Abstract
Small cell lung cancer (SCLC) is an aggressive lung cancer subtype in need of better therapies. Histone deacetylase inhibitors (HDIs) promote increased lysine acetylation in nucleosomal histones and are thought to relax chromatin, thereby allowing increased access of transcription factors and DNA damaging agents alike to DNA. We studied whether two HDIs, belinostat and romidepsin, could be effectively combined with cisplatin or etoposide (VP-16) for SCLC cells. Analysis of cell survival and synergy was performed using CalcuSyn mathematical modeling to calculate a combination index. Immunostaining of γH2AX was performed to evaluate persistence of DNA damage following simultaneous or sequential exposure. Based on CalcuSyn modeling, HDIs synergized with DNA damaging agents only when added simultaneously. An additive-to-antagonistic effect was seen with HDI pretreatment for 24 h or with addition after cisplatin or etoposide. Furthermore, pretreatment with HDIs resulted in normalization of cell cycle and reduced PARP degradation as compared with simultaneous treatment. The increase in γH2AX phosphorylation confirmed that simultaneous but not sequential treatment enhanced double-stranded DNA breaks. These results suggest that DNA relaxation is not required for synergy of HDIs with DNA damaging agents, and that scheduling of drug administration will be critical for rational development of clinical protocols.
Introduction
DNA methylation and post-translational modifications of nucleosomal histone proteins by acetylation are major epigenetic pathways that regulate gene expression.Citation1–Citation4 Two enzymes that work in opposition to alter the acetylation of the octomeric histone core of the nucleosome are histone deacetylase (HDAC) and histone acetyltransferase.Citation2,Citation3 It has become increasingly apparent that the activity of many transcription factors and some cytoplasmic proteins is also modulated by acetylation. Inhibitors of histone deacetylase (HDIs) have been shown to alter the expression of an estimated 4–12% of genes, presumably due in part to the relaxation of DNA that follows neutralization of the positive charge on histone lysines generated by acetylation and access to transcriptional regulators.Citation5 To date, the hierarchy of activities leading to cell death following HDI exposure has not been clarified. Previous studies have shown that HDIs induce cell growth arrest, differentiation and apoptosis in many cancer cell lines in vitro and in vivo.Citation6–Citation9 HDIs have been shown to potentiate cytotoxic therapy and radiation, although the mechanisms have not been fully elucidated.Citation10–Citation14 Synergy with DNA damaging agents has been thought related to increased access of these agents to DNA, although inhibition of DNA synthesis and repair has also been reported in reference Citation15.
Lung cancer remains a leading cause of cancer death worldwide among every ethnic group. SCLC accounts for about 10–12% of all lung cancer casesCitation16 and is rarely resectable at presentation due to its disseminated nature, involving many organs, including adrenal glands and the brain. These tumors usually respond to the combination of cisplatin and VP-16 but rapidly progress with drug-resistant disease.Citation16 Therapeutic maneuvers have been disappointing with median survivals of 6 to 12 mo. The rapid proliferation rate and disseminated nature of these tumors argues that improved upfront therapy has the potential to prolong survival of these patients.
Belinostat (PXD101) and romidepsin (depsipeptide, FK228) are novel inhibitors of histone deacetylase. Romidepsin was recently approved for the treatment of cutaneous T-cell lymphoma and peripheral T-cell lymphoma in patients who received at least one prior systemic therapy. Belinostat is currently undergoing phase I and II clinical trials.Citation17–Citation19 These compounds induce hyperacetylation of histones H3 and H4, increase p21 levels and induce G1 cell cycle arrest in some cells.Citation8,Citation20 The aim of the present study was to establish an optimal schedule and rationale for combining HDIs with conventional DNA damaging agents. We investigated the effects of these compounds alone and in combination with two DNA damaging agents that constitute the conventional treatment of SCLC. Using suspension and adherent SCLC cell lines, we found that the anticancer effect was dependent on the schedule of drug administration. Whereas some previous studies suggested that pretreatment with HDIs sensitize cancer cells to chemotherapeutic agents, presumably because sequential treatment may facilitate access to DNA,Citation10–Citation13,Citation21,Citation22 we found that only simultaneous treatment is synergistic. Molecular mechanisms responsible for this effect are considered.
Results
Synergy of HDAC inhibitors with DNA damaging agents.
To assess the effect of combination therapy, cell survival was determined using combinations of drugs added simultaneously or with a 24-h delay, which represents an approximate time for one cycle of cell division for cell lines used in our study. The combination index (CI) was modeled by CalcuSyn software to determine the synergy or antagonism of any given drug interaction. A CI = 1 indicates an additive effect, whereas < 1 indicates a synergistic and > 1 an antagonistic effect. Both survival curves and mathematical modeling showed that synergy is systematically achieved when the drugs are administered simultaneously. shows a representative experiment using H146 cells treated with belinostat, VP-16 or cisplatin. Remarkably, simultaneous treatment resulted in synergy for all fractions affected and for all drug concentrations. In contrast, only additive-to-antagonistic effects were seen when cells were pretreated with HDI for one cell cycle, with a 24 h delay, and consistent antagonism was observed when cells were exposed to DNA damaging agents prior to belinostat or romidepsin. The simultaneous schedule was also superior to the sequential approach using romidepsin (Figs. S1A, B and C). Similar results were obtained using other adherent and non-adherent SCLC cell lines, H446, H82 and H562 (Figs. S2A and B and data not shown), with CI below 1 observed consistently in simultaneous but not sequential treatment schedules for all SCLC cell lines examined. Thus, combination therapy is synergistic for all fractions affected with all drug concentrations when administered simultaneously, but only additive-to-antagonistic using other schedules of drug administration.
