Abstract
Emerging evidence suggests that Sirtuin 6 (SIRT6) functions as a longevity assurance gene by promoting genomic stability, regulating metabolic processes and attenuating inflammation. Here, we examine the effect of SIRT6 activation on cancer cells. We show that SIRT6 overexpression induces massive apoptosis in a variety of cancer cell lines but not in normal, non-transformed cells. This cell death requires the mono-ADP-ribosyltransferase but not the deacetylase activity of SIRT6 and is mediated by the activation of both the p53 and p73 apoptotic signaling cascades in cancer cells by SIRT6. These results suggest that SIRT6 is an attractive target for pharmacological activation in cancer treatment.
Introduction
Sirtuin 6 (SIRT6) is a mammalian homolog of the yeast Sir2 deacetylase. SIRT6 represents an interesting connection between longevity assurance and tumor suppression. Sirt6-knockout mice develop a multisystemic disorder with complete penetrance that, in many respects, resembles premature aging.Citation1 At the molecular level, SIRT6-deficient cells are characterized by dysfunctional telomeres,Citation2 genomic instability,Citation1 inefficient base excision and double-strand break repairCitation1,Citation3–Citation5 as well as hyperactive NFκB signalingCitation6 and aberrant glucose homeostasis.Citation7 Intriguingly, many of the molecular deficiencies that characterize SIRT6-knockout and -knockdown cells are also representative of cancer cells. In particular, genomic instability and imbalanced glucose homeostasis have emerged as hallmark features of many types of cancer.Citation8 AneuploidyCitation9,Citation10 and overactive NFκB signaling are observed in a large number of tumors as well.Citation11,Citation12
SIRT6 has two biochemical activities: deacetylase and mono ADP ribosyltransferase, both of which are dependent on the availability of NAD+. Although SIRT6 was first identified as a mono-ADP ribosyltransferase enzyme,Citation13 most studies to date have focused on the deacetylase activity of SIRT6. We recently identified PARP1 as the first substrate for SIRT6 mono-ADP ribosylation, wherein SIRT6 mono-ADP-ribosylates PARP1 to promote DNA double-strand break repair in response to oxidative stress.Citation5
The short lifespan of Sirt6-knockout mice has made it difficult to directly assess whether SIRT6 functions as a tumor suppressor. However, several other studies have indicated that SIRT6 may play a role in attenuating and even antagonizing tumor development. The SIRT6 chromosomal locus is mutated in a large number of acute myeloid leukemias;Citation14 higher levels of SIRT6 expression are correlated with lower incidences of relapse and recurrence in certain types of breast cancer,Citation15 and SIRT6 was identified as an interacting partner of the GCIP tumor suppressor.Citation16 Moreover, in the absence of SIRT6, cells transition from aerobic respiration to glycolysis to meet the cellular energy requirements; this changeover is reminiscent of the Warburg effect, wherein cancer cells rely on a similar transition for survival.Citation17,Citation18 Lastly, in the absence of SIRT6, cells exhibit global H3K56 hyperacetylation: a recent report indicated that the degree of total H3K56 acetylation correlates with tumorigenicity and tumor grade.Citation19
Given that SIRT6 appears to impact on multiple pathways related to tumor survival and development, we hypothesized that SIRT6 may function to suppress cancer cell growth or promote cancer cell death. Here we report, that SIRT6 overexpression is selectively toxic to multiple types of cancer cells and suggest that the gene may be an attractive target for genetic or pharmacological modulation. This killing is mediated by the mono-ADP ribosyltransferase activity of SIRT6 and the subsequent activation of the p53 and p73 apoptotic signaling cascades in cancer cells.
Results
SIRT6 overexpression selectively kills cancer cells.
