Abstract
Phosphorylation has long been recognized as the key mediator of protein signaling. New modes of signaling regulation are emerging with the development of specific chemical probes and application of high-throughput mass spectrometry technologies. Using biotin-tagged chemical probes for protein oxidation, mass spectrometry and functional assays, our group has recently reported isoform-specific oxidation of Akt2 in response to PDGF signaling. The studies included here investigate the functional consequence of oxidation on Akt2-mediated cell migration and cell cycle. Akt2-KO MEFs transduced with WT and Cys124Ser Akt2 were used as the model system for these studies. The implications of these findings on disease pathology are discussed.
Introduction
PKB/Akt kinases dictate and relay major signaling pathways to modulate a range of biological processes such as cell survival, proliferation, differentiation, migration and metabolism. These processes are mediated by overlapping as well as distinct functions of the three Akt isoforms, Akt1, Akt2 and Akt3. Mitogens, cytokines and other extracellular cues activate PI3K/Akt pathway. PI3K catalyzes production of the phosphoinositides, PIP3 and PIP2 that bind the PH domain of Akt and recruit it to the plasma membrane. PDK1 phosphorylates Akt at a threonine site in the catalytic kinase domain at position 308, 309 and 305 in Akt1, Akt2 and Akt3, respectively. mTOR/Rictor/Sin1 complex phosphorylates Akt at a serine site in the C-terminal tail at position 473, 474 and 472 in Akt1, Akt2 and Akt3, respectively. Citation1–Citation3 Phosphorylation at both sites is required for full activation of Akt kinase.
There is extensive literature on Akt kinases and isoform-specific Akt1, Akt2 and Akt3 knockout mice have been characterized as well.Citation4,Citation5 The current dogma is that Akt1 functions in cell migration and proliferation, while Akt2 is involved in regulation of cell metabolism. These functional differences are attributed to variations in their tissue expression, temporal activation, subcellular location and substrate-specific signaling.Citation6–Citation8
Our group recently reported the first posttranslational modification that distinguishes between the three Akt isoforms. Isoform-specific oxidation of Akt2 was shown to occur in response to PDGF and other extracellular signaling cues capable of inducing reactive oxygen species (ROS). Oxidation resulted in loss of Akt2 kinase activity. Mass spectrometry and site-directed mutagenesis pointed to oxidation at Cys124 in Akt2 as a trigger for intramolecular disulfide bond formation and selective inhibition of its kinase activity.Citation9 Cys124 is located within the linker region that connects the PH and kinase domains in Akt2 and is not conserved in Akt1 and Akt3 isoforms. The extent of PDGF-induced oxidation of Akt2 was estimated at 45–66% of total phosphorylated Akt2. Furthermore, a functional relationship between PDGF-dependent redox regulation of Akt2 and physiological outputs such as glucose-uptake was demonstrated using Akt2-KO MEFs that were transduced with Akt2 WT or Cys124Ser mutant.
Here, we determined the consequence of Akt2 oxidation on cell migration and cell cycle distribution under conditions of normal growth and starvation and discuss the broad implications of these cumulative findings. The data suggests that accumulation of intracellular oxidants promotes migration of Akt2 WT MEFs but not of the Akt2 Cys124Ser MEFs. Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs show distinct cell cycle distribution upon starvation with increased G1-S transition for the Akt2 Cys124Ser MEFs. These differences were not evident when the starved cells were rescued with serumsupplemented media.