Cell cycle analysis.
Belinostat and romidepsin effects on the cell cycle were examined after treatment with each agent alone or in combination with cisplatin and VP-16. Cells treated with belinostat or romidepsin alone did not show a substantial effect on cell cycle, nor was there an obvious G1 arrest as described in other cell types (). Notably, HDIs administered over a wide concentration range also failed to induce G1 arrest in H146 and H526 cells (data not shown). VP-16 induced accumulation in S phase at 24 h and an almost complete shift to G2/M at 48 h (), consistent with previous reports.Citation23,Citation24 Simultaneous exposure to VP-16 with belinostat or romidepsin resulted in a persistence of VP-16-induced S-G2/M arrest at 24 h and 48 h and an increase in apoptotic Sub-G1 fraction from 3.5% to 9.5% and 1.7% to 7.8%, respectively. In contrast, when HDIs were administered 24 h prior to DNA damaging agents, there was a decrease in VP-16-induced S-G2/M arrest, with some normalization of the cell cycle profile more apparent at 72 h for romidepsin ( and B, bottom part). Pretreatment with belinostat and romidepsin prior to a 48 h VP-16 exposure also resulted in only 11.7% and 28.1% of cells in G2/M phase, respectively, as compared with 70% of cells treated with VP-16 alone. Similar effects were observed when cells were treated with HDIs and cisplatin (data not shown). Comparison of the cell cycle response of the SCLC lines with that of other cancer cells is shown by analysis of PC3 and MCF-7 cells, where combination therapy increases G2/M arrest in these cells (Fig. S3 and data not shown). Although there was no increase in sub-G1 in PC3 or MCF-7 cells at drug concentrations used for these experiments, normalization of cell cycle was also apparent in sequential but not in simultaneous treatment schedules (Fig. S3). Thus, administration of HDIs prior to DNA damaging agents appears to have a protective effect on cell cycle and is consistent with the antagonism observed in the cell survival assays.
Analysis of PARP degradation.
To assess PARP degradation as a marker of apoptosis,Citation25 we used immunoblot analysis. We confirmed that PARP degradation is most efficient when the drugs are added simultaneously () as compared with all sequential schedules (). Notably, complete degradation of full-length PARP was seen with simultaneous treatment, whereas pretreatment with belinostat for 24 h before addition of VP-16 (BEL 48 h→VP-16 24 h) showed persistence of some full-length protein (). Similar results were observed when cells were treated with the HDIs and cisplatin (Fig. S4a). Thus, PARP degradation is incomplete when cells are pretreated with either drug type, indicating a protective anti-apoptotic effect.
Effect of combination therapy on HDI-induced histone H3 acetylation.
HDIs increase the amount of acetylated histone H3, which has been shown to precede HDI-induced cell death. We examined whether histone H3 acetylation (H3Ac) is affected by treatment with belinostat or romidepsin, and whether combination with DNA damaging agents changes H3 acetylation. shows that there were no significant changes in belinostat- or romidepsin-induced H3Ac hyperacetylation in the presence of VP-16 regardless of the schedule of administration. Similar results were observed when cells were treated with HDIs and cisplatin (Fig. S4B). Quantitative analyses of H3 acetylation from 3–4 independent experiments, normalized to control samples, confirmed the lack of changes and are shown as a bar graph in and Figure S4B. Thus, it is unlikely that H3 acetylation is responsible for synergy or antagonism among these agents.
HDIs do not alter interactions of VP-16 and cisplatin with DNA.
VP-16 is known to cause double-strand DNA damage through inhibition of topoisomerase II, an enzyme that cleaves both strands of DNA during transcription, replication and mitosis.Citation26 Inhibition of topoisomerase II prevents rejoining of the DNA strands and leads to protein-linked DNA double-strand breaks.Citation27 To measure the effect of VP-16, we used an alkaline filter elution assay that traps topoisomerase II on DNA double-strand breaks.Citation13,Citation27 DNA-protein crosslinks were quantified in cells treated with VP-16 in the absence or presence of belinostat. No statistically significant changes in DNA strand breaks were observed whether VP-16 was administered simultaneously or was delayed for 24 h after addition of belinostat ().
To determine whether cisplatin binding to DNA was increased when combined with HDIs, we measured DNA platination following exposure of cells to cisplatin alone or in combination with the HDIs in simultaneous and sequential schedules. No differences in platination were detected with either schedule, whether cells were treated for 3 h in high cisplatin concentration or for 24 h at 16 µM, a concentration used for all other studies ( left and right parts). These results argue against the notion that DNA accessibility to DNA damaging agents is increased by HDIs following histone hyperacetylation.
Simultaneous treatment with DNA damaging agents increases γH2AX phosphorylation.