To test our hypothesis that SIRT6 expression may be antagonistic to tumor growth and development, we overexpressed the gene in six primary, non-transformed human cell lines and six cancerous human cell lines. The control group of non-transformed cell lines was comprised of primary fibroblast cell lines (HCA2, WI-38 and IMR-90), an immortalized fibroblast cell line (HCA2-hTERT) and primary mammary epithelial cell lines (HMEC1, HMEC2). The cancer cell lines included a transformed cell line (GP2-293), a cervical carcinoma (HeLa), a fibrosarcoma (HT1080), primary breast tumor cell lines (HCC70, HCC1954) and a metastatic breast tumor cell line (MDA-MB-231). Each cell line was transfected with equal amounts of a SIRT6-expressing or a control plasmid. Transgene expression was similar for each line 24 hours after transfection (). Cell survival was measured relative to control 72 hours post-transfection.
We observed that SIRT6 overexpression was not toxic to primary human fibroblasts and exhibited only minor toxicity in primary human mammary epithelial cells (). However, SIRT6 overexpression induced massive cell death in each of the cancer cell lines we tested. Between 65–90% of the cancer cells receiving the SIRT6 transgene died (). Annexin V staining revealed that this death was driven by apoptosis (). Cumulatively, these results indicate that SIRT6 overexpression is selectively cytotoxic to multiple cancer cell lines but not to non-transformed cell lines.
Mono-ADP ribosyltransferase activity is required for killing cancer cells.
SIRT6 has two biochemical activities: deacetylase and mono-ADP ribosyltransferase. To determine which of these activities is required to drive apoptosis in cancer cells, we overexpressed mutant versions of SIRT6, defective for either or both of these activities in normal fibroblasts (HCA2) and in cancer cell lines (HeLa and HT1080). Ser56→Tyr56 (S56Y) lacks both activities; R65A lacks deacteylase activity but retains mono-ADP-ribosyltransferase activity, and G60A lacks mono-ADP-ribosyltransferase activity but retains deacetylase activity.Citation5 Overexpression of wild-type SIRT6 or any of these mutants did not affect viability in the non-transformed fibroblast cells. In cancer cells, however, overexpression of R65A but not G60A was sufficient to drive apoptosis ( and B). This suggests that the mono-ADP-ribosyltransferase activity of SIRT6 is required to drive apoptosis in cancer cells, whereas the deacetylase activity is dispensable for this function. The only known in vivo target of SIRT6 mono-ADP-ribosylation is PARP1, wherein mono-ADP-ribosylation functions to activate PARP1.Citation5 Pretreating the cancer cells with the PARP1 inhibitor PJ-34 failed to protect against SIRT6-driven apoptosis (), and overexpression of PARP1 did not drive apoptosis in cancer cells (), arguing that SIRT6 mono-ADP-ribosylates a different target protein to drive apoptosis in cancer cells.
Cancer cell killing is dependent on p53/p73 activity.
To gain additional insight into the biochemical cascade that SIRT6 initiates to drive apoptosis in cancer cells, we assessed whether the SIRT6-mediated killing of cancer cells was dependent on any of the classic apoptotic pathways. In HeLa cells, the prototypical regulator of apoptosis, p53, is targeted for degradation by the E6 oncoprotein, suggesting that an auxiliary apoptotic pathway mediates SIRT6-driven cytotoxicity in cancer cells. One such auxiliary apoptotic pathway operates through p73. When we co-transfected SIRT6 with a p73 dominant-negative plasmid, we observed that SIRT6-driven cytotoxicity was severely dampened (), suggesting that SIRT6 operates partially through p73 signaling to drive apoptosis in HeLa cells. Interestingly, we found that blockade of p73 in HT1080 cells, which express wild-type p53, was insufficient to prevent SIRT6-driven apoptosis. Only when both p53 and p73 were inhibited in HT1080 cells, were we able to prevent SIRT6-driven apoptosis (). This suggests that overexpression of SIRT6 is capable of driving apoptosis through either p53 or p73 in cancer cells.
SIRT6 induces apoptosis in cancer cells via ATM pathway.