Complexity in the Regulation of Cell Migration by Akt1 and Akt2 Isoforms
Migration is a fundamental homeostatic event that is integral to physiological processes such as embryogenesis, wound healing, tissue repair, immune surveillance as well as to a variety of vascular and inflammatory conditions including cancer, arthritis and others.Citation10 Regulation of cell migration by PI3K/Akt pathway and the role of individual Akt isoforms have been extensively studied in cancer cells and fibroblasts. A migrating cell typically forms a leading edge by restructuring actin through its polymerization. PI3K signaling pathways are largely known to generate an internal gradient through phosphoinositides that aid in the development of a leading edge by defining the cell polarity.Citation11,Citation12 This event is closely followed by adherence to the substratum initiated by integrins and relayed to the Rho GTPases by other adhesive signals such as FAKs, talin, vinculin, and a number of other cellular proteins. Finally, retraction of the rear body of the cell occurs through disruption of the adhesion at the rear and contraction of the cytoskeletal network.
Akt2 and cell migration in cancer.
Studies by Toker et al. report increased migration and invasion of the breast cancer cells by Akt1 knock down through a mechanism that involves ubiquitin-mediated degradation of NFAT.Citation13 Methyltransferase EZH2 was also proposed to promote migration in glioblastoma cells in a process that could be mediated by downregulation of Akt.Citation14,Citation15 In vitro and in vivo studies conducted by Slamon et al. on the other hand, demonstrate the opposite—enhanced motility, aggressive metastases and invasion in the breast and ovarian cancer cells overexpressing Akt2 kinase, through upregulated β1 integrin signaling.Citation16 Additional reports in breast cancer cells demonstrate reduced migration and invasion with decreased Akt2.Citation17–Citation20 These reports indicate activation of different signaling pathways downstream of Akt2 leading to a range of biological response depending on the cell/tissue type and stimuli.Citation21 The discrepancy in these findings could be explained by the presence of other mutations in cancer that might affect the association of Akt1 and Akt2 with their binding partners and route signaling toward cell migration differently in one cancer type vs. other or when compared with the fibroblasts.Citation22,Citation23
Akt2 and cell migration in fibroblasts.
In fibroblasts, studies using Akt1 and Akt2-KO MEFs by Field et al. have shown that Akt2-KO MEFs develop dorsal ruffles and migrate faster than the Akt1-KO MEFs.Citation24 Mechanistically, this has been shown to occur through Akt1-dependent activation of Cdc42 and Rac, and increased actin phosphorylation, which promotes polymerization of actin.Citation25–Citation28 Further, the inhibition of Pak1 by Akt2 was shown to suppress migration induced by Akt1.Citation24 This is consistent with other reports that proposed a scaffolding role for Pak1 in Akt1 activation by PDK1.Citation29 These selective isoform effects were attributed to differences in the N-terminal PH and linker domains of Akt1 and Akt2. Since Cys124 is located in the linker domain of Akt2 and oxidation was shown to inhibit Akt2 activity, the objective of our follow up studies was to determine whether oxidation of Akt2 would promote cell migration.
Cell migration was studied using the standard transwell migration method and the wound healing/scratch method in Akt2-KO MEFs transduced with Akt2 WT and Akt2 Cys124Ser constructs (). Increased intracellular H2O2 was achieved by inhibition of catalase with 3-AT. The results show that in the presence of 3-AT, there was a significant increase in the migration of Akt2 WT MEFs, compared with the untreated Akt2 WT MEFs. Migration of the Akt2 Cys124Ser MEFs was inhibited and was independent of the cellular oxidation state (). Similar results were obtained using the wound healing/scratch assay (). The efficiency of viral infection with Akt2 WT and Akt2 Cys124Ser constructs in the Akt2-KO MEFs was assessed using western immunoblotting ().
These data imply that combined phosphorylation and oxidation occur downstream of PDGF receptor activation to facilitate the cytoskeleton reorganization and cell migration. PDGF-induced oxidation of Akt2 or activation of the intracellular sources of ROS by other mechanisms could relieve the inhibition of Pak1 and promote migration in this system.