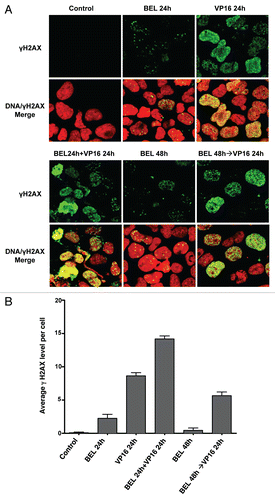
Phosphorylation of histone H2A on serine 139, termed γH2AX, by kinases sensing the double-strand DNA break is a sensitive marker of DNA damage.Citation28–Citation30 As shown in , phosphorylated γH2AX foci are noted 24 h following exposure to VP-16 alone. Their number and intensity are increased with simultaneous exposure to belinostat for 24 h, but are significantly less if VP-16 is added after exposure to belinostat. Similar data were obtained using cisplatin (data not shown). Thus, regardless of the type of DNA damage, simultaneous treatment potentiates the generation of DNA double-strand breaks, whereas pretreatment with HDIs is less toxic.
Discussion
Histone deacetylase inhibitors entered the anticancer armamentarium with the documentation of activity in the treatment of cutaneous and peripheral T cell lymphomas.Citation19 However, outcomes in solid tumors have been disappointing, and attention has turned to identifying combinations that would exploit the unique activity of HDIs in these tumors.Citation17–Citation19 Multiple mechanisms of action of HDIs have been described, making it difficult to choose the best combination strategy. The present study describes schedule-dependent synergy of HDIs in combination with the two most active agents for small cell lung cancer, VP-16 and cisplatin. A significant increase in cytotoxicity was observed with simultaneous exposure to HDIs and DNA damaging agents, in contrast to merely additive or less-than-additive effects when the HDIs were added prior to or following the chemotherapeutics. The cytotoxic effects of the combination could not be explained by increased accessibility of VP-16 or cisplatin to DNA, and the differences in scheduled drug administration were confirmed by cell cycle analysis, γH2AX phosphorylation and PARP cleavage.
We used computational analysis of cell survival calculated by combination index (CI) based on the multiple drug effect equation of Chou-Talalay.Citation31 A unique feature of this method for determination of synergy or antagonism is that a broad range of doses among the drugs can be used to evaluate cytotoxicity and to discriminate merely additive effects. The latter cannot be performed arithmetically because of the S-shaped dose-response curve in cytotoxicity assays. Our analysis showed that HDIs synergized most effectively with DNA damaging agents when added simultaneously, but were antagonistic when cells were pretreated with cisplatin or VP-16. In addition, synergy was reduced to only additive effects when cells received HDIs 24 h prior to DNA damaging agents. The cell survival curves and CI also show that either visual interpretation of cell survival or examination of a limited dose range for each combination can be misleading.
A number of reports have identified increased cytotoxicity resulting from addition of HDIs to classical chemotherapeutic agents without a clear definition of the underlying mechanisms or the optimal schedule for their administration.Citation11,Citation14,Citation15,Citation32,Citation33 One proposed explanation has been that the increased acetylation of lysine in the nucleosomal histone core and consequent relaxation of the chromatin allow increased access to drugs that have DNA as their target.Citation11,Citation15,Citation19,Citation34 Arguing against this hypothesis as the sole explanation is the convincing evidence of synergy between HDIs and radiation.Citation13,Citation14,Citation35 Our data also showed synergy only when HDIs were used simultaneously with VP-16 or cisplatin, arguing against the relaxed chromatin hypothesis. A second postulated mechanism is through increased release of reactive oxygen species (ROS) contributing to DNA damage.Citation21,Citation36,Citation37 The reported downregulation of DNA repair proteins, such as Ku70, Ku86 and RAD50, following HDAC inhibition in some cell types could contribute to the increase in damage by either mechanism.Citation38
We have previously shown that combining DNA damaging drugs in different schedules or sequences with agents directed at molecular targets, such as HSP90 inhibitors, can result in substantial differences leading to synergy or antagonism.Citation39,Citation40 Similarly, a number of reports described sequence-specific differences in the activity of combinations with HDIs. Although previous reports address some of these issues, most studies are incomplete. Some models suggest that prior treatment with HDIs increase cell sensitivity to other drugs,Citation11,Citation21,Citation22 whereas others find antagonismCitation32,Citation41,Citation42 or no schedule dependence.Citation36 Kim et al. found that 1-hour pretreatment with trichostatin A or SAHA increased sensitivity to a variety of anticancer agents targeting DNA, including VP-16, camptothecin, cisplatin and doxorubicin. However, the short pulse of HDI followed by 1-hour exposure to DNA damaging agents is a very different schedule from the 24-h pretreatment employed in the current study.
Cell cycle analysis suggested that pretreatment of SCLC cells with HDIs protects cells from the cytotoxic effects of DNA damaging agents. It is possible that this schedule impairs accumulation in G2/M because of earlier checkpoints induced by HDIs, including a reduction in replication fork velocity recently reported by Conti et al. Cell context, including the frequent loss in small cell lung cancer of functional cell cycle gatekeepers, such as p16, p21, p53 or Rb,Citation44,Citation45 may also influence the final cell cycle status following HDIs. In addition, PARP degradation was an independent parameter that also supported maximal effects with simultaneous administration. The amount of cleaved PARP was consistently reduced when belinostat or romidepsin were added prior to cisplatin or VP-16. The decrease in cleaved PARP was more apparent at 72 h after sequential treatment with romidepsin.
