Abstract
Comment on: Kozarova A, et al. Cell Cycle 2011; 10:1669-78.
Knocking down hYVH1/DUSP12 in human cells results in cell cycle arrest, as revealed by the striking increase in cells with G0/G1 DNA content following siRNA treatment. This result was further reiterated by a complementary experiment, in which ectopic hYVH1/DUSP12 expression led to arrest at the next checkpoint, with a decrease in G0/G1 population and a matched increase in G2/M cell population.Citation1
These interesting observations are outlined in the May 15th issue of Cell Cycle in a report by Kozarova et al. Together, they unveil a new dramatic role for hYVH1/DUSP12 at two critical checkpoints of the cell cycle that comes on the heels of a newly identified role of YVH1/DUSP12 in eukaryotes ribosome biogenesis.
hYVH1/DUSP12Citation2 is member of the dual-specificity phosphatases (DUSPs), a heterogeneous group that belongs to the protein tyrosine phosphatase (PTP) super family.Citation3 Based on sequence similarity, the DUSP group should be subdivided in seven subgroups: slingshots, PRLs, CDC14s, PTENs, myotubularins, MKPs and atypical dual-specificity phosphatases. hYVH1/DUSP12 belongs to the latter.Citation4
Until recently, little was known about the function of hYVH1/DUSP12, but recent reports from distinct groups demonstrated a role of yeast YVH1 in the control of ribosome assembly.Citation5,Citation6 More specifically, two groups have shown that YVH1 plays a role in the maturation of 60S particles assembly, and this function is conserved from yeast to human.Citation7
There is growing evidence that during normal conditions or exposure to cellular insults, ribosome biogenesis and cell cycle checkpoint pathways are strategically coordinated in order to assess cell size and DNA integrity prior to cell cycle progression.Citation8 Disruption of this relationship (e.g., during tumourigenesis) permits cells to proliferate and survive under conditions of cellular stress and/or genomic damage. Although the connection between ribosome biogenesis and cell cycle progression has gained much attention recently, the mechanistic details of this relationship remain poorly defined.
The remarkable observations made by Kozarova et al. raise the provocative suggestion of a complex but prominent function for hYVH1/DUSP12 in the regulation of cell proliferation in eukaryotic cells. In fact, if we integrate the new results with the role in ribosome biogenesis, we are lead to propose that hYVH1/DUSP12 plays a strategic role by networking two fundamental steps in cell physiology: cell cycle progression and ribosome assembly. In addition, because hYVH1/DUSP12 orthologs are extremely conserved throughout evolution, it is plausible that this strategic function might be a general mechanism of synchronizing cell division with fully functional protein translation machinery.
hYVH1/DUSP12 has been shown to be amplified in several cancers.Citation2 It is therefore increasingly important to further characterize and dissect all the roles of hYVH1/DUSP12 and their intersections, as it appears that analyzing its functions in details could lead to the discovery of new and essential mechanisms supporting tumor growth.
In summary, the report by Kozarova et al. is a further indication calling attention to the DUSPs, a group of enzymes with conserved function that, when mutated, can cause diverse diseases, such as cancer (PTEN), miopathy (MTM1) and epilepsy (Laforin).Citation3–Citation4
MicroRNAs are small 20–22 nucleotide RNAs that repress translation of specific protein coding genes. Over the last decade, our knowledge about the role of microRNAs in human diseases, including cancer, has grown exponentially. The connection between microRNAs and human disease began with the seminal discovery that these small RNAs were conserved in humans.Citation1 Subsequent studies indicated that microRNAs were involved in virtually every biological and developmental process examined.2 Nine years ago, the first connection between altered microRNA expression and cancer was reported, demonstrating that specific microRNAs were either deleted or down regulated in the majority of B-cell lymphomas.Citation3 Since that time, there has been overwhelming evidence demonstrating the connection between altered microRNA expression and cancer. The expression of microRNAs has been found to be altered in every malignancy examined.Citation4 Their expression patterns can distinguish tumor types and tissue types at least as well as, if not better than, mRNA expression patterns, highlighting their potential as biomarkers for cancer. Expression levels of microRNAs are associated with prognosis and therapeutic outcome in several cancer types, indicating that they may be useful as prognostic classifiers for cancer.Citation5,Citation6 Polymorphisms in microRNAs and microRNA binding sites are associated with cancer incidence.Citation7 MicroRNAs have a functional role in the initiation, progression and maintenance of various malignancies. For example, the over-expression of a single microRNA can induce malignancies in mice that are dependent on that microRNA.Citation8 Progress is being made on targeting microRNAs for therapeutic purposes. The reintroduction of tumor suppressor microRNAsCitation9 or inhibition of oncogenic microRNAsCitation10 have both shown potential in preclinical models. Therefore, microRNAs may be clinically useful biomarkers and therapeutic targets for cancer.
In order to translate breakthroughs in microRNA research into clinical practice, we must identify avenues of research that can address fundamental questions and issues relevant to the diagnosis, prognosis and treatment of cancer. Can microRNA expression patterns help identify patients at high risk of relapse to guide the decision on which patients should receive more intense screening or adjuvant therapy? Can their expression patterns help diagnose and classify subtypes of malignancies to help predict which patients will respond well to specific therapies? Can microRNAs be targeted therapeutically to treat various types of cancer, and how do we know which microRNAs would be most effective to target for any given patient? In their publication in the June 1st issue of Cell Cycle, Elcie Chan and colleagues address this area by using the expression of microRNAs in addition to mutations in specific microRNA binding sites to identify subgroups of melanoma patients.Citation11
In the United States alone, there are approximately 68,000 new cases of melanoma and 8,700 deaths annually. There are four subtypes of melanoma, including superficial spreading, nodular, lentigo maligna and acral lentiginous melanoma. Chan et al. identified microRNAs that were differentially expressed between cell lines derived from different subtypes of melanoma and are the first to report that microRNA expression patterns of acral lentiginous melanoma differ from other subtypes of melanoma. The differences in microRNA expression may be responsible for some of the biological differences between these subtypes. The authors also characterize a polymorphism in the binding site for the let-7 microRNA, which is located in the 3′UTR of KRAS. The authors have previously reported that this polymorphism inhibits the ability of let-7 to regulate the translation of KRAS and is associated with an increased risk for lung cancer. In the present study, they demonstrate that 25% of the non-acral lentiginous samples have this polymorphism suggesting its association with an increased risk of melanoma.