Akt2 and Regulation of Cell Cycle under Conditions of Starvation
Multimodal regulation of G1-S and G2-M transition by Akt kinase at transcriptional and translational levels is well established. Akt inhibits degradation of cyclin D1 and c-myc proteins and translocates p21 and p27 to the cytoplasm to regulate G1-S transition.Citation30 Isoform-specific contribution of the Akt kinases in the regulation of cell cycle have also been investigated.Citation31,Citation32 The PH domain of Akt1 was shown to be important for both, nuclear translocation and for rapid G1-S transition in the cell cycle. Knocking down Akt1 delays induction of the S phase. This further correlates with reduced proliferation of the MEFs. While the nuclear translocation of Akt1 is believed to regulate cell cycle progression, the signaling mechanisms activated by Akt1 toward achieving this transition are still under investigation. Cyclin A is critical for inducing transition of the myoblasts to the S phase and Akt1 is known to upregulate the expression of cyclin A. Interestingly, Akt2 downregulates cyclin A in the myoblasts and arrests the cells in the G2-M phase. Since transition through G2-M phase is critical for differentiating cells, Lamb et al. proposed that Akt2 could be a key player in regulating this process.
To investigate the contributions of Akt2 WT and the oxidation resistant Akt2 Cys124Ser to the cell cycle progression, Akt2-KO MEFs transduced with Akt2 WT and Akt2 Cys124Ser were serum-starved overnight, followed by the addition of serum-supplemented medium for additional 24 h. Cell cycle analysis was performed before and after the addition of the serum-supplemented medium. Under conditions of serum starvation, the MEFs transduced with Akt2 WT or Akt2 Cys124Ser constructs were capable of decreasing the distribution of cells in the G1 phase with the Akt2 C124S mutant decreasing it the most (). The MEFs transduced with Akt2 Cys124Ser also appeared to maintain a higher proportion of cells in the S phase, the most active phase involving DNA synthesis. Serum starvation is known to induce oxidative stress and also to inhibit phosphorylation of the signaling proteins. Oxidative stress was found to prevent the entry into the cell cycle while the antioxidant treatment was shown to promote the G1-S transition.Citation33 Based on the results included in , expression of Akt2 WT and Akt2 Cys124Ser in the Akt2-KO MEFs could protect the cells from this stress and help maintain a relatively higher proportion of cells in the S phase. Addition of serum-containing medium to serumstarved MEFs rescued the cells from the G0/G1 arrest and abrogated the differences in the cell cycle distribution between the Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs (). These observations raise the possibility of an unexplored scope of cell cycle regulation by the Akt2 isoform under conditions of stress. The signaling mechanism could involve Skp2 which was shown to promote the transition of cells from G1 to S phase by destabilizing and degrading p27, the inhibitor of CDK2.Citation34 Akt kinases are known to regulate Skp2 activity through mechanisms that may involve downregulation of tuberin.Citation30 Future studies will address whether there is an isoform-specific effect of Akt2 on Skp2 activity to investigate this potential mechanism of cell cycle regulation. Thus far, the data suggests that the presence of Akt2 confers a survival advantage to cells under conditions of stress such as that induced by serum starvation despite the presence of Akt1, which does not completely compensate for the loss of Akt2.
Concluding Remarks and Implications of Redox-Dependent Akt2 Signaling
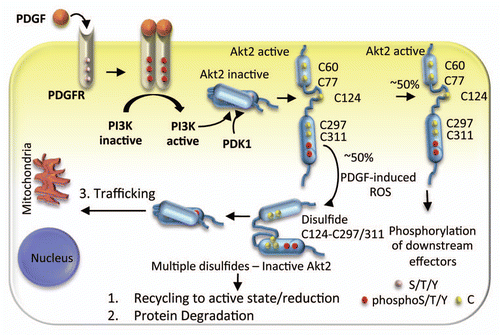
Our recently published findings and the studies presented here collectively point to Akt2 oxidation as a mechanism that fine-tunes Akt signaling with consequences on various cellular functions such as migration, glucose uptake and cell cycle. An integrated view of PDGF-signaling through Akt2 is thus emerging, where both phosphorylation and protein oxidation contribute to mediate critical cellular functions (). Upon activation of the PDGF receptor, Akt2 is phosphorylated at the membrane through the mechanisms described earlier. We propose that parallel or subsequent activation of NADPH oxidase results in accumulation of the oxidant species at the membrane, and oxidation of ∼50% of the phosphorylated Akt2.9 Further studies are required to determine whether the oxidized inactive Akt2 is (1) targeted to degradation; (2) recycled into the active pool by a yet to be determined reductase; or (3) trafficked to other subcellular compartments where reduction of disulfide bonds can occur to regenerate active Akt2. It will also be important to determine whether the uniquely positioned Cys124 in Akt2 can contribute to the development of a distinct phenotype in vivo.