Despite the observed synergy of the combintion in cell survival and apoptosis, we found no evidence for enhanced histone acetylation or an increase in DNA-protein cross-links indicative of topoisomerase II trapping by VP-16 when combined with HDIs. These results are consistent with an earlier report from one of the authors showing that pretreatment with trichostatin A did not increase the overall number of DNA-protein cross-links despite increased apoptosis and decreased cell survival.Citation11 In addition, we found no evidence for increased platinum adducts in cells treated with HDIs and cisplatin. This is in contrast to the studies of Rabik et al., in which platination increased after pretreatment with O6-benzylguanine, an inhibitor of DNA repair.Citation46
Consistent with cell survival data, double-strand DNA (dsDNA) breaks markedly increased in cells treated simultaneously with belinostat and VP-16 as detected by γH2AX foci formation. These results are supported by a report of persistent γH2AX in cells treated by radiation in the presence of vorinostat.Citation38 However, we observed a reduction in γH2AX foci formation when cells were pretreated with HDIs prior to VP-16. Phosphorylation of H2AX, termed γH2AX, is a marker of persistent dsDNA breaks. H2AX is an adaptor molecule that maintains repair factors in proximity to DNA lesions; its C terminus is phosphorylated in response to the formation of dsDNA breaks.Citation11,Citation28,Citation38 Hyperacetylation of HSP90 and a subsequent delay in DNA repair may also contribute to the maximal efficacy of simultaneous treatment.Citation40 The possible role of HSP90 inhibition following HDIs will require additional studies.
One of the challenges in conducting a clinical trial when combining an HDI with DNA damaging agents will be to achieve optimal DNA damage in a given tumor tissue. Our results suggest that pretreatment with HDIs mitigates the cytotoxicity of DNA damaging agents in SCLC without increasing their access to DNA. The synergy in inducing cell death and dsDNA breaks by simultaneous treatment is most likely due to failed DNA repair rather than the previously postulated increase in accessibility of DNA to chemotherapeutic agents. Thus, the data presented here show that simultaneous addition of HDI with cisplatin or VP-16 is superior to alternative schedules and supports the development of clinical trials using these combinations for SCLC. In addition, γH2AX foci formation may be a useful marker for monitoring these clinical trials. A phase I clinical trial combining belinostat with both cisplatin and VP-16 for patients with SCLC is ongoing.
Materials and Methods
Cell lines and drugs.
SCLC cell lines H82, H146, H526 and H446 were obtained from American Type Culture Collection; vials were replaced in less than 3 mo. Cells were cultured in RPMI 1640 (GIBCO, Grand Island, NY) supplemented with 10% fetal bovine serum (GIBCO, Grand Island, NY), 2 mM glutamine (BioFluids, Rockville, MD) and 100 units/L penicillin/streptomycin (BioFluids, Rockville, MD) in humidified 5% CO2 atmosphere at 37°C. HDAC inhibitors belinostat and romidepsin (Cancer Therapy Evaluation Program, NCI), were dissolved in DMSO at 10 mM and 100 µg/mL, respectively, stored in aliquots at −20°C and diluted in media immediately before use. Etoposide (VP-16) and cisplatin (Sigma, St. Louis, MO) were dissolved in DMSO at 10 mM and stored at −20°C.
Cell survival assay.
Cells were seeded in 96-well plates at 20,000 cells per well. Serial dilutions of drugs in medium were added in sextuplicate wells. Dose-response curves to a single drug following 48, 72 or 96 h were generated to determine the range of concentrations to be used for combination studies. For interaction studies, drugs were added simultaneously or sequentially (with the second compound introduced 24 h later) to a final volume of 100 µL. Cytotoxicity was measured using the Cell Titer 96 Aqueous One Solution Cell Proliferation Assay (Promega, Madison, WI), a colorimetric method for determining the number of viable cells based on reduction of a tetrazolium compound [3-(4,5-dimethyl-thiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) by metabolically active cells. After 72 h of exposure to a single drug or 96 h following sequential addition, 20 µL of MTS reagent were added to each well, and the plates were incubated in a humidified 37°C incubator with 5% CO2 for 2 h to 4 h. Absorbance at 490 nM was recorded using a 96-well plate reader. Data were averaged and normalized against the untreated cells to generate dose-response curves.
Analysis of drug interaction.
Drug combinations were evaluated using CalcuSyn (BIOSOFT, Ferguson, MO) software based on the multiple drug-effect equation of Chou-Talalay.Citation31 This method defines the expected effect of multiple agents used in combination and quantifies synergy or antagonism by determining how much the combination effect differs from the expected additive effect. Values of fraction affected (Fa) represent the level of effect or survival loss achieved by each drug individually or in combination. Experimental data were entered into the CalcuSyn interface to calculate a combination index (CI), a quantitative measure of the degree of drug interactions for a corresponding Fa. Serial CI values over a wide range of drug-effect levels (Fa) were then calculated, and the concentrations for each agent were chosen based on synergy. Once those concentrations were selected, a fixed ratio between HDIs and DNA damaging agents was chosen for all experiments. These data were used to generate Fa-CI plots, from which synergy, additivity or antagonism can be evaluated: CI < 1, CI = 1 and CI > 1, respectively.
Cell cycle analysis by flow cytometry.
Cell cycle distribution was determined by analyzing DNA content after propidium iodide staining. Cells were harvested, fixed in 70% ice-cold ethanol, washed with PBS and stained with 10 mg/mL propidium iodide containing 10 µg/mL RNase A. DNA content was analyzed using a FACScan flow cytometer (Becton Dickinson, San Jose, CA). Data were collected with Cell Quest Pro software (Becton Dickinson, San Jose, CA) from no fewer than 10,000 cells and analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Protein gel blot.