Future areas of interest will be to determine if these microRNA expression patterns can be used in clinically useful ways to further stratify melanoma patients into those who will be more receptive to particular therapies or those who will be in longer remission following surgery. Since acral lentiginous tumors are more common in darker skinned populations, it will be interesting to determine whether the same differences between acral lentiginous melanomas are seen in different ethnicities, since Chan and colleagues examine a Caucasian population. It is important to determine whether tumors that harbor mutations in the let-7 binding site of KRAS define a clinically relevant subgroup of melanoma. Do these patients respond well to specific inhibitors? Does this polymorphism lead to increased risk of melanoma, and, if so, can we use this information in an actionable way to help prevent the disease?
Altered microRNA expression is undeniably linked to cancer. Continued research in these areas should lead to novel breakthroughs to help prevent, diagnose and treat cancer.
References
- Pasquinelli AE, et al. Nat Rev Mol Cell Biol 2008; 9:219 - 230
- Calin GA, et al. Proc Natl Acad Sci USA 2002; 99:15524 - 15529
- Volinia S, et al. Proc Natl Acad Sci USA 2006; 103:2257 - 2261
- Schetter AJ, et al. Jama 2008; 299:425 - 436
- Yanaihara N, et al. Cancer Cell 2006; 9:189 - 198
- Ryan BM, et al. Nat Rev Cancer 2010; 10:389 - 402
- Medina PP, et al. Nature 2010; 467:86 - 90
- Trang P, et al. Mol Ther 2011; 19:1116 - 1122
- Lanford RE, et al. Science 2010; 327:198 - 201
- Chan E, et al. Cell Cycle 2011; 10:1845 - 1852
MicroRNA expression profiling has been used to classify cancers and predict progression and responses to therapies. Typically, microRNA expression in cancer cells closely approximates, but is distinct from, untransformed cell of origin.Citation1 During cancer progression, the expression of most tissue-specific microRNAs is maintained and thus can be used to identify the tissue of origin, whereas the expression of other microRNAs change in a characteristic manner that can be used to identify the presence of cancer. A recent report by Slack, Weidhaas and colleagues challenges this concept and suggests novel insights into the biology of an uncommon subtype of melanomas.Citation2
Consistent with previous microRNA expression profiling data in melanomas,Citation1,Citation3,Citation4 Chan et al. identified several differentially expressed microRNAs between melanocytes and melanomas. However, when corrected for multiple comparisons, statistically significant changes in microRNA expression were not discernable between melanocytes and melanomas, perhaps implying the existence of subtle changes in microRNA regulation leading to large biological differences when melanocytes undergo transformation into melanomas. Although globally microRNA expression was unchanged in melanomas relative to melanocytes, a subset of KRAS variant melanomas (with mutations in the let-7 microRNA binding site)Citation5 demonstrated statistically significant changes in microRNA expression relative to melanocytes. Chan et al. report a high frequency (25%) of mutations in the melanoma oncogene KRAS and a statistically significant inverse association of KRAS variants with miR-137 expression in melanomas arising on sun-exposed epithelial surfaces. Thus, the authors' data is consistent with a model in which loss of let-7-mediated repression upregulates KRAS, which upregulates the transcriptional regulator MECP2, which increases transcriptional repression of miR-137, which results in de-repression of predicted miR-137 target genes such as CDK6.
Other potentially important implications in melanoma biology became apparent when the more common melanomas were compared to the relatively rare acral lentiginous melanomas that arise on non-sun-exposed epithelial surfaces, such as the palms of hands, soles of feet and mucous membranes. Acral melanoma, the subtype that claimed the life of Bob Marley at the age of 36, most frequently affects Asians and people with dark skin. Six upregulated microRNAs (miR-31, miR-142-3p, miR-214, miR-218, miR-486, and miR-650) and one downregulated microRNA (miR-362) distinguish acral from non-acral melanomas, implying that the mechanisms of melanocyte transformation in acral melanomas may be significantly different from sun-exposed melanomas. It is likely that the altered expression of these seven differentially expressed microRNAs may discern the genetic and/or environmental susceptibility to acral versus non-acral melanomas.
c-KIT is a melanoma oncogene with a critical role in melanocyte developmentCitation6 and is mutated in approximately one third of acral melanomas.Citation7 Early analyses suggested that melanomas were not responsive to the receptor tyrosine kinase inhibitor imatinib mesylate (Gleevec).Citation8,Citation9 However, more recent data indicates that melanomas with particular c-KIT mutations are responsive to Gleevec.Citation10 Notably, c-KIT is a predicted target of miR-218 (as well as miR-137).Citation11 It would therefore be interesting and clinically relevant to test whether changes in miR-218 (or miR-137) or their predicted binding sites in the c-KIT 3′ untranslated region affect Gleevec sensitivity of acral melanomas. Further research into detection of microRNA binding site variants in melanoma oncogenes may reveal new pathways of melanomagenesis and predict drug responses.
