Abstract
Double-strand DNA breaks detected in different phases of the cell cycle induce molecularly distinct checkpoints downstream of the ATM kinase. p53 is known to induce arrest of cells in G1 and occasionally G2 phase but not S phase following ionizing radiation, a time at which the MRN complex and cdc25-dependent mechanisms induce arrest. Our understanding of how cell cycle phase modulates pathway choice and the reasons certain pathways might be favored at different times is limited. In this report, we examined how cell cycle phase affects the activation of the p53 checkpoint and its ability to induce accumulation of the cdk2 inhibitor p21. Using flow cytometric tools and centrifugal elutriation, we found that the p53 response to ionizing radiation is largely intact in all phases of the cell cycle; however, the accumulation of p21 protein is limited to the G1 and G2 phase of the cell cycle because of the activity of a proteasome-dependent p21 turnover pathway in S-phase cells. We found that the turnover of p21 was independent of the SCFskp2 E3 ligase but could be inhibited, at least in part, by reducing hdm2, although this depended on the cell type studied. Our results suggest that there are several redundant pathways active in S-phase cells that can prevent the accumulation of p21.
Introduction
Ionizing radiation induces double-strand DNA breaks, which activate distinct molecular pathways to induce cell cycle arrest depending on whether the cell is in the G1, S or G2 phase of the cell cycle.Citation1 In G1 cells, a p53-dependent transcriptional program induces cell cycle arrest in part by activating expression of p21, a cdk inhibitor that targets cdk2-containing complexes. In response to genotoxic stress, ATM- and chk2-dependent phosphorylation of hdm2 inhibits its ability to regulate p53 in three ways: reducing its E3-ubiquitin ligase for p53, preventing the binding of hdm2 to p53 (which can also block the transactivation function of p53) and by inhibiting the ability of hdm2 to promote nuclear export of p53.Citation2 Additionally, hdm2 can promote the proteasome-dependent but ubiquitin-independent degradation of p21. Phosphorylation of hdm2 may affect this activity as well. The absence of p21 weakens p53-dependent G1 arrest in a variety of different cell lines and primary cells, both mouse and human.Citation3–Citation8 In S-phase cells, checkpoints are triggered by multiple mechanisms involving both the inhibition of cdc25A, which removes an inhibitory phosphorylation on cyclin-cdk complexes, and the MRN complex.Citation9–Citation12 In G2 cells, inactivation of cdc25 prevents the activation of cyclin B-cdc2, and in some cell types, p53-dependent accumulation of p21 can also play a role.Citation4
Although there is extensive data for the involvement of p21 in causing G1 arrest following DNA damage, cells might have evolved mechanisms that prevent p21 accumulation in S phase, because p21 can affect DNA repair and the ability of a cell to restart DNA synthesis.Citation13–Citation15 Thus, we were interested in determining whether the accumulation of p21 was prevented in S-phase cells responding to DNA damage. In this report, we show that an hdm2-dependent mechanism reduces accumulation of p21 in S-phase cells. We suggest that this might prevent p21 from inhibiting PCNA ubiquitination and recovery from DNA damage.
Results
Accumulation of p21 was reduced in S-phase cells exposed to ionizing radiation.
We set out to determine a collection of cells in which we could investigate p53-dependent p21 accumulation following exposure to ionizing radiation. In MCF7 cells, Darzynkeiwicz had reported that p53 accumulated throughout the cell cycle, but accumulation of p21 was restricted to cells in G1 and G2 phase of the cell cycle following an 8–16 h treatment with camptothecin.Citation16 However, it was unclear whether this phenomena was limited to MCF7 cells, whether it was due to the extended length of time that the cells were in the presence of the drug, or whether the antibodies used in the laser scanning analysis were capable of detecting p21 species or complexes that formed in S-phase cells. To avoid these caveats, we revisted these results and began our analysis by screening a diverse collection of five transformed cell lines at 3 and 6 h following exposure to different doses of ionizing radiation (ranging from 1 Gy to 20 Gy). In a B-lymphoblast cell line (TK6), a colorectal carcinoma cell line (HCT116), a mammary epithelial adenocarcinoma cell line (MCF7) and a lung carcinoma cell line (A549), both p53 and p21 protein accumulated within 6 h post-irradiation (). In contrast, neither p53 nor p21 accumulated in the glioma cell line U87 in this time. Similar results were obtained at 3 h and with 5 Gy or 20 Gy as well (data not shown). These cell lines, as well as others that are discussed below, were subsequently used interchangeably in the experiments that followed.