Cell pellets were suspended in lysis buffer (10 mM HEPES, pH 7.4, 1% SDS, 1% Triton X-100) with a protease inhibitor cocktail (Sigma, St. Louis, MO) and phosphatase inhibitors (Roche Diagnostic, Indianapolis, IN), and sonicated for 40 sec in a water bath (Misonic, Farmingdale, NY). Protein concentrations were measured using the BCA Protein Assay (Pierce, Rockford, IL). Twenty µg denaturated protein were loaded in each lane onto precast 4–12% Bis-Tris NuPAGE gels, subjected to electrophoresis and transferred onto 0.2 µM pore size nitrocellulose membranes (Invitrogen, Carlsbad, CA). Membranes were stained with 0.1% Ponceau S (Sigma, St. Louis, MO) and checked for comparable loading. After blocking with 5% non-fat dry milk in PBS for 1 h at room temperature, the membranes were incubated with the following primary antibodies: antiacetylated histone H3 K9 lysine (H3Ac), (1:3,000, Upstate Biotechnology, Lake Placid, NY), anti-poly adenosine diphosphateribose polymerase (PARP), anticleaved-PARP (1:1,000, Cell Signaling Technology, Danvers, MA) and antiglyceraldehyde phosphate dehydrogenase (GAPDH) (1:3,000, American Research Products, MA) overnight at 4°C. Membranes were probed with the IRDye 800CW goat anti-mouse or IRDye 680 goat anti-rabbit secondary antibodies, (1:10,000, LI-COR, Lincoln, NE) and visualized using Odyssey System (LI-COR).
Alkaline elution assay.
DNA damage was detected using alkaline elution assays as described previously in reference Citation27. Briefly, H146 cells were radiolabeled with [3H] thymidine (1.0 µCi/mL) for 72 h and chased overnight (16 h) with radioisotope-free medium before drug treatment. Cells were treated with 3 Gy radiation using a cesium-137 chamber at 1.7 Gy/min or incubated with 1 µM VP-16 for 2 h or with 1 µM belinostat for 24 h and harvested in ice-cold HBSS. DNA single-strand breaks were detected using DNA denaturing (pH 12.1) alkaline elution performed under deproteinizing conditions described previously in references 13 and 27. Following alkaline elution, filters were incubated at 65°C with 1 M HCl for 45 min and 0.04 M NaCl added for an additional 45 min. Radioactivity in each fraction was measured by liquid scintillation (Packard Instruments, Meridien, CT). The data were normalized to control untreated cells, and results from three independent experiment ares presented.
Analysis of DNA platination.
To detect the formation of DNA platinum adducts, we measured elemental platinum using an atomic absorption spectrometer. Following incubation with 100 µM cisplatin for 3 h or 16 µM for 24 h, cells were collected, washed three times in ice-cold PBS, and the DNA was extracted using DNA elution reagent DNAzol (Invitrogen, Carlsbad, CA) according to the manufacturer's protocol. Rehydrated DNA was analyzed for platinum content on a Perkin Elmer Analyst 800 atomic absorption spectrometer as described in reference Citation47. Platinum was measured in triplicate 20 µL aliquots, and the concentration was determined using an elemental platinum standard curve. Elemental platinum standards (0.05–2.5 µM) were prepared by serial dilution using High-Purity Standards's platinum standard (1 mg/mL). Platinum levels were normalized to DNA concentration. Assays were performed three times for each drug combination on different days.
Analysis of γH2AX phosphorylation by immunofluorescence.
Cells were treated as indicated, immobilized on glass slides by cytospin and stained for γH2AX as described in reference Citation48. Briefly, cells were fixed for 20 min with 4% paraformaldehyde in PBS (pH 7.4) and washed twice with PBS. After incubation for 20 min in 70% ethanol and washing with PBS, the cells were incubated in blocking buffer [8% bovine serum albumin in PBS] for 1 h before addition of a primary antibody against γH2AX for 2 h (Millipore, CA). Slides were incubated for an additional hour with the Alexa488-conjugated secondary antibody (Alexa Fluor® 488 goat anti-mouse IgG, Molecular Probes, Invitrogen, CA). Cells were stained with 0.5 mg/mL propidium iodide (PI) in the presence of 100 mg/mL RNase A (Sigma, MO) for 15 min in the dark. Finally, slides were mounted with Vectashield anti-fade mounting media (Vector Laboratories, Inc., Burlingame, CA). Images were taken using a Nikon Eclipse TE-300 confocal microscope.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Belinostat is synergistic with either cisplatin or VP-16 when added simultaneously. Cells were treated with increasing concentrations of drugs as indicated. Control cells received media alone, and the data was normalized to these untreated cells. The ordinate indicates concentrations of each one of the drugs. The upper part shows survival curves of cells treated simultaneously for 96 h with belinostat, cisplatin or V-P16 alone or in combination as indicated. The middle part corresponds to cell survival following belinostat alone for 96 h, cisplatin or VP-16 for 72 h or in combination. The bottom parts show the results using a reverse order of administration: cisplatin or VP-16 for 96 h and belinostat for 72 h. The right hand graphs show mathematical modeling of the data, with ordinate showing fraction affected (Fa). Note that simultaneous treatment (upper part) shows synergistic effects with combination index CI < 1 at all fractions affected and for all drug combinations. One representative experiment from 3–5 independent experiments, each one performed in six replicas, is shown here. Similar data for romidepsin are shown in Figure S1.