References
- Lu J, et al. Nature 435:834 - 838
- Chan E, et al. Cell Cycle 2011; 10:1845 - 1852
- Gaur A, et al. Cancer Res 67:2456 - 2468
- Mueller DW, et al. J Invest Dermatol 2009; 129:1740 - 1751
- Paranjape T, et al. Lancet Oncol 2011; 12:377 - 386
- Fleischman RA, et al. Proc Natl Acad Sci USA 1991; 88:10885 - 10889
- Curtin JA, et al. J Clin Oncol 2006; 24:4340 - 4346
- Ugurel S, et al. Br J Cancer 2005; 92:1398 - 1405
- Wyman K, et al. Cancer 2006; 106:2005 - 2011
- Hodi FS, et al. J Clin Oncol 2008; 26:2046 - 2051
- Lewis BP, et al. Cell 2005; 120:15 - 20
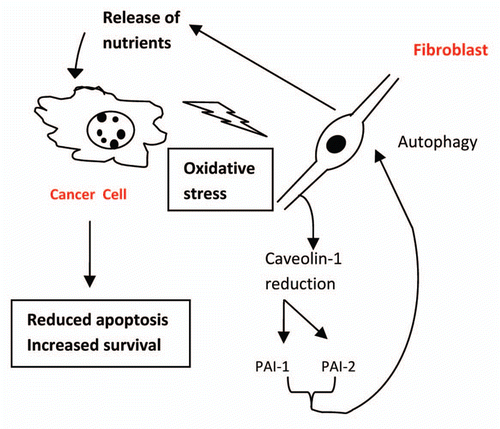
Though not particularly new in concept, at least in terms of its existence, the crosstalk between normal stromal cells and transformed epithelial cells has come to be seen, in recent years, as a major determinant of carcinoma initiation and progression.Citation1 Thus, probably through the elaboration and release of various molecules, including growth factors and/or reactive oxygen species, carcinomas are able to modify their local environment, and this modification includes induction of a more “activated” state in adjacent fibroblasts.Citation2 Lisanti and colleagues have come up with a novel twist on how this communication between stromal and transformed entities might facilitate cancer development and progression. Their work suggests that a lack, or downregulation, of caveolin-1 expression in the stromal compartment, specifically in fibroblasts, induces authophagy in these cells, leading to the liberation of recycled nutrients, which can then be utilized by adjacent carcinoma cells, as indicated by increased oxidative metabolism in mitochondria, to promote decreased apoptosis and increased cell survival.Citation3,Citation4 in their recent report,Citation5 they suggest, based on the identification of PAI-1 and PAI-2 as upregulated proteins in caveolin-1-null (-/-) fibroblasts relative to their wild-type controls, that these inhibitors of plasminogen activator somehow play an intermediary role in this induction of fibroblastic autophagy. These carcinoma cell-fibroblast interactions are summarized by the cartoon depicted in and suggest that PA1-1 and PAI-2 in tumor-associated fibroblasts might represent a logical target for anticancer therapy.
The concept of “the autophagic tumor stroma model of cancer cell metabolism”Citation4 has interesting possibilities that need to be explored. It is known, for example, that loss of caveolin-1 function in stromal cells “leads to a disorganized stromal compartment that, in turn, indirectly promotes abnormal growth and differentiation of adjacent epithelium.”Citation6 It certainly would seem plausible then that the caveolin-1-null (-/-) cells should impact upon tumor development and even progression, as they do in the manner outlined in the report by Castello-Cros et al.Citation5 Indeed, these authors previously have shown that loss of stromal caveolin-1 is sufficient to promote tumor growth.Citation7 This is true even if the evidence for lack of caveolin-1 expression being associated with poor prognosis in breast cancer is not perhaps as unequivocal as the authors suggest.Citation8 What is more difficult to assess is whether the proposed mechanism of autophagy, with consequent nutrients release, truly does lie behind the impact of “activated” caveolin-1 deficient fibroblasts on cancer cell survival. Surely a corollary of this hypothesis would be that, in caveolin-1-deficient cancers, autophagy of stromal cells would lead, over time, to a gradual reduction in the fibroblastic content of the primary tumor? The fact that another, independent group already has shown that caveolin-1-deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibilityCitation9 further suggests that a loss of caveolin-1 expression should be associated with a gradual reduction in stromal content. If nutrients released by these autophagic fibroblasts were so important for increased growth/decreased apoptosis of cancer cells, might it not be expected that a limited fibroblast compartment, caused as a consequence of increased autophagy, would be an indicator of poor-prognosis cancers? How then to explain the apparent higher malignancy of breast tumors with an abundant desmoplastic reaction?
Interestingly, idiopathic pulmonary fibrosis is associated with a marked reduction in caveolin-1,Citation10 which then appears to be associated with induction of the pro-fibrotic cytokine, TGFβ1. Castello-Cros et al.Citation5 comment that caveolin-1 knockdown in fibroblasts is sufficient to activate the TGFβ signaling pathway, and that this pathway already is known to be capable of regulating PAI-1 expression. With the established pleiotropic effects of TGFα on cancer, such that the inhibitory impact of this cytokine on tumor cell growth becomes stimulatory in the later stages of disease, it seems possible that the impact of caveolin-1 reduction in fibroblasts might act through the known mechanisms of TGFβ, and this might occur without the need to invoke an autophagy component to the process. Certainly the evidence for autophagy in the present reportCitation5 is somewhat indirect.
Whatever the eventual molecular mechanism(s) driving tumor progression turn out to be, the report by Castello-Cros et al.Citation5 adds to the burgeoning literature indicating that the stromal compartment of cancers is both a major mediator of overall tumor behavior and a bona fide target for therapeutic interventions. Future investigations in this area can only increase our understanding of how the complex microenvironments of tumors are modulated.
Figures and Tables
Figure 1 Nature of cancer cells.