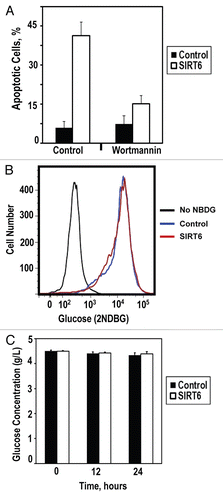
The p53 and p73 tumor suppressors are the downstream effectors of pro-apoptotic signaling cascades. We hypothesized that SIRT6 activates an upstream signal rather than p53 or p73 directly to initiate apoptosis in cancer cells. Two common characteristics of cancer cells are a heightened genomic instability and a hyperglycemic phenotype characterized by increased glucose intake. Because SIRT6 promotes genomic stability by catalyzing base excision and double-strand break repair and attenuates glucose intake through its interactions with HIF1α, we hypothesized that overexpression of SIRT6 may drive apoptosis in cancer cells through either of these pathways. To assess whether signaling through DNA repair and DNA damage response pathways was required for SIRT6 to promote apoptosis in cancer cells, we examined whether the ATM kinase inhibitor wortmannin attenuated SIRT6 cytotoxicity in cancer cells. We observed that blockade of ATM signaling strongly reduced SIRT6-driven apoptosis in cancer cells (), suggesting that ATM and perhaps the DNA damage response are involved in mediating SIRT6 cytoxicity. Contrastingly, we did not observe a role for glucose metabolism or intake in the mediation of SIRT6-driven apoptosis in cancer cells. Overexpression of SIRT6 did not alter the rate of 2-NBDG, a fluorescently-labeled glucose analog, intake in HeLa or HT1080 cells () or the rate at which glucose was depleted from the media (). In agreement with these observations, we noted that growing HeLa cells in glucose-free media did not induce massive apoptosis in these cells within 72 hours (data not shown).
Discussion
We have provided the first evidence that SIRT6 overexpression is selectively toxic to cancer cell lines but not non-transformed cell lines. This toxicity is a consequence of SIRT6-driven apoptosis. This cancer-specific cytotoxicity is dependent on the mono-ADP ribosyltransferase activity of SIRT6. Mechanistically, SIRT6 appears to promote apoptosis through either p53 or p73 signaling and requires ATM to initiate this response.
We observed that a broad spectrum of cancer cell lines were sensitive to SIRT6 overexpression. This may stem from the ability of SIRT6 to stimulate apoptosis redundantly, through both the p53 and the p73 programmed cell death pathways. While inactivating mutations in p53 are the single most common type of genetic lesion observed in cancer, loss of function mutations in p73 are far less common.Citation20
p53 and p73 are typically executors of upstream pro-apoptotic signals. The precise mechanism by which SIRT6 overexpression selectively activates these programmed cell death pathways in cancer cells remains unclear, however. One hypothesis was that SIRT6 overexpression would shut down or attenuate glycolysis and glucose uptake in cancer cells, thereby denying the cancer cells their primary energy source. However, we were unable to detect significant changes in glucose intake prior to cancer cell death after SIRT6 overexpression, suggesting that an alternative mechanism drives apoptosis in these cells.
The dependency of killing on ATM signaling and SIRT6 mono-ADP ribosylation activity provides some hints as to how SIRT6 overexpression may selectively induce cancer cell death. One possibility is that SIRT6 potentiates or activates a dormant DNA damage response (DDR) in cancer cells. The oncogene expression and rapid proliferation that characterize many cancer cells typically induces large amounts of DNA damage. Normally, this type of damage is recognized by ATM and other members of the DDR pathway, and cells enter into cell cycle arrest or undergo either p53- or p73-dependent apoptosis. In cancer cells, however, the DDR signaling is often subverted to avoid cell death. It is possible that SIRT6 overexpression sensitizes cancer cells to DNA damage. Exactly how SIRT6 would exert this type of effect remains unclear, but it is likely that SIRT6 mono-ADP-ribosylates a target protein to achieve these ends. To date, the only known, in vivo, target of SIRT6 mono-ADP ribosylation is PARP1. Our data indicates that SIRT6 overexpression induces apoptosis independently of PARP1, however. This suggests that there are additional targets for SIRT6 mono-ADP ribosylation in vivo.