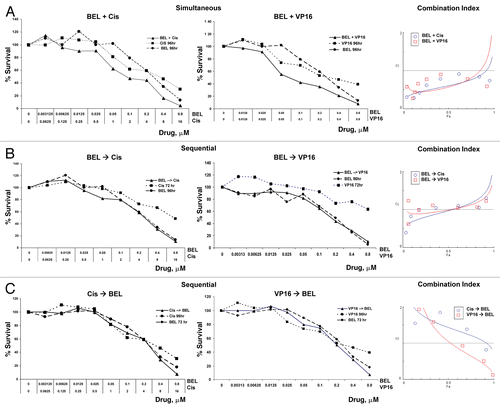
Figure 2 Effect of HDAC inhibitors in combination with VP-16 on cell cycle in H146 cells. (A) Cells were treated with belinostat (BEL) (0.3 µM), VP-16 (4 µM) or combination (BEL + VP-16). Top part shows control untreated cells or cells treated with a single drug for the indicated times. The bottom part shows simultaneously treated cells (left) and sequentially treated cells with belinostat added for 24 h prior to VP-16 (right), as indicated. (B) Same conditions as above using romidepsin (DP) at 1 ng/mL. The numbers in each part represent percent cells in each cell phase of cell cycle. A representative example of three independent experiments for each condition is shown here.
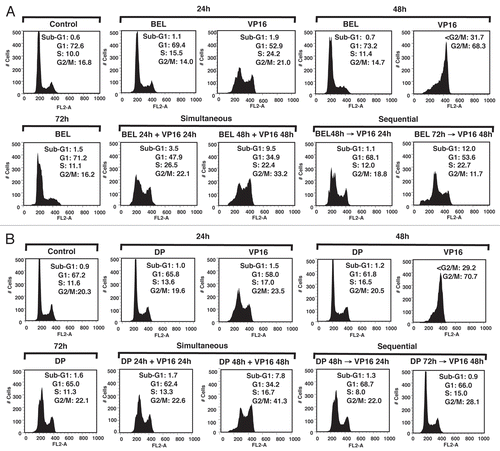
Figure 3 PARP degradation but not H3 acetylation depends on the schedule of drug administration. H146 cells were treated with belinostat (0.3 µM) or romidepsin (1 ng/mL) and VP-16 (4 µM) for 24 h and 48 h as indicated. Protein gel blot shows degradation of PARP, appearance of cleaved PARP (89 kD), acetylated H3 (17 kD) and GAPDH used as a loading control (37 kD). (A) Effect of belinostat, romidepsin, VP-16 and simultaneous treatment. (B) Effect of belinostat, romidepsin, VP-16 or combination added sequentially, HDI→VP-16, with a 24-h delay (HDI for 48 h and VP-16 for 24 h). (C) H3 acetylation in H146 cells treated with belinostat (0.3 µM) or romidepsin (1 ng/mL) and VP-16 (4 µM). Bar graph represents quantitative analysis of H3 acetylation from immunoblots as in (A and B). Results are mean ± SD from three to four independent experiments. Data are presented for both belinostat (BEL) and romidepsin (DP) as indicated. For this and the remaining figures, the (+) symbol indicates simultaneous exposure, while the (→) symbol indicates sequential exposure.
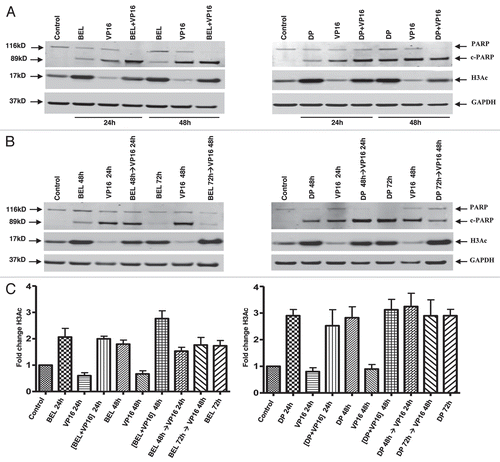
Figure 4 Accessibility of DNA to VP-16 and cisplatin following exposure to HDIs. (A) DNA damage detection by alkaline elution assay. H146 cells were treated simultaneously for 24 h with belinostat and VP-16, left part, or pretreated for 24 h with belinostat, followed by VP-16 for 2 h (sequential), right part. Error bars indicate SD from three or more biological replicas. (B) Detection of DNA platination. H146 cells were treated either with 100 µM cisplatin alone or in combination with 0.3 µM belinostat or 1 ng/mL romidepsin for 3 h or 24 h (left part) as indicated. Right part shows cells exposed to the same drugs with 16 µM of cisplatin for 24 h with or without belinostat or romidepsin for 24 h or 48 h. DNA platination was arbitrarily assigned to one for samples treated with cisplatin alone and data normalized to those values. Results are shown with SD from three independent experiments.
Figure 5 Imunofluorescence detection of γH2AX in H146 cells. (A) H146 cells were treated with 0.3 µM belinostat or 10 µM VP-16 alone or with different schedules of combinations as follows: simultaneous treatment with belinostat and VP-16 for 24 h (BEL 24 h + VP-16 24 h) or pretreatment with belinostat for 24 h followed by VP-16 for 24 h (BEL 48 h→VP-16 24 h). Representative confocal microscope images (X40) are shown. (B) Quantitation of average γH2AX fluorescence signal per cell from six random fields in one of three independent experiments. Standard t-test was used for statistical analysis **p < 0.01.