References
- Bhowmick NA, et al. Nature 2004; 432:332 - 337
- Kalluri R, et al. Nat Rev Cancer 2006; 6:392 - 401
- Martinez-Outschoorn UE, et al. Cell Cycle 2010; 9:4297 - 4306
- Pavlides S, et al. Cell Cycle 2010; 9:3485 - 3505
- Castello-Cros R, et al. Cell Cycle 2011; 10:2021 - 2034
- Young G, et al. Exp Mol Path 2008; 84:131 - 14
- Trimmer C, et al. Cancer Biol Ther 2011; 11:383 - 394
- Elsheikh SE, et al. Br J Cancer 2008; 99:327 - 334
- Bosch M, et al. Curr Biol 2011; 681 - 686
- Wang XM, et al. J Exp Med 2006; 2895 - 2906
Autophagy is emerging as an important cellular process in tumor progression. However, whether the activation of autophagy promotes or suppresses tumor development is dependent upon a number of factors, particularly the malignancy of the cell.Citation1 Recently, Lisanti and colleagues identified another layer of complexity in the autophagic system, which they term the “autophagic tumor stroma model of cancer metabolism.”Citation2 In this model, autophagy is activated in stromal cells, producing amino acids and other metabolites, which are then consumed by neighboring tumor cells, increasing metastasis. So, here, autophagy is indeed a tumor-promoting mechanism, albeit in a paracrine manner.
Thus far, the Lisanti group has linked this autophagic stromal model of malignancy to the loss of caveolin-1 (Cav-1) and the activation of HIF1α.Citation2,Citation3 In their publication in a recent issue, “Matrix remodeling stimulates stromal autophagy, ‘fueling’ cancer cell mitochondrial metabolism and metastasis,” the factors that control stromal fibroblast autophagy have been extended to include plasminogen activator inhibitor type 1 and type 2 (PAI-1 and PAI-2).Citation4 In mammary-derived stromal fibroblasts, the loss of Cav-1 led to an increase in PAI-1 and PAI-2 expression. Additionally, the overexpression of PAI-1 and PAI-2 in fibroblasts promoted stromal autophagy and increased mitochondrial biogenesis and metastasis in a human tumor cell line, making PAI-1 and PAI-2 mediators of Cav-1-induced autophagy.
This “reverse Warburg” model of tumor metabolism has been gaining support in the last few years. The classic Warburg model proposes that tumor cells are defective in respiration and mitochondrial metabolism and, instead, are dependent upon the glycolytic conversion of glucose to lactate. The “reverse” model proposes that at least one subset of tumor cells use respiration as a primary source of ATP production, and that inhibiting respiration can inhibit tumor development. Varied mechanisms can cause this dependence upon respiration. For example, Sonveaux et al. observed that lactic acid produced by hypoxic tumor cells may then be consumed by neighboring oxygenated tumor cells.Citation5 The Lisanti group has reported a similar model, except that in this case, it is cancer-associated fibroblasts that produce the lactic acid that drives tumor respiration and malignancy (much like the autophagic exchange described in a previous issue).Citation6 A signaling mechanism that drives such a switch from glycolysis to respiration has also been identified by Fogal et al., in which the cell surface/mitochondrial protein p32/C1QBP increases oxidative phosphorylation and tumorgenicity.Citation7
A major question that emerges from the current study is how do the autophagic stromal fibroblasts stimulate mitochondrial biogenesis in the surrounding tumor cells? As the fibroblast cells in this and previous studies are genetically manipulated, the obvious hypothesis is that a component of the Cav-1/PAI-1 signaling system acts to promote mitochondrial biogenesis in neighboring cells. Alternately, the tumor cells may directly sense the presence of metabolites produced by autophagy and increase their mitochondrial mass in order to take advantage of the nutrient-rich environment. The addition of metabolomic and metabolic flux studies to future work will help to shed light on these mechanisms.
As our understanding of metabolic signaling in cancer increases, we will have increasing chances to manipulate these metabolic programs to our advantage. Most directly, metabolic enzymes tend to be druggable, allowing us to inhibit metabolic pathways that are needed for the unrestrained growth of tumors. More intriguingly, understanding state-specific metabolic needs may allow us to inhibit specific malignant states. For example, in the report being discussed here, PAI-1/2-overexpressing fibroblasts did not increase the proliferation of tumor cells in vivo but, instead, increased metastasis to the lungs. Further identifying the signaling or metabolic pathways that mediate this increase in metastasis may allow us to inhibit metastasis-specific metabolic states, an outcome of great clinical importance. While inhibiting the Warburg effect in tumor cells has thus far had limited clinical utility, expanding our knowledge of cancer metabolism beyond the Warburg effect may yet allow the metabolic manipulation of tumor progression.
References
- Mathew R, et al. Curr Opin Genet Dev 2011; 21:113 - 119
- Pavlides S, et al. Cell Cycle 2010; 9:3485 - 3505
- Chiavarina B, et al. Cell Cycle 2010; 9:3534 - 3551
- Castello-Cros R, et al. Cell Cycle 2011; 10:2021 - 2034
- Sonveaux P, et al. J Clin Invest 2008; 118:3930 - 3942
- Bonuccelli G, et al. Cell Cycle 2010; 9:3506 - 3514
- Fogal V, et al. Mol Cell Biol 2010; 30:1303 - 1318
The identification of candidate tumor suppressors in frequently deleted and/or microsatellite instable chromosomal regions is often elusive. This was also the case with 17q.21, which shows recurrent loss of heterozigosity (LOH) in several human neoplasias. For obvious reasons, previous studies on 17q21 had concentrated on BRCA1. However, evidences from genetic linkage analysis suggested the existence of an additional tumor suppressor locus within this region. Indeed, exhaustive mapping showed that 53% of primary human lung carcinomas displayed LOH within a chromosomal region distal to BRCA1.Citation1 The Spinophilin (PPP1R9B) locus is precisely located in this genomic position and, indeed, the Spinophilin protein is absent from a significant number of human lung tumors when assessed by immunohistochemistry.Citation2
Spinophilin (also known as Neurabin 2) is a scaffold protein that interacts with over 30 partners, including the tumor suppressor p14ARF.Citation3 Interestingly enough, Spinophilin also regulates retinoblastoma protein (pRb) function by modulating the activity of protein phosphatase 1 (PP1). In this context, PP1-mediated dephosphorylation of pRb activates its growth suppressive function.Citation4 Since p53 and pRb pathways are functionally impaired in most human cancers, it was tempting to speculate that the putative tumor suppressor properties of Spinophilin might be funnelled through either of these pathways.