This newly discovered redox sensitivity of Akt2 has broad biological implications. Wound healing, for example, is a well-known redox-dependent process. Damage to the intact epithelial barrier induces inflammatory response, and initiates repair signaling for the development of new tissue.Citation35 Low but persistent levels of ROS are maintained around the site of wound during and after the inflammatory response to promote wound healing.Citation36,Citation37 Under these conditions, Akt2 oxidation could be one of the critical redox players promoting migration of the cells toward the lesion.
Besides wound healing, there are multiple biological events that are both redox-regulated and Akt2-dependent. For instance, Akt2 regulates differentiation of myoblasts to myotubes and both, oxidative stress and Akt2 knock down are suggested to impair this differentiation process.Citation38–Citation41 We reported in our earlier findings that insulin-stimulated ROS inhibits Akt2 only in the C2C12 myoblasts and not in the differentiated myotubes. We have also observed lack of differentiation in the C2C12 myoblasts when subjected to oxidative stress with 3-AT treatment. These suggest that active Akt2 is essential for regulating the differentiation to myotubes and that oxidation could prevent this process by inhibiting Akt2. Akt2 also has been reported to regulate survival signaling pathways and metabolism in the terminally differentiated cardiomyocytes.Citation42,Citation43 Redox stress accompanies ischemia and reperfusion-based injuries in cardiomyocytes.Citation44 Hence, inhibition of Akt2 under these conditions could underlie the impaired survival of cardiomyocytes. Similar redox-based and Akt2-dependent regulation could be involved in the development of diabetes. Studies have shown that the obesity-associated ROS are critical to the progression of type-2 diabetes.Citation45,Citation46 Coupled with the findings of insulin resistance and diabetes-like syndrome in the Akt2-KO mice,Citation47–Citation50 it is reasonable to suggest that Akt2 inactivation due to oxidation could be an important factor in the development of type-2 diabetes in obese patients.
Hence, on a broader note, our studies implicate redox-impaired Akt2 signaling in a range of clinical conditions including some discussed here. Most importantly, it has become clear that oxidation of signaling proteins and the consequence of oxidation on protein activity need to be considered when signaling that leads to specific physiological outcomes involves production of reactive oxygen species.