Protocols that induce cancer cell death but do not harm healthy tissues are naturally of intense scientific and medical interest. Here, we have provided evidence that SIRT6 overexpression or hyperactivation may constitute such a therapy. Several cellular and animal studies have indicated that dietary restriction and glucose deprivation are sufficient to increase the cellular titer of SIRT6.Citation21 It will be interesting to see if cancer cells exhibit a similar response to starvation and, if so, whether the subsequent increase in SIRT6 levels is lethal. There is evidence that at least a subset of human cancers are sensitive to dietary restriction,Citation22 and it is possible that SIRT6 plays a role in mediating this effect.Citation21 Chemical intervention also represents an intriguing method for stimulating SIRT6 activity. SIRT6 is a member of the sirtuin gene family, and several lines of evidence indicate that the activity of the founding member of this family in mammals, SIRT1, can be stimulated by small molecules.Citation23 Analogously, it may be possible to synthesize compounds that can stimulate SIRT6 activity. Any such compounds could potentially be useful in treating cancer and a variety of other age-related diseases. We propose, therefore, that genetic or pharmacological activation of SIRT6 may constitute a valuable new strategy for treating and attenuating neoplastic disease.
Methods
Plasmids.
SIRT6 was cloned into the backbone of pDsRed2-N1 as described previously in reference Citation5. The S56Y, G60A and R65A mutant SIRT6 plasmids were produced via site-directed mutagenesis,Citation5 and confirmed with sequencing.Citation5 The control pMaxGFP plasmid was obtained from Amaxa. The PARP1 plasmid was purchased from Origene (SC119157). The p53 and p73 dominant-negative plasmids were purchased from Addgene (16436, 22978).Citation24,Citation25
Cell culture.
All cell lines were grown in monolayer at 37°C in 3% O2, 5% CO2 and 97% relative humidity in HERA Cell 240 incubators on treated polystyrene cell culture plates (Corning). Human fibroblast cell lines were maintained in MEM (ATCC) supplemented with 15% FBS; FBS, (Gibco) and 1x Pen/Strep (Gibco). Human mammary epithelial cells were maintained in in MEBM (Lonza) and supplemented with MEGM SingleQuots (Lonza), which contains BPE, hEGF, insulin, hydrocortisone and GA-1000. Human cervical carcinoma, fibrosarcoma and transformed GP2-293 cell lines were maintained in DMEM (Gibco) supplemented with 10% FBS (Gibco), 1x Pen/Strep (Gibco) and 1x nonessential amino acids (Gibco). Breast carcinoma cell lines were maintained in MEBM (Lonza) and supplemented with MEGM SingleQuots (Lonza), which contain BPE, hEGF, insulin, hydrocortisone and GA-1000. Cells were split regularly using standard tissue culture techniques.
Protein gel blots.
Exponentially growing cells were transfected with either a control plasmid or a SIRT6-expressing plasmid 24 hours prior to harvesting. Once harvested, the cells were resuspended in PBS pH 7.4 (Gibco) with Complete Protease Inhibitor Mixture (Roche) and lysed by mixing with Laemmli sample buffer (BioRad) containing 5% 2-mercaptoethanol (J.T. Baker) followed by boiling for 10 min with vortexing every 5 min. The protein concentration of each sample was determined using the DC protein assay (BioRad). Equal amounts of protein were loaded for each sample. The proteins were separated on a 10% SDS/PAGE and then blotted onto a nitrocellulose membrane (Biorad) and blocked in TBST-T with 1.25% dried milk. The membranes were probed with antibodies against SIRT6 (Abcam) and Tubulin (Abcam).