Ferrer et al. have recently addressed this possibility in vivo using genetically modified mice.Citation5 Spinophilin deficient animals showed a mild phenotype and developed spontaneous tumors, albeit with long latency. Yet, tumor susceptibility and overall tumor burden were significantly increased when combined with a p53-deficient background. This is in good agreement with clinical data, since Spinophilin loss in human tumors is concurrent with p53 mutations, resulting in high-grade lesions and poor prognosis.Citation2 To further confirm this synergistic effect, Ferrer and colleagues went on to combine Spinophilin loss with a p53 mutation observed in human breast cancer. Mice expressing p53R172H in the mammary glands display negligible levels of spontaneous tumorigenesis.Citation6 Yet, it predisposes these mice to the effect of cancer promoting insults, developing faster and more aggressive tumors if treated with a carcinogen or crossed to oncogene expressing strains. Interestingly, a similar effect was observed with Spinophilin deficiency. The presence of p53R172H boosted the appearance of preneoplastic and tumorigenic lesions even in Spinophilin heterozygotes, reinforcing the notion that Spinophilin exerts tumor suppressive properties that only become evident in a p53-deficient background.Citation5 This is, therefore, suggestive of partially redundant functions in their tumor suppression properties.
It is yet unclear how this genetic interaction between Spinophilin and p53 is brought about in molecular terms. Spinophilin is known to increase PP1-mediated dephosphorylation of doublecortin,Citation7 suggesting that it could equally enhance PP1 activity onto pRb. If this is the case, Spinophilin deficiency could result in hyperphosphorylated pRb and aberrant activation of E2F-dependent transcription. Importantly, proper control of this transcriptional program is essential to prevent replicative stress.Citation8 It is therefore possible that, upon loss of Spinophilin, hypophosphorylated pRb triggers an initial period of unscheduled proliferation, causing replicative stress and eventually a p53-mediated cell cycle arrest.
The pRb pathway controls several aspects of stem cell biology, including the tight control of self-renewal characteristics of progenitor cells.Citation9 It is tempting to speculate that loss of Spinophilin may result in a partial expansion of certain progenitor pools. This may explain the preneoplastic abnormalities associated with increased cell proliferation found in Spinophilin-null mice. Activation of a p53-mediated arrest may further limit expansion of these cells. Elimination of this additional barrier may, therefore, explain the synergistic effect observed in terms of tumor formation in Spinophilin-null animals when p53 function is impaired.
In addition to the important physiological functions carried out by Spinophilin in the nervous system,Citation7 Ferrer and colleagues have now unveiled its tumor suppresor functions insinuated by the genetic linkage studies. Further work will be required to substantiate the molecular basis of its p53-dependent tumor protective function in vivo.
References
- Abujiang P, et al. Oncogene 1998; 17:3029 - 3033
- Molina-Pinelo S, et al. J Pathol 2011; 225:73 - 82
- Sarrouilhe D, et al. Biochimie 2006; 88:1099 - 1113
- Nelson DA, et al. J Biol Chem 1997; 272:4528 - 4535
- Ferrer I, et al. Cell Cycle 2011; 10:1948 - 1955
- Li B, et al. Oncogene 1998; 16:997 - 1007
- Feng J, et al. Proc Natl Acad Sci USA 2000; 97:9287 - 9292
- Bueno MJ, et al. Mol Cell Biol 2010; 30:2983 - 2995
- Galderisi U, et al. Oncogene 2006; 25:5250 - 5256
As they get immortalized and progressively transformed, cancer cell precursors must accumulate and accommodate genetic and epigenetic alterations that allow them to acquire all hallmarks of malignancy. At least at the conceptual level, it appears attractive to design drugs that target or reverse these (epi) genetic hallmarks of cancer. Tumor cells often exhibit DNA methylation of potential tumor suppressor genes, and so-called demethylating agents are the first epigenetic therapeutics that have been clinically approved for the therapy of myelodysplastic syndrome (MDS), a preneoplastic stage of acute myeloid leukemia (AML). Clinical trials are on the way to explore the therapeutic utility of such demethylating agents, in particular azacitidine (AZA) and 2′-deoxy-5-azacitidine (DAC), for the treatment of MDS and other malignant hematopoietic diseases.Citation1
In the July 15th issue of Cell Cycle, Cluzeau et al.Citation2 explore the effects of azacidine on SKM1 cells, which represent malignant myeloblasts from a patient with secondary AML (i.e., AML after MDS). These cells undergo both autophagy and apoptosis in response to AZA,Citation2 in line with the finding that chronic myeloid leukemia (CML) cell lines also can manifest autophagy and, later, apoptosis in response to DAC.Citation3 These results indicate that different demethylating agents can activate cellular stress pathways that culminate in the manifestation of macroautophagy, a lysosomal degradation pathway for the bulk degradation of cytoplasmic material (hitherto referred to as “autophagy”), as well as in apoptosis, a subroutine of programmed cell death that is classically accompanied by caspase activation.Citation2,Citation3 Accordingly, caspase inhibition reduced AZA-induced cell death.Citation2
The question was then whether autophagy also contributed to cell death induced by demethylating agents. One of the hallmarks of autophagy is the lipidation of an essential autophagy protein, LC3, leading to an increase in its electrophoretic mobility that shifts from the normal, cytosolic form of LC3 (LC3I) to the autophagosome-bound from of LC3 (LC3II).Citation4 AZA clearly induced the autophagy-associated lipidation of LC3. Small interfering RNA-mediated knockdown of LC3 provoked an increase in cell death in response to AZA, in line with the idea that AZA-induced autophagy is a cytoprotective rather than cytotoxic mechanism.Citation4 Accordingly, systematic studies revealed that cytotoxic anticancer agents capable of inducing autophagy usually do so as a “side effect,” meaning that autophagy inhibition does not prevent cancer cell killing and often actually accelerates cell death in response to established and experimental anticancer agents.Citation5 Conversely, autophagy-stimulatory agents often are cytoprotective and actually can prolong cellular and organismal life span.Citation6,Citation7 Thus, the observation that autophagy does not contribute to AZA-induced cell killing suggests that AZA operates as do other, conventional anticancer agents through the induction of cellular stress that triggers compensatory mechanisms (including autophagy) before a threshold of damage is attained and the cells activate the intrinsic pathway of apoptosis.