We next measured whether there were cell cycle phase-specific differences in p21 and p53 accumulation by three independent assays: centrifugal elutriation followed by immunoblotting, cell sorting followed by immunoblotting and dual-staining flow cytometry. TK6 cells were fractionated into G1-, S- and G2-phase enriched populations by centrifugal elutriation, irradiated at different doses ranging from 1 to 20 Gy and allowed to grow for an additional 3 to 6 h prior to analyzing their progression in the cell cycle and measuring the accumulation of p53 and p21 (). Representative flow cytometric profiles of the G1- and S-phase cells before and after 2 or 20 Gy irradiation are shown in the figure. Similar results were obtained at 1, 5 and 10 Gy as well (data not shown). In all cases, irradiation delayed progression to the next phase of the cell cycle, with greater delays occurring with increasing dose. We observed an increase in the amount of p53 in all cell cycle phases following IR, but p21 expression increased maximally in G1- and G2-phase cells. Similar results were seen at both 3 and 6 h and with 2 Gy, 5 Gy and 20 Gy of ionizing radiation (data not shown). Because of the contaminating G1- and G2-phase cells in the S-phase population, we could not definitively determine whether the induction of p21 was prevented in S-phase cells. Furthermore, centrifugal elutriation could not be used to enrich adherent cells into different cell cycle phase fractions. Thus, we used flow sorting to isolate G1-, S- and G2-phase populations of HCT116 () and TK6 cells following Hoescht staining. Under these conditions, p53 accumulated in a cell cycle phase-independent manner, whereas p21 accumulation was clearly limited to cells in G1 and G2 phase (). Additionally, when asynchronously growing TK6 cells were irradiated and doubly stained 3 or 6 h later with p21 antibodies and propidium iodide, the level of p21 increased in both G1- and G2-phase cells but not in S-phase cells (). A similar pattern of p21 accumulation in IMR90 lung fibroblasts transformed with H-Ras and adenoviral E1A, MCF7, A549 and HCT116 cells was also noted (data not shown). Again, this occurred irrespective of time post-irradiation (3 or 6 h) or dose of irradiation (from 1 to 20 Gy). In all instances, less p21 protein accumulated in S phase compared with the amount seen in G1 or G2 phase.
We next asked whether the inability to accumulate p21 in S-phase cells following irradiation was a property restricted to the five transformed cell lines. The flow cytometric analysis was repeated on two epithelial cell lines immortalized by the catalytic subunit of human telomerase (RPE-hTERT and BJ-hTERT), as well as a normal non-immortalized lung fibroblast strain, IMR90. We found that, similarly to transformed cells, these cell lines showed increases in both p53 and p21 protein expression in response to irradiation, but the accumulation of p21 only increased in G1 and G2-phase ( and data not shown). This suggests that the lack of p21 accumulation in S-phase after irradiation is a property shared by transformed, immortalized and normal cells alike.
To determine if this pattern was due to a direct downstream effect of the DNA damage response, we took advantage of the fact that nutlin-3 can increase p53 without affecting other functions of hdm2 or directly activating the DNA damage response.Citation17 Nutlin-3 fits into the small hydrophobic p53 binding pocket of hdm2, eliminating hdm2-induced turnover of p53. Thus, we took HCT116 cells and treated them with 1 or 3 µM nutlin-3 for 6 h and looked at the accumulation of hdm2, p53 and p21 protein. Both p53 and p21 increased following treatment of non-irradiated HCT116 cells; however, the increase in p21 was again greater in G1- and G2 phase compared with S phase (). Taken together, these results suggest that irrespective of how p53 was activated, p21 protein does not accumulate in this collection of S-phase cells.
p53 accumulation and transcriptional activity is cell cycle phase-independent.
After p53 protein is synthesized, it must accumulate in the nucleus, bind to the appropriate promoters and activate transcription. Blocking nuclear accumulation of p53 may contribute to the reduced p21 accumulation seen in S-phase cells. Genotoxic stress induces phosphorylation of p53 and hdm2 by ATM and chk2, ultimately leading to an increase in nuclear p53 and the transcription of p53 target genes. Other post-translational modifications of p53 can affect the profile of the transcriptional program but are not required for p53-dependent transcription.Citation17 Thus we began to address whether p53 was activated to a similar extent in S-phase and G1- and G2-phase cells. We found that the ability of elutriated cells to sense DNA damage, as measured by p53 and chk2 phosphorylation, was largely unaffected by cell cycle phase (). In addition, p53 entered the nucleus of S-phase A549 cells following ionizing radiation (). Similar results were also seen with MCF7 cells (data not shown). Furthermore, PUMA and p21 transcripts were present at nearly equivalent levels in irradiated G2- and S-phase TK6 cells enriched by cell sorting, directly demonstrating that p53 was activating transcription in a cell cycle phase-dependent manner (). Finally, we established that p53 levels were equivalent at all phases of the cell cycle following irradiation of TK6 (), RPE-hTERT () and BJ-hTERT cells (data not shown). Together, these data suggest that the lack of p21 accumulation in S-phase cells in any of these cell lines does not result from the inability of DNA damage to activate a p53 response.
Accumulation of p21 in S-phase cells is regulated at the level of proteasome-dependent turnover.
Many proteasome-dependent pathways, both ubiquitin-dependent and ubiquitinin-dependent, have been shown to suppress p21 accumulation in cycling cells. These pathways can be inactivated by signals that eventually lead to growth arrest. To begin determining whether S-phase cells have a greater capacity to degrade p21 protein, we prepared S-phase extracts from cells synchronized with hydroxyurea (, lower right part) or G1/G2-phase extracts from cells synchronized with nocodazole (, lower left part). These extracts were supplemented with rabbit reticulocyte lysate, and Citation35 S-methionine-labeled, in vitro-translated p21 protein was subsequently added. We observed that p21 protein was degraded more rapidly in extracts prepared from S-phase cells (t1/2 = 45.4 ± 15.8 min) compared with extracts prepared from G1- and G2-phase cells (t1/2 = 116.8 ± 36.9 min) ().