Transfections.
Cells were split to 5 × 105 cells per 10 cm plate 24 hours prior to transfection with 5 µg of either a control or SIRT6-expressing plasmid. Human fibroblasts, mammary epithelial cells and breast carcinoma cells were transfected using Amaxa Nucleofactor II electroporation. HCA2 cells were transfected using the U-20 program and NHDF solution; IMR-90, program X-001 and NHDF solution; WI-38, program V-001 and solution NHDF; HCA2-hTERT, program U-20 and solution NHDF; HMEC1/2, program Y-001 and solution V; HCC1954, program A-023, solution V; HCC70, program X-013, solution V; MDA-MB-231, program X-001, solution V. HeLa, HT1080 and GP2-293 cells were transfected using Fugene (Roche).
Cell survival and apoptosis assays.
Cells were split to a density of 5 × 105 cells per plate 24 hours prior to transfection with either a control or SIRT6-expressing plasmid. Cell survival was measured 72 hours after transfection by counting the adherent cells in each group using a Z2 particle counter (Beckman Coulter). The ration of SIRT6 transfected cells to cells transfected with a control plasmid represents the raw survival. These values were then normalized for transfection efficiency as described previously in reference Citation26. Apoptosis was measured by pooling the adherent and floating cells 72 hours after transfection, staining the cells with FITC-Annexin V and PI stain; the cells were sorted using FACS.
Glucose uptake assays.
To measure glucose uptake, cells were grown for 24 hours under normal conditions, after which 100 µM 2-NBDG (Invitrogen) was added to the media for 2 hours. Fluorescence was measured in a FACSCalibur Analyzer (BD). The depletion of glucose from the media was measured with an enzymatic assay (Sigma). Cells were grown under normal conditions and at the indicated time points the media was decanted, remaining cells and debris were removed by centrifugation and glucose concentration was quantified as prescribed by the manufacturer's protocol.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 SIRT6 overexpression is selectively cytotoxic to multiple cancer cell lines. (A) The indicated non-cancerous and cancer cell lines were transfected with a SIRT6-expressing vector or a control plamsid. Representative immunoblot of SIRT6 levels 24 h after transfection. SIRT6 expression is similar in cancer and normal cells. (B) Represenative images of cells taken 72 h post-transfection. (C) Quantification of SIRT6 toxicity. Cell survival 72 h post-transfection, measured as the number of adherent cells transfected with SIRT6 expressing vector compared to the number of adherent cells transfected with a control plasmid. (D) Quantification of SIRT6-induced apoptosis. Relative levels of apoptosis 72 h post-transfection, determined by Annexin V staining. Experiments were repeated three times and error bars show SD.
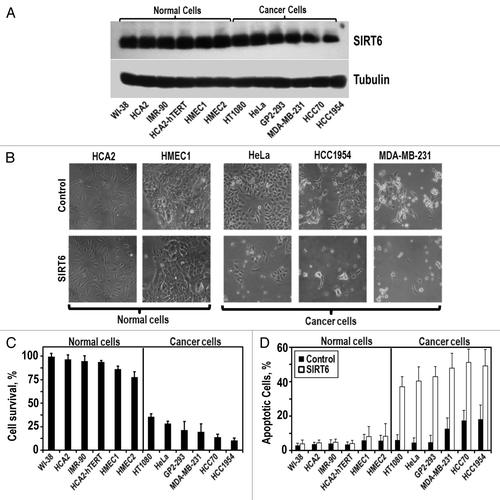
Figure 2 SIRT6 mono-ADP-ribosyltransferase activity is required for killing cancer cells. (A) Adherent cell count 72 h after transfection with indicated SIRT6-encoding vector relative to a control plasmid. (B) Quantification of apoptosis levels 72 h after transfection with a plasmid expressing wild-type SIRT6 or its mutants. (C) Pretreatment of HT1080 cells with the PARP1 inhibitor PJ-34 did not prevent SIRT6-induced apoptosis. (D) Overexpresison of PARP1 did not induce apoptosis in HT1080 cells. Experiments were repeated three times, and error bars show SD.