In a further twist of the story, Cluzeau et al. established SKM1 clones that were able to proliferate in the continuous presence of high AZA doses.Citation2 Even when AZA was transiently withdrawn from the cultures, these AZA-resistant cells failed to respond to re-challenge with AZA and, hence, were completely refractory to AZA-induced apoptosisCitation2 as well as to the AZA-induced reactivation of FOXO3a.Citation8 Intriguingly, the AZA-resistant cells manifested an increase in baseline autophagy, and this level of autophagy was not further enhanced by the re-addition of AZA. Moreover, knockdown of LC3 failed to restore the apoptotic response to AZA, suggesting that the elevated level of autophagy cannot explain AZA resistance. Although off-target effects of AZA (such as NFκB inhibition)Citation9 cannot be excluded in this model, it appears that both the chronic and the acute exposure of AML cells to AZA have a similar capacity to stimulate autophagy. Whether the induction of autophagy may contribute to AZA-mediated therapeutic effects other than cell killing, such as induction of senescence or terminal differentiation, needs to be addressed in future studies. Independently of these considerations, the AZA-refractory cells developed by Cluzeau et al.Citation2 constitute a welcome addition to the clinical investigator's armamentarium and, undoubtedly, will contribute to the elucidation of AZA resistance mechanisms that account for the relapse of MDS.
Figures and Tables
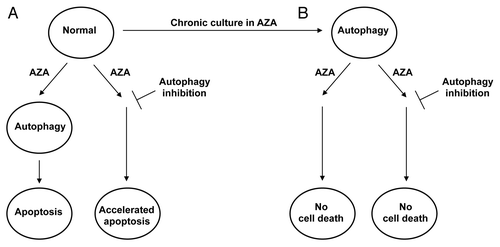
Figure 1 Coordinated regulation of autophagy and apoptosis by azacitidine. (A) Azacitidine (AZA) causes the induction of autophagy and apoptosis in acute myeloid leukemia (AML) cells, presumably through demethylation of DNA and perhaps through off-target effects. Inhibition of autophagy stimulates cell killing by AZA (B) Chronic exposure of the cells to AZA leads to a permanent state of elevated autophagy without cell death that is maintained upon transient withdrawal of AZA. Moreover, AZA-resistant cells fail to die in response to the readdition of AZA to the cells, even when autophagy is inhibited.
References
- Garcia-Manero G, et al. J Clin Oncol 2011; 29:516 - 523
- Cluzeau T, et al. Cell Cycle 2011; 10:2339 - 2343
- Schnekenburger M, et al. Biochem Pharmacol 2011; 81:364 - 378
- Kroemer G, et al. Mol Cell 2010; 40:280 - 293
- Shen S, et al. Oncogene 2011; In Press
- Eisenberg T, et al. Nat Cell Biol 2009; 11:1305 - 1314
- Morselli E, et al. J Cell Biol 2011; 192:615 - 629
- Thépot S, et al. Cell Cycle 2011; 10:2323 - 2330
- Fabre C, et al. Cell Cycle 2008; 7:2139 - 2145
The role of cellular senescence as a barrier to tumor formation is perhaps best illustrated by oncogene-induced senescence, or OIS. This phenomenon was first observed when an oncogenic form of H-RAS was expressed in normal fibroblasts.Citation1 The resulting enlarged, flattened morphology and accumulation of senescence-associated β-galactosidase (SA-β-gal) activityCitation2 as well as the inability to progress through the cell cycle signaled the discovery of OIS. Subsequently, other members of the RAS signaling pathway and nuclear proteins were shown to cause senescence when expressed as oncogenic versions. The loss of tumor suppressor genes can also induce senescence in normal cells. Importantly, mouse and human studies show that the senescence response in vivo curtails the development of cancer.Citation3 Complicating this tumor suppressive mechanism is the production by senescent cells of a plethora of secreted factors, known as the senescence-associated secretory phenotype (SASP),Citation4 the senescence secretomeCitation5 or the senescence-messaging secretome (SMS).Citation6 These factors may explain a paradoxical ability of senescent cells to positively impact the growth of adjacent tumor cells.Citation3
Clearly, the possibility of exploiting senescence for therapeutic benefit requires better mechanistic understanding. Work from Akakura et al. in the December 1st issue of Cell CycleCitation7 extends the understanding of senescence induction to include the scaffold protein SSeCKS/Gravin/Akap12, previously identified as a suppressor of v-Src oncogenicity and a scaffold for PKC and PKA. Interestingly, this group has also shown that knockout of Akap12 leads to prostatic hyperplasia,Citation8 consistent with its reduced expression in cells transformed by certain common oncogenes.