We also asked if the proteasome was required to keep p21 levels low during S phase. A 6-h treatment of asynchronously growing HCT116 cells with the proteasome inhibitor MG132 increased p53 and p21 mRNA levels to the amounts seen in irradiated cells, while LLM, a calpain inhibitor, had no effect (data not shown). However, unlike in the irradiated cells, the amount of p21 protein was equally high at all phases of the cell cycle in the MG132 treated cells (). Similar results were seen using two other proteasome inhibitors, lactacystin and ALLN (data not shown). Irradiation only had a modest effect on p21 accumulation in the MG132-treated cells (), and LLM treatment did not affect the cell cycle phase-dependent accumulation of p21 following irradiation (). Similar results were seen when RPE-hTERT cells were treated with MG132 (data not shown). Together, these results indicate that p21 protein is subject to pro-teosome-dependent turnover during all phases of the cell cycle, but apparently with greater efficiency in S-phase.
p21 turnover in S-phase cells is skp2-independent but involves hdm2.
SCFskp2 and hdm2 can contribute to p21 turnover (Bornstein, 2003; Jin, 2008). To examine the role that each of these two pathways played in S phase-specific turnover, we first treated HCT116 cells with pools of siRNAs designed to reduce skp2 and looked at p21 accumulation by dual-color flow cytometry. The amount of skp2 was significantly decreased at 48 h post-transfection ( and left). Consistent with this we observed an accumulation of p27 (, left), the best-documented substrate of the SCFskp2 E3 ligase.Citation18,Citation19 Dual-color flow cytometry for DNA content and accumulated p21 showed that the increase in p21 was most evident in G2 phase, with little change observed in S phase (, right). Similar results were obtained in RPEhTERT cells after knockdown of skp2 (). Hence, S-phase turnover of p21 in response to ionizing radiation appears to be independent of skp2; however, skp2 clearly played a role in reducing the accumulation of p21 in G2 cells.
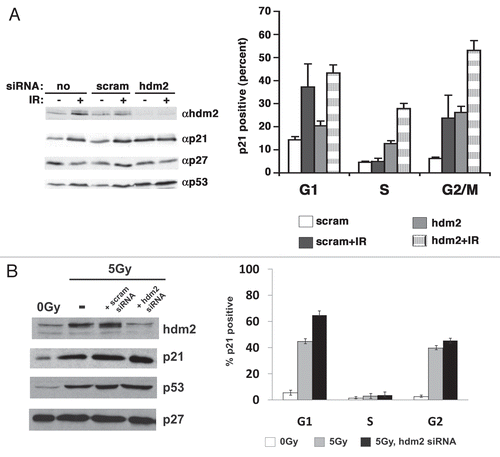
We next assessed the effect of reducing hdm2 on the level of p21 expression in S-phase cells following irradiation. The amount of hdm2 was significantly decreased at 24 h post-transfection, correlating with an increase in p53 (, left). By 48 h the cells were too sick to be used for experiments. Strikingly, when p21 expression was examined in cells treated with siRNA to hdm2 for 24 h, there was a nearly two-fold increase in p21 levels in S and G2 phase (, right). The level of p21 increased another 2-fold upon irradiation in S and G2 phase (, right). The amount of p53 induced by either reducing hdm2 or irradiating cells was equivalent, suggesting that the effect on p21 accumulation was not due to some change in the level of p53 protein. However, when we knocked down hdm2 in RPE-hTERT cells, there was no increase in p21 in S-phase cells following irradiation (). This was despite a strong increase in p53 levels across all cell cycle phases (data not shown). In addition, when we knocked-down either DDB1, cul5 or REGγ in RPE-hTERT, there was never a reproducibly strong induction of p21 in S phase following irradiation in such cells (data not shown). Hence, the mechanisms regulating p21 turnover in S phase are cell-type dependent. Consequently, the hdm2-and proteasome-dependent mechanism of p21 turnover in HCT116 may explain why the p53 growth arrest response, which is dependent on the accumulation of p21, is restricted to G1 cells and cells that fail to arrest have reduced viability and are lost.
Discussion
Our knowledge of the pathways that regulate p21 is extensive. A variety of mechanisms operate at the transcriptional, post-transcriptional and post-translational levels in different cell types and under different conditions to dictate the level of p21. In this manuscript, we demonstrate that hdm2-dependent p21 protein turnover can control p21 accumulation in S and G2 phase and prevent the elaboration of a p53-induced, p21-dependent cell cycle arrest in response to DNA damage at that time.
Using selective synchrony methods based on either cell volume (elutriation) or DNA content (cell sorting), we enriched populations of cells in each phase of the cell cycle and showed that p21 accumulation was curtailed in S phase in transformed, immortalized and normal cells following exposure to ionizing radiation. Ionizing radiation induces double-strand DNA breaks, leading to activation of chk2 and a number of downstream checkpoints dependent on p53, cdc25A, BRCA1 or Nbs1.Citation1 Our finding that p53-induced p21 accumulation was dependent on the phase of the cell cycle is consistent with the prior observation of Deptala and colleagues,Citation16,Citation20 who examined p53 and p21 accumulation in MCF7 cells treated with camptothecin. However, our results extend their finding by showing that p53 was in the nucleus and transcriptionally active in S-phase cells, but a proteolytic barrier prevented p21 protein accumulation.
