 456:524 - 528; PMID: 18931659; http://dx.doi.org/10.1038/nature07433
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 2010; 207:855 - 865; PMID: 20368578; http://dx.doi.org/10.1084/jem.20100244
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243 - 254; PMID: 20362325; http://dx.doi.org/10.1016/j.cell.2010.03.012
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 916; PMID: 10477747; http://dx.doi.org/10.1083/jcb.146.5.905
- Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J Cell Biol 2000; 151:1381 - 1390; PMID: 11134068; http://dx.doi.org/10.1083/jcb.151.7.1381
- Olive PL, Banath JP, Durand RE. Heterogeneity in radiation-induced DNA damage and repair in tumor and normal cells measured using the “comet” assay. Radiat Res 1990; 122:86 - 94; PMID: 2320728; http://dx.doi.org/10.2307/3577587
- Wang H, Zeng ZC, Bui TA, DiBiase SJ, Qin W, Xia F, et al. Nonhomologous end-joining of ionizing radiation-induced DNA double-stranded breaks in human tumor cells deficient in BRCA1 or BRCA2. Cancer Res 2001; 61:270 - 277; PMID: 11196174
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010; 17:688 - 695; PMID: 20453858; http://dx.doi.org/10.1038/nsmb.1831
- Tripathi V, Nagarjuna T, Sengupta S. BLM helicase-dependent and -independent roles of 53BP1 during replication stress-mediated homologous recombination. J Cell Biol 2007; 178:9 - 14; PMID: 17591918; http://dx.doi.org/10.1083/jcb.200610051
- Zhang J, Ma Z, Treszezamsky A, Powell SN. MDC1 interacts with Rad51 and facilitates homologous recombination. Nat Struct Mol Biol 2005; 12:902 - 909; PMID: 16186822; http://dx.doi.org/10.1038/nsmb991
- Shi W, Feng Z, Zhang J, Gonzalez-Suarez I, Vanderwaal RP, Wu X, et al. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis 2010; 31:994 - 1002; PMID: 20130019; http://dx.doi.org/10.1093/carcin/bgq035
- Mekeel KL, Tang W, Kachnic LA, Luo CM, DeFrank JS, Powell SN. Inactivation of p53 results in high rates of homologous recombination. Oncogene 1997; 14:1847 - 1857; PMID: 9150391; http://dx.doi.org/10.1038/sj.onc.1201143
- Bindra RS, Glazer PM. Repression of RAD51 gene expression by E2F4/p130 complexes in hypoxia. Oncogene 2007; 26:2048 - 2057; PMID: 17001309; http://dx.doi.org/10.1038/sj.onc.1210001
- Hegan DC, Lu Y, Stachelek GC, Crosby ME, Bindra RS, Glazer PM. Inhibition of poly(ADP-ribose) polymerase downregulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc Natl Acad Sci USA 2010; 107:2201 - 2206; PMID: 20133863; http://dx.doi.org/10.1073/pnas.0904783107
- Johnson BR, Nitta RT, Frock RL, Mounkes L, Barbie DA, Stewart CL, et al. A-type lamins regulate retinoblastoma protein function by promoting subnuclear localization and preventing proteasomal degradation. Proc Natl Acad Sci USA 2004; 101:9677 - 9682; PMID: 15210943; http://dx.doi.org/10.1073/pnas.0403250101
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis 2002; 23:687 - 696; PMID: 12016139; http://dx.doi.org/10.1093/carcin/23.5.687
- van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2001; 2:196 - 206; PMID: 11256071; http://dx.doi.org/10.1038/35056049
- Nakamura AJ, Redon CE, Bonner WM, Sedelnikova OA. Telomere-dependent and telomere-independent origins of endogenous DNA damage in tumor cells. Aging (Albany NY) 2009; 1:212 - 218; PMID: 20157510
- Hawtin RE, Stockett DE, Wong OK, Lundin C, Helleday T, Fox JA. Homologous recombination repair is essential for repair of vosaroxin-induced DNA double-strand breaks. Oncotarget 2010; 1:606 - 619
- Olive PL. Endogenous DNA breaks: gammaH2AX and the role of telomeres. Aging (Albany NY) 2009; 1:154 - 156; PMID: 20157507
- Han X, Feng X, Rattner JB, Smith H, Bose P, Suzuki K, et al. Tethering by lamin A stabilizes and targets the ING1 tumour suppressor. Nat Cell Biol 2008; 10:1333 - 1340; PMID: 18836436; http://dx.doi.org/10.1038/ncb1792
- Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 2010; 29:2598 - 2610
- Liu Y, Wang Y, Rusinol AE, Sinensky MS, Liu J, Shell SM, et al. Involvement of xeroderma pigmentosum group A (XPA) in progeria arising from defective maturation of prelamin A. FASEB J 2008; 22:603 - 611; PMID: 17848622; http://dx.doi.org/10.1096/fj.07-8598com
- Pendás AM, Zhou Z, Cadinanos J, Freije JM, Wang J, Hultenby K, et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat Genet 2002; 31:94 - 99; PMID: 11923874
- Liu B, Zhou Z. Lamin A/C, laminopathies and premature ageing. Histol Histopathol 2008; 23:747 - 763; PMID: 18366013
- Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY) 2009; 1:28 - 37; PMID: 19851476
- Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol 1999; 147:913 - 920; PMID: 10579712; http://dx.doi.org/10.1083/jcb.147.5.913
- Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res 2005; 3:531 - 539; PMID: 16254187; http://dx.doi.org/10.1158/1541-7786.MCR-05-0192
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9:493 - 501; PMID: 12649500; http://dx.doi.org/10.1261/rna.2192803
- Olive PL, Banath JP, Durand RE. Detection of etoposide resistance by measuring DNA damage in individual Chinese hamster cells. J Natl Cancer Inst 1990; 82:779 - 783; PMID: 2325148; http://dx.doi.org/10.1093/jnci/82.9.779
- Samper E, Goytisolo FA, Slijepcevic P, van Buul PP, Blasco MA. Mammalian Ku86 protein prevents telomeric fusions independently of the length of TTAGGG repeats and the G-strand overhang. EMBO Rep 2000; 1:244 - 252; PMID: 11256607; http://dx.doi.org/10.1093/emboreports/kvd051
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc 2006; 1:2315 - 2319; PMID: 17406473; http://dx.doi.org/10.1038/nprot.2006.339