Akap12-knockout (KO) mouse embryonic fibroblasts (MEFS) in culture undergo a rapid onset of senescence, as indicated by SA-β-gal induction and morphological changes, dependent on pRB but not p53. Importantly, activation of pRB requires PKCα and -δ, strongly indicating that the scaffold function of Akap12 is key to its ability to prevent pRB-induced senescence in normal cells, presumably through inhibition of ectopic PKC activity. Thus, the normal function of Akap12 as an inhibitor of pRB-induced senescence would seem to be that of an oncogene. However, it is important to note that it is most likely deregulated oncogenic signaling, in this case from PKC activity, that is ultimately needed for transformation. In certain cellular contexts, such senescence-inducing effects may predominate, and the retention of certain tumor suppressors or inhibition of oncogenes may thus be observed to have counterintuitive effects dependent on a particular tumor's genetic composition.
Interestingly, in addition to stimulating the PKC/MEK pathway and thus engaging a form of OIS, Akap12 loss also leads to downregulation of the Lats1/Warts kinase, a component of the Hippo pathway that regulates cytokinesis and genomic stability. This finding, which presages a study connecting Lats1 function to pRB,Citation9 indicates that Akap12 loss triggers multiple cellular stresses that result in a robust induction of cellular senescence. This is also true in vivo, since prostate glands derived from Akap12-KO mice show focal areas of senescent cells adjacent to hyperplastic regions, indicating that the senescence effect is not simply responsive to the added cellular stress of MEF culture.
This finding again provokes the question of whether senescence is truly a barrier to tumorigenesis, or if it indeed contributes to neoplasia by modifying the tumor environment or promoting progression in a cell-autonomous manner. Unfortunately, SASP expression was not evaluated, so a role for these factors in Akap12-KO cells awaits further study. However, it is most intriguing that Akap12-KO MEFs were found to be sensitive to transformation by activated ras. This result is surprising given the senescence observed in Akap12-KO MEFs, and the fact that activated ras itself induces senescence in wild-type MEFs, which, therefore, are resistant to transformation by activated ras alone. This finding implies that complementary pathways induced by Akap12 loss and ras hyperactivity may provide an effective “work around” to senescence induction by either oncogenic event alone. Thus, the identification of Akap12 loss and PKC dysregulation leading to senescence and hyperplasia produces an intriguing set of questions that may ultimately yield key information about the complex role of senescence in tumor growth in vivo.
References
- Serrano M, et al. Cell 1997; 88:593 - 602
- Dimri GP, et al. Proc Natl Acad Sci USA 1995; 92:9363 - 9367
- Rodier F, et al. J Cell Biol 2011; 192:547 - 556
- Coppe JP, et al. PLoS Biol 2008; 6:2853 - 2868
- Adams PD. Mol Cell 2009; 36:2 - 14
- Kuilman T, Peeper DS. Nat Rev Cancer 2009; 9:81 - 94
- Akakura S, et al. Cell Cycle 2010; 9:4656 - 4665
- Akakura S, et al. Cancer Res 2008; 68:5096 - 5103
- Tschop K, et al. Genes Dev 2011; 25:814 - 830
Although pathologists have recognized morphologic heterogeneity among cells within a tumor for over a century, this observation has recently taken on heightened clinical significance with the demonstrations by multiple laboratories that certain subpopulations of tumor cells have a greater capacity to form tumors in animal xenograft models and colonies in sphere-forming in vitro assays. In head and neck squamous cell carcinoma (HNSCC), these cells with “tumor-initiating capacity” have been found in the CD44+ compartment, similar to the tumor-initiating cells (also dubbed cancer stem cells) in other epithelial-derived malignancies.Citation1,Citation2 Of clinical importance, patients with HNSCC containing higher percentages of CD44+ cells within their tumors appear to have much more aggressive disease with a higher incidence of recurrence.Citation3 Partially accounting for this observation may be the fact that the CD44+ cells in HNSCC and breast carcinomas have been found to be more resistant to radiation therapy due to their greater capacity to handle oxidative stress.Citation4 In breast cancer models, CD44+ cells have been shown to have more mesenchymal-like features,Citation5,Citation6 leading some to hypothesize that epithelial-to-mesenchymal transition (EMT) may underlie the development of the more invasive and metastatic phenotype that has also been associated with this subpopulation.Citation6
In the June 15th issue of Cell Cycle, Basu et al.Citation7 demonstrated that a mesenchymal-like (E-cadherinlo vimentin+) subpopulation is present in HNSCC, and that this subpopulation is inherently more resistant to cisplatin (the chemotherapeutic agent most commonly used against this malignancy in the clinic), compared to the E-cadherinhi vimentin− subpopulation of cells. This E-cadherinlo vimentin+ mesenchymal-like subpopulation was consistently (but variably) found in both human HNSCC cell lines and primary human HNSCC tumor specimens, indicating that this heterogeneity is a common feature in this malignancy. When cisplatin was administered to mice implanted with primary human HNSCC xenografts, the mesenchymal-like subpopulation was markedly enriched, further supporting the authors' conclusions that these cells are resistant to cisplatin toxicity.
Interestingly, salinomycin (a drug identified in a high-throughput screen to find compounds with selective toxicity toward breast tumor-initiating cells)Citation8 had equal toxicity against both the E-cadherinlo vimentin+ and E-cadherinhi vimentin− subpopulations when tested in vitro. Furthermore, the administration of salinomycin to mice implanted with primary human HNSCC xenografts resulted in marked selective depletion of the vimentin+ cells, in contrast to what was seen with cisplatin. Whether this more selective depletion in vivo represents even greater sensitivity of the vimentin+ cells to salinomycin in vivo or an inhibition of their development through EMT remains to be determined. Nevertheless, it introduces a potential strategy for combination therapy in this disease.