The effect of cell cycle phase on p53 activation had been studied by a number of groups. Gottifredi et al. reported that a transcriptional blockade prevented p53-induced-accumulation of p21 but not other p53 targets in S-phase RKO cells.Citation21 These investigators also reported that a proteolytic mechanism could contribute in other cell types,Citation13,Citation14 but the molecular nature of this mechanism was not addressed. The translocation of p53 to the nucleus may also be regulated in some cells.Citation22
p21 protein turnover is regulated by a number of pathways, with both ubiquitin-dependent and ubiquitin-independent mechanisms contributing to its regulation in S-phase cells. These include a WISp39-associated chaperone pathway,Citation23 an SCFskp2-dependent pathwayCitation24–Citation26 and an hdm2-dependent pathway.Citation27,Citation28 Overwhelming cellular, biochemical and genetic evidence indicates that p27 is a bona fide substrate for SCFskp2 in S and G2 phase,Citation28–Citation34 and, although it is generally accepted that skp2 can regulate p21 in cycling cells as well, the evidence for this is largely drawn from the observation that p21 levels rise in quiescent serum-deprived, skp2-deficient mouse embryo fibroblasts induced to re-enter the cell cycle.Citation24 Our inability to define a similar S-phase role for skp2 may reflect cell type- or species-specific differences or, because we can see a strong G2-phase role for skp2 and a more modest one for G1-phase, could reflect the purity of the populations that were studied by Bornstein and colleagues. For example, the degree of synchronization obtained in mouse embryo fibroblasts by serum starvation/release protocols is not sufficient to eliminate G1 and G2 cell contamination, when skp2 promotes p21 turnover.
Hdm2 can regulate p21 accumulation in two ways. Indirectly, hdm2 ubiquitinates p53, targeting it for degradation; reducing hdm2 can increase p53-dependent transcription of p21. More directly, hdm2 can bind p21 and target it to the proteasome for degradation.Citation27,Citation28 We noted that neither p53-dependent transcription of p21 nor the accumulation of hdm2 was affected by cell cycle phase; however, in S-phase cells, unlike G1 and G2 cells, the p53-dependent accumulation of p21 was limited by hdm2. This raises an interesting question as to what cell cycle phase-dependent events are controlling the ability of hdm2 to promote p21 turnover.
Are their additional factors missing in G1 cells? Are there factors that are present in G1 cells preventing turnover? To address such questions in the future, we are attempting to develop an in vitro hdm2-p21 turnover system that will allow the biochemical identification of such factors (or modifications) and validate their effect in cellular systems. Additionally, given recent reports that the mTOR signaling environment could affect the outcome of p53 induction vis a vis reversible growth arrest or irreversible senescence,Citation35,Citation36 it is reasonable to speculate that cell cycle phase-specific differences in the signaling environment might also impact mdm2-dependent turnover of p21. Consistent with this, we note that LY294002 and PD98059 were able to inhibit G1 accumulation of p21 protein, although the accumulation of mdm2 mRNA and protein were unaffected (Bhakta R and Koff A, unpublished data). In contrast, neither of these compounds, nor rapamycin, induced S-phase accumulation of p21 in treated cells. This suggests that the activity of PI3-kinase and MEK pathways may be required to control hdm2-p21 turnover events.
Having such an S- and G2-phase dependent proteolytic mechanism is consistent with the findings that persistent blockade of cdk activity in S phase can lead to apoptosis (Shapiro 2006). Thus, the presence of the hdm2-dependent regulatory mechanism to eliminate p21 may allow for the dominance of cdc25-dependent mechanisms to inhibit kinases, a process which is more easily reversible. Thus, unraveling the pathways that prevent p21 turnover in S phase may allow us to consider therapeutic strategies that would drive a cell toward apoptosis through p21-mediated inhibition of cdk activity in S phase.
Material and Methods
Protease inhibitors.
MG132, N-Acetyl-L-leucyl-L-leucyl-L-methioninal (LLM) and N-Acetyl-L-leucyl-L-leucyl-L-norleucinal (ALLN) were acquired from Sigma and used at 50 µM.
Flow cytometry.
In order to simultaneously measure protein and DNA, cells were washed with phosphate buffered saline (PBS) and fixed in 1% paraformaldehyde at 4°C for 15 min. Cells were rinsed twice in PBS, permeabilized in 70% ethanol (added dropwise) and rotated overnight at 4°C. Following fixation, the cells were washed twice with 1% bovine serum albumin in PBS (BSA-PBS) and blocked in 5% goat serum (Vector) for 15 min at room temperature. After washing, primary antibody was added (diluted in BSA-PBS, in a total volume of 500 µL) and cells were incubated in the dark at room temperature for the indicated times: α-p21WAF1 (clone 2G12, Becton Dickinson) at 1:300, 3 h; α-p53 (clone DO-7, Becton Dickinson) at 1:300, 3 h. Parallel isotype-matched antibodies were used as negative controls. Cells were washed twice in BSA-PBS and FITC-conjugated goat anti-mouse secondary antibody (Dako) added at a 1:60 ratio diluted in BSA-PBS for 1 h at room temperature. Following antibody staining, cells were rinsed twice with BSA-PBS, resuspended in a solution containing 5 µg/mL propidium iodide and 100 µg/mL RNase and processed using a FACScan flow cytometer (Becton Dickinson). Data was analyzed on FlowJo version 6.0 software (Tree Star).
Western blot analysis.