Additional material
Download Zip (4 MB)Acknowledgments
The authors wish to thank William Telford and Robert Robey for assistance with FACS analysis, Lois Bangiolo for assistance with immunoblot analysis and Mollie Wright for advice on drug interaction studies. This work was supported by the Intramural Research Program at the National Cancer Institute, NIH.
References
- Felsenfeld G, Groudine M. Controlling the double helix. Nature 2003; 421:448 - 453; PMID: 12540921
- Becker PB, Hörz W. ATP-dependent nucleosome remodeling. Annu Rev Biochem 2002; 71:247 - 273; PMID: 12045097
- Holbert MA, Marmorstein R. Structure and activity of enzymes that remove histone modifications. Curr Opin Struct Biol 2005; 15:673 - 680; PMID: 16263263
- Turner BM. Reading signals on the nucleosome with a new nomenclature for modified histones. Nat Struct Mol Biol 2005; 12:110 - 112; PMID: 15702071
- Minucci S, Pelicci P. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6:38 - 51; PMID: 16397526
- Xu WS, Parmigiani R, Marks P. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541 - 5552; PMID: 17694093
- Zhou Q, Melkoumian Z, Lucktong A, Moniwa M, Davie J, Strobl J. Rapid induction of histone hyperacetylation and cellular differentiation in human breast tumor cell lines following degradation of histone deacetylase-1. J Biol Chem 2000; 275:35256 - 35263; PMID: 10938272
- Schrump DS. Cytotoxicity mediated by histone deacetylase inhibitors in cancer cells: mechanisms and potential clinical implications. Clin Cancer Res 2009; 15:3947 - 3957; PMID: 19509170
- Sikandar S, Dizon D, Shen X, Li Z, Besterman J, Lipkin SM. The class I HDAC inhibitor MGCD0103 induces cell cycle arrest and apoptosis in colon cancer initiating cells by upregulating Dickkopf-1 and non-canonical Wnt signaling. Oncotarget 2010; 1:596 - 605; PMID: 21317455
- Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche T, et al. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst 2007; 99:1107 - 1119; PMID: 17623797
- Kim MS, Blake M, Baek J, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res 2003; 63:7291 - 7300; PMID: 14612526
- Fuino L, Bali P, Wittmann S, Donapaty S, Guo F, Yamaguchi H, et al. Histone deacetylase inhibitor LAQ824 downregulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine and epothilone B. Mol Cancer Ther 2003; 2:971 - 998; PMID: 14578462
- Zhang Y, Jung M, Dritschilo A. Enhancement of radiation sensitivity of human squamous carcinoma cells by histone deacetylase inhibitors. Radiat Res 2004; 161:667 - 674; PMID: 15161353
- Camphausen K, Tofilon P. Inhibition of histone deacetylation: a strategy for tumor radiosensitization. J Clin Oncol 2007; 25:4051 - 4056; PMID: 17827453
- Glaser KB. HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 2007; 74:659 - 671; PMID: 17498667
- Puglisi M, Dolly S, Faria A, Myerson J, Popat S, O'Brien M. Treatment options for small cell lung cancer—do we have more choice?. Br J Cancer 2010; 102:629 - 638; PMID: 20104223
- Steele NL, Plumb J, Vidal L, Tjørnelund J, Knoblauch P, Rasmussen A, et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res 2008; 14:804 - 810; PMID: 18245542
- Bates SE, Zhan Z, Steadman K, Obrzut T, Luchenko V, Frye R, et al. Laboratory correlates for a phase II trial of romidepsin in cutaneous and peripheral T-cell lymphoma. Br J Haematol 2010; 148:256 - 267; PMID: 19874311
- Lane AA, Chabner B. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol 2009; 27:5459 - 5468; PMID: 19826124
- Romanov VS, Abramova MV, Svetlikova SB, Bykova TV, Zubova SG, Aksenov ND, et al. p21(Waf1) is required for cellular senescence but not for cell cycle arrest induced by the HDAC inhibitor sodium butyrate. Cell Cycle 2010; 9:3945 - 3955; PMID: 20935470
- Rosato RR, Almenara J, Maggio S, Coe S, Atadja P, Dent P, et al. Role of histone deacetylase inhibitor-induced reactive oxygen species and DNA damage in LAQ-824/fludarabine antileukemic interactions. Mol Cancer Ther 2008; 7:3285 - 3297; PMID: 18852132
- Kurz EU, Wilson S, Leader K, Sampey B, Allan W, Yalowich J, et al. The histone deacetylase inhibitor sodium butyrate induces DNA topoisomerase II alpha expression and confers hypersensitivity to etoposide in human leukemic cell lines. Mol Cancer Ther 2001; 1:121 - 131; PMID: 12467229
- Tanaka T, Halicka HD, Traganos F, Seiter K, Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle 2007; 6:371 - 376; PMID: 17297310
- Schonn I, Hennesen J, Dartsch D. Cellular responses to etoposide: cell death despite cell cycle arrest and repair of DNA damage. Apoptosis 2010; 15:162 - 172; PMID: 20041303
- Kaufmann SH, Desnoyers S, Ottaviano Y, Davidson N, Poirier G. Specific proteolytic cleavage of poly(ADP-ribose) polymerase: an early marker of chemotherapy-induced apoptosis. Cancer Res 1993; 53:3976 - 3985; PMID: 8358726