The authors' finding that HNSCC contains a subpopulation of mesenchymal-like cells that are resistant to cisplatin also sheds light on a well-described clinical phenomenon found in HNSCC patients. It has long been observed that patients' tumors that respond well to cisplatin induction chemotherapy also display a good response to radiation therapy; whereas, those that do not respond well to cisplatin also do not fare well with radiation.Citation9 This observation was, in fact, the backbone of a pivotal prospective trial for advanced laryngeal squamous cell carcinoma.Citation10 An understanding of the relationship between cisplatin sensitivity and radiation sensitivity, however, has remained elusive. In light of the study by Basu et al., one can speculate that the mesenchymal-like cisplatin-resistant cells represent the CD44+ cancer stem cell population, which has previously been shown to be resistant to radiation therapy in HNSCC.Citation4
References
- Prince ME, et al. Proc Natl Acad Sci USA 2007; 104:973 - 978
- Al-Hajj M, et al. Proc Natl Acad Sci USA 2003; 100:3983 - 3988
- Joshua B, et al. Head Neck 2011; In press
- Diehn M, et al. Nature 2009; 458:780 - 783
- Fang X, et al. Oncogene 2011; In press
- Mani SA, et al. Cell 2008; 133:704 - 715
- Basu D, et al. Cell Cycle 2011; 10:2008 - 2016
- Gupta PB, et al. Cell 2009; 138:645 - 659
- al-Sarraf M, et al. Cancer Invest 1995; 13:41 - 53
- The Department of Veterans Affairs Laryngeal Cancer Study Group. N Engl J Med 1991; 324:1685 - 1690
GTPases are a well-known family of proteins involved in a wide variety of crucial regulatory processes in the cell. A structurally distinct and highly conserved subfamily of GTPases called GPN-loop GTPases has recently been described in archae and eukaryotes. The new family of G proteins is characterized by a conserved Gly-Pro-Asn motif inserted onto the GTPase core-fold.Citation1 The new study by Alonso et al. published in the June 1st issue of Cell Cycle provides evidence that the GPN-loop GTPase, yGPN1, is involved in the regulation of sister chromatid resolution in yeast cells.Citation2
The authors used S. cerevisiae strains in which the endogenous copy of yGPN1 was deleted by homologous recombination, and yGPN1 expression was reconstituted by a rescue plasmid coding for yGPN1 under the control of an inducible promoter. In the absence of yGPN1, there was a decrease of the growth rate, and the majority of the cells had an unusually large bud. Flow cytometric analysis showed a severe S-phase delay in asynchronously cycling cells under yGPN1 repression as well as in cells allowed to proceed to the next cell cycle following mitotic synchronization. Compared to wild-type cells, microscopic analysis of cells repressed for yGPN1 at G2/M phase revealed a greater number of doublets, with a large bud containing a nucleus whose position is adjacent to the neck or whose nucleus is passing from mother to daughter cell. In contrast, induction of yGPN1 expression resulted in an increase in the proportion of cells with a large bud containing one nucleus each.
The characteristic “bow-tie” morphology of nuclei in cells repressed for yGPN1, was suggestive of chromatids that were still attached and not yet separated into two distinct entities. Therefore, the authors carried out a GFP-based assay for sister chromatid cohesion in cells synchronized in mitosis under conditions of yGPN1 induction. The large majority of mitotically arrested cells under yGPN1 repression had a single GFP spot, whereas in cells induced to express yGPN1, there was a noticeable increase in the cells with two GFP spots, indicating that the presence of yGPN1 is correlated with sister chromatid resolution and therefore opening of the cohesin ring.
Cohesin ring opening is needed on three different occasions. The first is during the loading of the cohesin ring to the DNA, a process that involves transient opening of the hinge domains of SMC1 and SMC3.Citation3 The second is during S phase and is needed for the replication complex to go through.Citation4 Indeed, the severe S-phase delay observed in cells that are repressed for yGPN1 expression is most likely due to a slowing of the replication process and is consistent with an yGPN1's role in cohesin ring opening. The third time the ring needs to be opened is following spindle assembly checkpoint satisfaction in order for sister chromatids to be segregated equally during anaphase in the two daughter cells.Citation5 Although the mechanisms for cohesion ring opening are different during replication and anaphase, yGPN1 could play a role in the control of these two mechanisms. It would be interesting to elucidate in the future whether yGPN1 is involved in the prophase mechanism of cohesin removalCitation6 or in the separase-dependent opening of the cohesin ring due to proteolytic cleavage of the α-kleisin Scc1p.Citation7 Does the spindle assembly checkpoint exert an influence on the yGPN1 activity and vice versa?
The reported phenotypic analysis of yeast cells depleted or overexpressing yGPN1 is consistent with the authors' proposal for a role of yGPN1 in sister chromatid regulation. But are there other evidences? Using genome analysis and from evolutionary point of view, the appearance of the GPN-loop GTPases family of proteins coincides with the appearance of genes coding for proteins that are involved in DNA replication and chromosome segregation during cell division.Citation1 Indeed, the protein-protein interaction network reported here for yGPN1 in S. cerevisiae revealed a plethora of possible interacting proteins that are involved in DNA replication, chromosome segregation and sister chromatid cohesion. Their ability to interact with yGPN1 and the impact of their function on the GTPase activity of yGPN1 remains to be shown.
Although the data presented in the Alonso et al. paper do not prove that yGPN1 is directly involved in sister chromatid cohesion, the authors open exciting new avenues for exploring the functions and the regulation of the activities of GPN-loop GTPases family of proteins in DNA replication and sister chromatid cohesion.
References
- Gras S, et al. EMBO Reports 2007; 8:569 - 575
- Alonso B, et al. Cell Cycle 2011; 10:1828 - 1837
- Gruber S. Cell 2006; 127:523 - 537
- Skibbens RV. Trends Cell Biol 2010; 20:507 - 513
- Pines J, et al. Nat Cell Biol 2001; 3:E3 - E6
- Waizenegger IC, et al. Cell 2000; 103:399 - 410
- Uhlmann F, et al. Nature 1999; 400:37 - 42