Protein lysates were prepared and analyzed as we previously described in reference Citation28 and Citation37. Lysates (80 µg) were resolved by sodium dodecyl sulfate (SDS) PAGE and transferred onto PVDF membranes (Millipore, Bedford, MA). Primary antibodies were added overnight at room temperature, diluted in TNT (25 mM Tris base; 150 mM NaCl; 0.5% Tween-20). Following washing in TNT, the appropriate secondary antibodies conjugated to horseradish peroxidase were added (Jackson Laboratories), diluted 1:5,000 in TNT for 1 h at room temperature. Membranes were washed, Supersignal Pico ECL chemiluminescent reagent (Pierce) added and the membranes exposed to film (Kodak, XAR). The following primary antibodies from Santa Cruz were used: p21 (F5; 1:200), hdm2 (SMP14; 1:200), cyclin B1 (GNS1; 1:1,000), skp2 (H435; 1:200). The p53S15P, chk2T68P and chk2 were all obtained from Cell Signaling Technologies and used at 1:500. The p53 antibody (clone DO-7) was obtained from Becton Dickinson and used at 1:200.
Isolation of cell cycle-phase specific populations (cell sorting).
HCT116 and TK6 cells at an approximate density of 6 × 105 cells/mL were aliquoted and incubated with 3 µg/mL of Hoechst 33,342 at 37°C for 30 min. The cells were then pelleted and resuspended in 5 mL of media containing Hoechst 33,342 and placed on ice for sorting. Sorts were completed within 3 h.
RnA gel blotting.
Total RNA was collected using the RNeasy kit (Qiagen, Valencia, CA) and resuspended in diethyl pyrocarbonate (DEPC)-treated water. The samples were lyophilized and resuspended in 20 mM morpholine-propanesulfonic acid, 5 mM sodium acetate and 1 mM EDTA, 6.5% formaldehyde and 25% formamide heated to 55°C for 15 min and then chilled on ice. RNA was resolved on a 0.6%-agarose formaldehyde gel containing 20 ng/mL ethidium bromide (EtBr) at 100 V for 3 h. The RNA was blotted to Hybond-N+ membrane (Amersham Biosciences) and RNA crosslinked to the membrane with UV light for 12 sec (Stratalinker; Stratagene).
cDNAs containing the entire open reading frame of p21 and PUMA were used to generate probes using the Prime-It Random Primer Labeling kit (Stratagene, La Jolla, CA). QuickHyb hybridization solution (Stratagene, La Jolla, CA) was used to pre-hybridize the membrane for 30 min at 68°. A mixture of 1 × 107 CPM of probe and 100 µg of sonicated salmon sperm DNA were boiled for 5 min and added to the hybridization buffer for 2 h. The membrane was then washed with 2x SSC containing 0.1% SDS twice for 15 min at room temperature and once for 35 min with 0.1x SSC containing 0.1% SDS at 68°C. For detection of the specific RNA, the membrane was exposed to XMR film (Eastman Kodak, Chedex, France) overnight at −80°C.
Laser scanning cytometry.
Adherent cells were plated in 4-well chamber slides (Nalge Nunc) at a density of 85,000 cells/well and allowed to attach overnight. Following irradiation, the cells were fixed using 1% paraformaldehyde for 15 min at 4°C and subsequently permeabilized in 80% ethanol at 4°C overnight. Fixed slides were initially rinsed in BSA-PBS twice for 5 min and then blocked with 5% goat serum for 15 min. Either α-p53-FITC-conjugated antibody or an IgG2b-FITC isotype (1 mg/mL) diluted in BSA-PBS was carefully dropped onto the slide and then covered with a layer of parafilm to allow even distribution of the antibody along the slide. After a 2 h incubation at room temperature in the dark, slides were thoroughly rinsed with PBS and placed in a PI (5 µg/mL)/RNase (100 µg/mL) solution overnight at 4°C. The slides were then mounted with cover-slips using PBS and analyzed under a laser-scanning microscope (CompuCyte). Analysis included identification of 100 individual S-phase cells.
siRNA transfection.
SMARTpools of four siRNAs specific for human skp2 or hdm2 were obtained from Dharmacon. Approximately 1 × 106 HCT116 cells were resuspended in 81.8 µl of nucleofector solution V, 18.2 µl supplement and 8 µl 20 µM siRNA (2 µg) and electroporated using the Nucleofector device as recommended by the manufacturer (Amaxa). Cells were harvested either 24 (hdm2) or 48 (skp2) hours after electroporation.