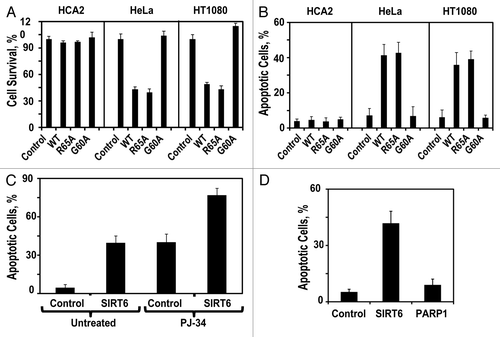
Figure 3 SIRT6 induces apoptosis via the p53 and p73 cell death pathways. (A) HeLa cells were transfected with plasmids encoding p53 dominant-negative fragment (p53DN) or p73 dominant-negative fragment (p73DN). Blockade of p73 signaling is sufficient to prevent SIRT6-induced apoptosis. (B) HT1080 cells were transfected with indicated expressing plasmids. Inhibition of both p53 and p73 signaling is required to prevent SIRT6-induced apoptosis. Experiments were repeated three times, and error bars show SD.
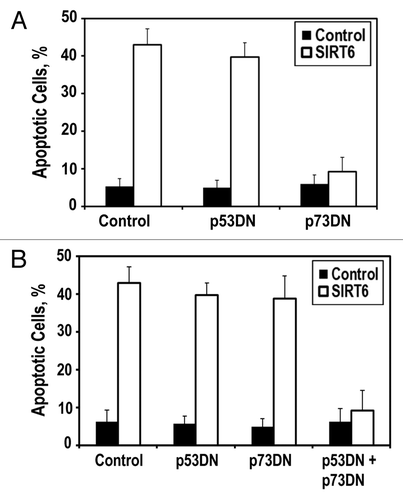
Figure 4 SIRT6 induces apoptosis in cancer cells via ATM pathway. (A) Cancer cells were pretreated with DMSO or ATM inhibitor wortmannin prior to transfection with the SIRT6-encoding vector or a control plasmid. Pretreatment with ATM inhibitor attenuated SIRT6-mediated apoptosis. (B) Glucose intake was measured in cancer cells. Transfection with SIRT6-encoding vector did not alter the kinetics of glucose intake compared to control. (C) Glucose depletion from the media was measured in cancer cells transfected with the SIRT6-encoding vector or a control plasmid. No significant changes were observed within 24 h of transfection. Experiments were repeated three times, and error bars show SD.
Acknowledgments
We thank Lai Chan for technical help. This work was supported by grants from the National Institutes of Health AG031227 and AG27237 to V.G., and grants from the Ellison Medical Foundation to V.G. and A.S.