- Nitiss JL. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009; 9:338 - 350; PMID: 19377506
- Kohn KW. DNA filter elution: a window on DNA damage in mammalian cells. Bioessays 1996; 18:505 - 513; PMID: 8787538
- Bonner WM, Redon C, Dickey J, Nakamura A, Sedelnikova O, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer 2008; 8:957 - 967; PMID: 19005492
- Sokolov MV, Dickey JS, Bonner WM, Sedelnikova OA. gammaH2AX in bystander cells: not just a radiation-triggered event, a cellular response to stress mediated by intercellular communication. Cell Cycle 2007; 6:2210 - 2212; PMID: 17881892
- Nakamura AJ, Rao VA, Pommier Y, Bonner WM. The complexity of phosphorylated H2AX foci formation and DNA repair assembly at DNA double-strand breaks. Cell Cycle 2010; 9:389 - 397; PMID: 20046100
- Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 2010; 70:440 - 446; PMID: 20068163
- Sato T, Suzuki M, Sato Y, Echigo S, Rikiishi H. Sequence-dependent interaction between cisplatin and histone deacetylase inhibitors in human oral squamous cell carcinoma cells. Int J Oncol 2006; 28:1233 - 1241; PMID: 16596240
- Hajji N, Wallenborg K, Vlachos P, Nyman U, Hermanson O, Joseph B. Combinatorial action of the HDAC inhibitor trichostatin A and etoposide induces caspase-mediated AIF-dependent apoptotic cell death in non-small cell lung carcinoma cells. Oncogene 2008; 27:3134 - 3144; PMID: 18071312
- Piekarz RL, Bates S. Epigenetic modifiers: basic understanding and clinical development. Clin Cancer Res 2009; 15:3918 - 3926; PMID: 19509169
- Miller KM, Tjeertes JV, Coates J, Legube G, Polo SE, Britton S, et al. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 2010; 17:1144 - 1151; PMID: 20802485
- Bruzzese F, Rocco M, Castelli S, Di Gennaro E, Desideri A, Budillon A. Synergistic antitumor effect between vorinostat and topotecan in small cell lung cancer cells is mediated by generation of reactive oxygen species and DNA damage-induced apoptosis. Mol Cancer Ther 2009; 8:3075 - 3087; PMID: 19887547
- Yu C, Friday B, Lai J, McCollum A, Atadja P, Roberts L, et al. Abrogation of MAPK and Akt signaling by AEE788 synergistically potentiates histone deacetylase inhibitor-induced apoptosis through reactive oxygen species generation. Clin Cancer Res 2007; 13:1140 - 1148; PMID: 17317822
- Munshi A, Tanaka T, Hobbs M, Tucker S, Richon V, Meyn R. Vorinostat, a histone deacetylase inhibitor, enhances the response of human tumor cells to ionizing radiation through prolongation of gammaH2AX foci. Mol Cancer Ther 2006; 5:1967 - 1974; PMID: 16928817
- Robles AI, Wright M, Gandhi B, Feis S, Hanigan C, Wiestner A, et al. Schedule-dependent synergy between the heat shock protein 90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin and doxorubicin restores apoptosis to p53-mutant lymphoma cell lines. Clin Cancer Res 2006; 12:6547 - 6556; PMID: 17085670
- Koll TT, Feis S, Wright M, Teniola M, Richardson M, Robles A, et al. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol Cancer Ther 2008; 7:1985 - 1992; PMID: 18645008
- Bevins RL, Zimmer S. It's about time: scheduling alters effect of histone deacetylase inhibitors on camptothecin-treated cells. Cancer Res 2005; 65:6957 - 6966; PMID: 16061681
- de Ruijter AJ, Leen R, Hoebink J, Caron H, van Kuilenburg A. Antagonistic effects of sequential administration of BL1521, a histone deacetylase inhibitor and gemcitabine to neuroblastoma cells. Cancer Lett 2006; 233:240 - 246; PMID: 15907366
- Conti C, Leo E, Eichler G, Sordet O, Martin M, Fan A, et al. Inhibition of histone deacetylase in cancer cells slows down replication forks, activates dormant origins and induces DNA damage. Cancer Res 2010; 70:4470 - 4480; PMID: 20460513
- Harr MW, Willey J. The interactive transcript abundance index [c-myc*p73alpha]/[p21*Bcl-2] correlates with baseline level of apoptosis and response to CPT-11 in human bronchogenic carcinoma cell lines. Int J Oncol 2007; 30:1553 - 1560; PMID: 17487378
- Kaye FJ. Molecular biology of lung cancer. Lung Cancer 2001; 34:35 - 41; PMID: 11720739
- Rabik CA, Fishel M, Holleran J, Kasza K, Kelley M, Egorin M, et al. Enhancement of cisplatin [cis-diammine dichloroplatinum (II)] cytotoxicity by O6-benzylguanine involves endoplasmic reticulum stress. J Pharmacol Exp Ther 2008; 327:442 - 452; PMID: 18664592
- McGahan MC, Tyczkowska K. The determination of platinum in biological materials by electrothermal atomic absorption spectroscopy. Spectrochimia Acta 1987; 42:665; PMID: 509738
- Zhang YW, Jones T, Martin S, Caplen N, Pommier Y. Implication of checkpoint kinase-dependent upregulation of ribonucleotide reductase R2 in DNA damage response. J Biol Chem 2009; 284:18085 - 18095; PMID: 19416980