Figures and Tables
Figure 1 Cell cycle phase-dependent accumulation of p21 in cells after induction of p53. (A) p53 and p21 protein accumulation following ionizing radiation. The indicated asynchronously growing transformed cells were irradiated and extracts prepared 6 h later and p21 and p53 were detected by immunoblot. Cell types are indicated above each autoradiograph, (−) no irradiation, (+) 10 Gy. (B) TK6 lymphoblasts were grown and separated into cell cycle phase enriched populations by centrifugal elutriation prior to irradiation. Top part, propidium iodide staining was used to monitor the position of the cells in the cell cycle; Bottom part, the amount of p21 and p53 was detected by immunoblot. The dose or ionizing radiation and the time after treatment prior to harvest and analysis is indicated. (C) Asynchronously growing HCT116 cells were irradiated, allowed to grow for an additional 3 h and then stained for 30 min with Hoescht 33,342 and sorted into populations. S-phase synchrony and S-phase contamination in G1 and G2 populations was assessed by a 15 min pulse of BrdU (data not shown). Extracts were prepared for immunoblot as indicated in each part. Cyclin B1 is a loading and synchronization control. This experiment was repeated four times with different doses or ionizing radiation in different cell lines, all with similar results. A representative example is shown. (D) Asynchronously growing TK6 cells were treated as indicated and incubated for an additional 3 h, after which they were fixed and stained with either an antibody to p21 or a control isotype as indicated in the parts. Antibody staining is on the y-axis and DNA content on the x-axis. The median level of p21 expression at five points along the DNA profile was determined and plotted. This graph shows the mean and standard deviation compiled from six independent experiments performed at each indicated dose. (E) Immortalized RPE-hTERT epithelial cells were irradiated with 5 Gy and stained as described in (D). In these parts, the line indicates above which no cells were detected by isotype staining. The y-axis in the graph indicates the percent p21-positive cells after subtracting the cells below the gate in the flow cytometric plots. The graphed data was compiled from three independent experiments. (F) Nutlin-3 did not induce S-phase accumulation of p21. In the top part, extracts were prepared from HCT116 cells treated as described above each lane and the amount of hdm2, p53 and p21 determined by immunoblot. In the bottom part, cells were stained as described in (E) and the percentage of p21 positive cells in each cell cycle phase determined.
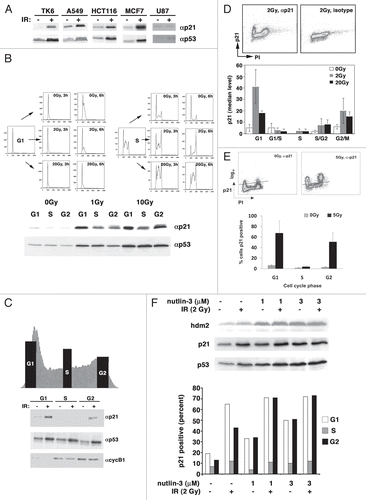
Figure 2 Transcriptionally active p53 accumulates in all phases of the cell cycle following irradiation. (A) Centrifugal elutriation. As described in the legend to , we looked at the expression of serine15-phosphorylated p53, chk2, threonine68-phosphorylated chk2 and hdm2 by immunoblot. (B) Asynchronously growing A549 cells were plated onto coverslips and p53 immunofluorescence carried out. The percentage of S-phase cells with nuclear p53 was plotted as a function of time. This experiment was repeated with MCF7 cells. (C) Enriched populations of TK6 cells were obtained by cell sorting as described in the legend to and the expression of p21 and PUMA mRNA determined by RNA gel blotting. Ethidium bromide staining of 28S and 18S rRNA is a loading control. (D) TK6 cells were irradiated with 5 Gy and assessed for p53 expression in different cell cycle phases by flow cytometry as desribed in the legend to . p53 expression before and after irradiation is also depicted graphically (lower part). This is a representative image from one experiment. (E) Immortalized RPE-hTERT epithelial cells were irradiated with 5 Gy and assessed for p53 expression as described in the legend to .
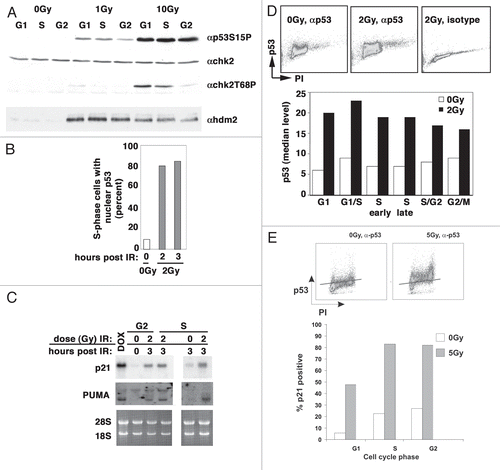
Figure 3 Proteasome activity inhibits p21 turnover in S phase. (A) Asynchronously growing TK6 cells were treated with either nocodazole or hydroxyurea and subsequently released to enter G1/G2 and S phase, respectively. Representative flow profiles for the synchronized populations are shown (up per left). Extracts were prepared from these enriched populations and tested for the ability to degrade in vitro-translated p21. Degradation reactions were prepared as outlined in the Materials and Methods section and incubated at 30°C for the times indicated above each lane prior to separation by SDS-PAGE and detection by autoradiography, a representative example of which is shown in the lower left. On the right, the top graphs combine data from four independent experiments using different preparations of substrate and extract, and the bottom part is from a representative experiment in which a very rapid time course was examined. (B) HCT116 cells were exposed to ionizing radiation and immediately treated for 6 h with either MG132 or LLM. After treatment, cells were fixed and stained with an antibody to p21, which was detected with a secondary antibody conjugated to FITC, prior to counterstaining of DNA with propidium iodide. Representative flow cytometric profiles are shown with treatment conditions indicated above the dot-plots. The graph is compiled from at least three independent experiments for each condition.