References
- Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006; 124:315 - 329; PMID: 16439206; http://dx.doi.org/10.1016/j.cell.2005.11.044
- Michishita E, McCord RA, Berber E, Kioi M, Padilla-Nash H, Damian M, et al. SIRT6 is a histone H3 lysine 9 deacetylase that modulates telomeric chromatin. Nature 2008; 452:492 - 496; PMID: 18337721; http://dx.doi.org/10.1038/nature06736
- McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging (Albany NY) 2009; 1:109 - 121; PMID: 20157594
- Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010; 329:1348 - 1353; PMID: 20829486; http://dx.doi.org/10.1126/science.1192049
- Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011; 332:1443 - 1446; PMID: 21680843; http://dx.doi.org/10.1126/science.1202723
- Kawahara TL, Michishita E, Adler AS, Damian M, Berber E, Lin M, et al. SIRT6 links histone H3 lysine 9 deacetylation to NFkappaB-dependent gene expression and organismal life span. Cell 2009; 136:62 - 74; PMID: 19135889; http://dx.doi.org/10.1016/j.cell.2008.10.052
- Zhong L, D'Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 2010; 140:280 - 293; PMID: 20141841; http://dx.doi.org/10.1016/j.cell.2009.12.041
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646 - 674; PMID: 21376230; http://dx.doi.org/10.1016/j.cell.2011.02.013
- Pathak S, Multani AS, Furlong CL, Sohn SH. Telomere dynamics, aneuploidy, stem cells and cancer [review]. Int J Oncol 2002; 20:637 - 641; PMID: 11836581
- Storchova Z, Pellman D. From polyploidy to aneuploidy, genome instability and cancer. Nat Rev Mol Cell Biol 2004; 5:45 - 54; PMID: 14708009; http://dx.doi.org/10.1038/nrm1276
- Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 1999; 18:6938 - 6947; PMID: 10602468; http://dx.doi.org/10.1038/sj.onc.1203221
- Chiao PJ, Na R, Niu J, Sclabas GM, Dong Q, Curley SA. Role of Rel/NFkappaB transcription factors in apoptosis of human hepatocellular carcinoma cells. Cancer 2002; 95:1696 - 1705; PMID: 12365017; http://dx.doi.org/10.1002/cncr.10829
- Liszt G, Ford E, Kurtev M, Guarente L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 2005; 280:21313 - 21320; PMID: 15795229; http://dx.doi.org/10.1074/jbc.M413296200
- Mahlknecht U, Ho AD, Voelter-Mahlknecht S. Chromosomal organization and fluorescence in situ hybridization of the human Sirtuin 6 gene. Int J Oncol 2006; 28:447 - 456; PMID: 16391800
- McGlynn LCJ, Edwards J, Shiels P. Evaluating the role of sirtuins 5, 6 & 7 in breast cancer. Cancer Research 2009; 69:3
- Ma W, Stafford LJ, Li D, Luo J, Li X, Ning G, et al. GCIP/CCNDBP1, a helix-loop-helix protein, suppresses tumorigenesis. J Cell Biochem 2007; 100:1376 - 1386; PMID: 17131381; http://dx.doi.org/10.1002/jcb.21140
- Zhong L, Mostoslavsky R. SIRT6: A master epigenetic gatekeeper of glucose metabolism. Transcr 2010; 1:17 - 21; PMID: 21327158; http://dx.doi.org/10.4161/trns.1.1.12143
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 1033; PMID: 19460998; http://dx.doi.org/10.1126/science.1160809
- Das C, Lucia MS, Hansen KC, Tyler JK. CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 2009; 459:113 - 117; PMID: 19270680; http://dx.doi.org/10.1038/nature07861
- Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2:371 - 386; PMID: 15280445
- Kanfi Y, Shalman R, Peshti V, Pilosof SN, Gozlan YM, Pearson KJ, et al. Regulation of SIRT6 protein levels by nutrient availability. FEBS Lett 2008; 582:543 - 548; PMID: 18242175; http://dx.doi.org/10.1016/j.febslet.2008.01.019
- Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci 2010; 31:89 - 98; PMID: 20097433; http://dx.doi.org/10.1016/j.tips.2009.11.004
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003; 425:191 - 196; PMID: 12939617; http://dx.doi.org/10.1038/nature01960
- Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Suppression of human colorectal carcinoma cell growth by wild-type p53. Science 1990; 249:912 - 915; PMID: 2144057; http://dx.doi.org/10.1126/science.2144057
- Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, Liu W, et al. Role for the p53 homologue p73 in E2F-1-induced apoptosis. Nature 2000; 407:645 - 648; PMID: 11034215; http://dx.doi.org/10.1038/35036614
- Hine CM, Seluanov A, Gorbunova V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc Natl Acad Sci USA 2008; 105:20810 - 20815; PMID: 19106292; http://dx.doi.org/10.1073/pnas.0807990106