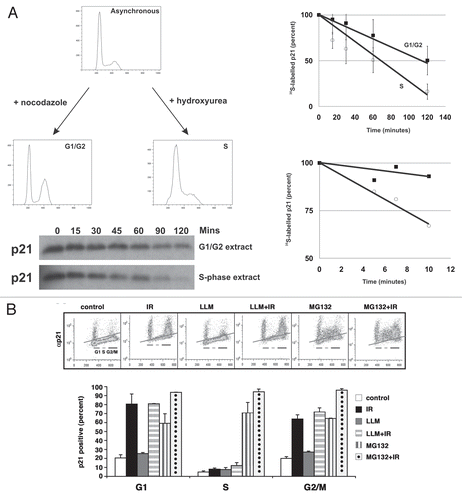
Figure 4 Skp2 does not contribute to p21 turnover in S-phase cells. (A) HCT116 cells were transfected with either a scrambled siRNA or a SMARTpool skp2 siRNA by electroporation. Cells were harvested 48 h later and processed. Left part, immunoblot; Right part, cell cycle phase dependent accumulation of p21 as measured by flow cytometry. The graph compiles the average and standard deviation of three independent experiments. Cells received 5 Gy of ionizing radiation. (B) Similar to (A); however, we used RPE-hTERT epithelial cells.
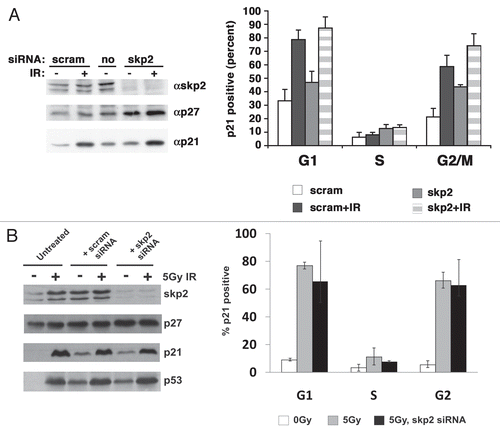
Figure 5 hdm2 contributes to p21 turnover in S-phase HCT116 cells but not RPE-hTERT. This is exactly as described in the legend to , except hdm2 was knocked down instead of skp2.
Acknowledgments
The authors thank Jacob Jacobberger (Case Western Reserve University) and Frank Traganos (Brander Cancer Institute, New York Medical College) for advice with the p21 dual-color flow cytometry; Gloria Juan and Carlos Cordon-Cardo (MSKCC, Department of Pathology) for assistance with the analysis of p53 localization; and Vincent Sahi, Patrick Anderson and Cris Bare (MSKCC, Flow Cytometry Core Facilities) for developing the conditions for cell sorting. We also thank Stephen Jones (University of Massachussetts/Worcester), Carol Prives (Columbia University), Vanesa Gottifredi (Fundacion Instituto Leloir) and Pengbo Zhao (Weill College of Medicine, Cornell University) for helpful discussions as our work progressed and Roshni Mody-Bhakta and Carmen Carneiro, former members of the laboratory who provided the initial insight and preliminary data on the S phase-dependent regulation of p21. This work was supported by grants to Andrew Koff from the NCI (CA89563) and Golfers Against Cancer Foundation. Additional support was provided by an Institutional Core Grant to Memorial Sloan-Kettering Cancer Center (NCI). Daniel Ciznadija was supported by fellowships from the Brain Tumor Center (Memorial Sloan-Kettering Cancer Center) and the Joel A. Gingras Jr. Basic Research Fellowship from the American Brain Tumor Association.
References
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39 - 85; PMID: 15189136; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723
- Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol 2003; 13:49 - 58; PMID: 12507556; http://dx.doi.org/10.1016/S1044-579X(02)00099-8
- Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T, Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature 1995; 377:552 - 557; PMID: 7566157; http://dx.doi.org/10.1038/377552a0
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 1501; PMID: 9822382; http://dx.doi.org/10.1126/science.282.5393.1497
- Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82:675 - 684; PMID: 7664346; http://dx.doi.org/10.1016/0092-8674(95)90039-X
- di Pietro A, Vries EG, Gietema JA, Spierings DC, de Jong S. Testicular germ cell tumours: the paradigm of chemo-sensitive solid tumours. Int J Biochem Cell Biol 2005; 37:2437 - 2456; PMID: 16099193; http://dx.doi.org/10.1016/j.biocel.2005.06.014
- Hastak K, Agarwal MK, Mukhtar H, Agarwal ML. Ablation of either p21 or Bax prevents p53-dependent apoptosis induced by green tea polyphenol epigallo-catechin-3-gallate. FASEB J 2005; 19:789 - 791; PMID: 15764647
- Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187 - 5190; PMID: 7585571
- Bartek J, Lukas C, Lukas J. Checking on DNA damage in S phase. Nat Rev Mol Cell Biol 2004; 5:792 - 804; PMID: 15459660; http://dx.doi.org/10.1038/nrm1493
- Falck J, Petrini JH, Williams BR, Lukas J, Bartek J. The DNA damage-dependent intra-S phase checkpoint is regulated by parallel pathways. Nat Genet 2002; 30:290 - 294; PMID: 11850621; http://dx.doi.org/10.1038/ng845
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat Rev Mol Cell Biol 2000; 1:179 - 186; PMID: 11252893; http://dx.doi.org/10.1038/35043058
- Nakanishi K, Taniguchi T, Ranganathan V, New HV, Moreau LA, Stotsky M, et al. Interaction of FANCD2 and NBS1 in the DNA damage response. Nat Cell Biol 2002; 4:913 - 920; PMID: 12447395; http://dx.doi.org/10.1038/ncb879
- Gottifredi V, McKinney K, Poyurovsky MV, Prives C. Decreased p21 levels are required for efficient restart of DNA synthesis after S phase block. J Biol Chem 2004; 279:5802 - 5810; PMID: 14597617; http://dx.doi.org/10.1074/jbc.M310373200
- Soria G, Podhajcer O, Prives C, Gottifredi V. P21Cip1/WAF1 downregulation is required for efficient PCNA ubiquitination after UV irradiation. Oncogene 2006; 25:2829 - 2838; PMID: 16407842; http://dx.doi.org/10.1038/sj.onc.1209315
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003; 114:599 - 610; PMID: 13678583; http://dx.doi.org/10.1016/j.cell.2003.08.001
- Deptala A, Li X, Bedner E, Cheng W, Traganos F, Darzynkiewicz Z. Differences in induction of p53, p21WAF1 and apoptosis in relation to cell cycle phase of MCF-7 cells treated with camptothecin. Int J Oncol 1999; 15:861 - 871; PMID: 10536167
- Thompson T, Tovar C, Yang H, Carvajal D, Vu BT, Xu Q, et al. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J Biol Chem 2004; 279:53015 - 53022; PMID: 15471885; http://dx.doi.org/10.1074/jbc.M410233200
- Bloom J, Pagano M. Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin Cancer Biol 2003; 13:41 - 47; PMID: 12507555; http://dx.doi.org/10.1016/S1044-579X(02)00098-6
- Pagano M. Control of DNA synthesis and mitosis by the Skp2-p27-Cdk1/2 axis. Mol Cell 2004; 14:414 - 416; PMID: 15149588; http://dx.doi.org/10.1016/S1097-2765(04)00268-0
- Darzynkiewicz Z, Crissman H, Jacobberger JW. Cytometry of the cell cycle: cycling through history. Cytometry A 2004; 58:21 - 32; PMID: 14994216; http://dx.doi.org/10.1002/cyto.a.20003
- Gottifredi V, Shieh S, Taya Y, Prives C. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proc Natl Acad Sci USA 2001; 98:1036 - 1041; PMID: 11158590; http://dx.doi.org/10.1073/pnas.021282898
- Komarova EA, Zelnick CR, Chin D, Zeremski M, Gleiberman AS, Bacus SS, et al. Intracellular localization of p53 tumor suppressor protein in gamma-irradiated cells is cell cycle regulated and determined by the nucleus. Cancer Res 1997; 57:5217 - 5220; PMID: 9393737
- Jascur T, Brickner H, Salles-Passador I, Barbier V, El Khissiin A, Smith B, et al. Regulation of p21(WAF1/CIP1) stability by WISp39, a Hsp90 binding TPR protein. Mol Cell 2005; 17:237 - 249; PMID: 15664193; http://dx.doi.org/10.1016/j.molcel.2004.11.049
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem 2003; 278:25752 - 25757; PMID: 12730199; http://dx.doi.org/10.1074/jbc.M301774200
- Wang W, Nacusi L, Sheaff RJ, Liu X. Ubiquitination of p21Cip1/WAF1 by SCFSkp2: substrate requirement and ubiquitination site selection. Biochemistry 2005; 44:14553 - 14564; PMID: 16262255; http://dx.doi.org/10.1021/bi051071j
- Yu ZK, Gervais JL, Zhang H. Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc Natl Acad Sci USA 1998; 95:11324 - 11329; PMID: 9736735; http://dx.doi.org/10.1073/pnas.95.19.11324
- Jin Y, Lee H, Zeng SX, Dai MS, Lu H. MDM2 promotes p21waf1/cip1 proteasomal turnover independently of ubiquitylation. EMBO J 2003; 22:6365 - 6377; PMID: 14633995; http://dx.doi.org/10.1093/emboj/cdg600
- Zhang Z, Wang H, Li M, Agrawal S, Chen X, Zhang R. MDM2 is a negative regulator of p21WAF1/CIP1, independent of p53. J Biol Chem 2004; 279:16000 - 16006; PMID: 14761977; http://dx.doi.org/10.1074/jbc.M312264200
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661 - 672; PMID: 15130491; http://dx.doi.org/10.1016/S1534-5807(04)00131-5
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 2000; 19:2069 - 2081; PMID: 10790373; http://dx.doi.org/10.1093/emboj/19.9.2069
- Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, et al. A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell 2001; 7:639 - 650; PMID: 11463388; http://dx.doi.org/10.1016/S1097-2765(01)00210-6
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, et al. The cell cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 2001; 3:321 - 324; PMID: 11231585; http://dx.doi.org/10.1038/35060126
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM. A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 2001; 413:323 - 327; PMID: 11565035; http://dx.doi.org/10.1038/35095083
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, et al. Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell 2005; 20:9 - 19; PMID: 16209941; http://dx.doi.org/10.1016/j.molcel.2005.09.003
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 352; PMID: 20606252
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 2010; 107:9660 - 9664; PMID: 20457898; http://dx.doi.org/10.1073/pnas.1002298107
- Nguyen H, Gitig DM, Koff A. Cell-free degradation of p27(kip1), a G1 cyclin-dependent kinase inhibitor, is dependent on CDK2 activity and the proteasome. Mol Cell Biol 1999; 19:1190 - 1201; PMID: 9891053