Abbreviations
| 3-AT | = | 3-amino-1,2,4-triazole |
| PDGF | = | platelet-derived growth factor |
| PH | = | pleckstrin homology |
| ROS | = | reactive oxygen species |
| FAK | = | focal adhesion kinase |
| PI3K | = | phosphatidyl inositol-3-kinases |
| PIP3 | = | phosphoinositide-3,4,5-trisphosphate |
| PIP2 | = | phosphoinositide-4,5-bisphosphate |
| PDK1 | = | phosphoinositide-dependent kinase 1 |
| mTOR | = | mammalian target of rapamycin |
| DCF | = | 2′,7′-dichlorodihydrofluorescein diacetate |
| ROS | = | reactive oxygen species |
| WT | = | wild type |
| KO | = | knockout |
| MEF | = | murine embryonic fibroblast |
Figures and Tables
Figure 1T ranswell and scratch/wound healing cell migration assays. (A) Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs were serum-starved for 16 h in the presence and absence of 3-AT (10 mM). Next day, the cells were dissociated, normalized and 2.5 × 104 cells/well were added to the transwell plates. Serum-free DMEM (with and without 3-AT) was added to the upper chamber and serum-containing DMEM (with and without 3-AT) along with PDGF-BB (20 ng/ml) was added to the lower chamber. The cells were incubated overnight for about 16 h at 37°C and stained with crystal violet (1%). The cells from 4 random fields were counted with the Olympus confocal microscope using 20X objective. The p value for WT Akt2 MEFs ± 3-AT was <0.001 and for Cys124Ser Akt2 MEFs was ± is 0.473. (B) In a parallel experiment, Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs were serum-starved for 16 h in the presence and absence of 3-AT (10 mM). Next day, the cells were lysed and the cell-free extracts were normalized. These lysates were separated on SDS-PAGE and probed for Akt2 expression using western immunodetection. Additionally, the lysates were probed for the β-actin control. (C) Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs were serum-starved for 16 h in the presence and absence of 3-AT (10 mM). The starvation medium was then replaced with a complete medium containing serum with and without 3-AT (10 mM) and a scratch was made using a 200 µl pipet tip. Phase images were collected around the scratch first, within an hour of generating the scratch and 36 h later using the Olympus microscope under a 4X objective.
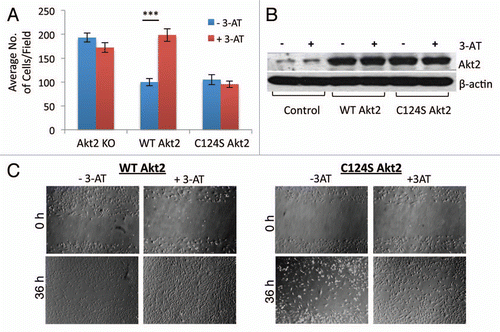
Figure 2 Cell cycle analysis. (A) Akt2-KO, Akt2 WT and Akt2 Cys124Ser MEFs were serum-starved for 16 h. Next day, one set of cells was dissociated with trypsin and the cells were resuspended in 1 ml propidium iodide stain (1 µg/ml propidium iodide, 0.06% NP40 and 37 µg/ml RNase A). The data was collected with Accuri C6 Flow Cytometer and analyzed using the ModFit LT software. (B) The serum-free culture medium in the second set was replaced with the complete medium and the cells were incubated further for 24 h at 37°C. Then the cells were trypsinized and resuspended in 1 ml propidium iodide stain and analyzed as in (A).
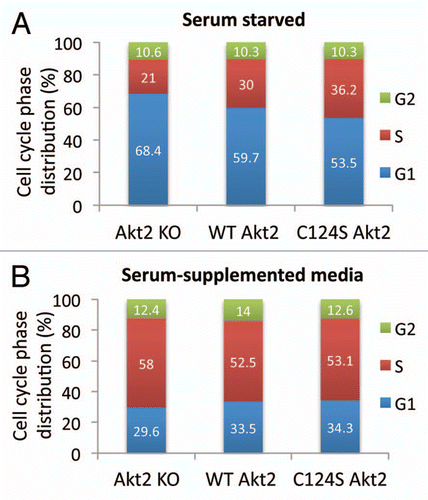
Figure 3 Proposed model of Akt2 signaling induced by PDGF. PDGF stimulates PI3K as well as ROS. PI3K-derived phosphoinositides bind the PH domain of Akt2 and translocates it to the plasma membrane where PDK1 phosphorylates Akt2. In addition to Cys124 in the linker region, the PH and the kinase domains each have two redox sensitive sites Cys60, Cys77 and Cys297, Cys311. The PDGF-induced ROS oxidizes ∼50% Akt2 and induces Cys124-Cys297/Cys311 disulfide bond formation. This disulfide could direct Akt2 to another subcellular location, target Akt2 for degradation or could be reduced to regenerate active Akt2.
Acknowledgments
Financial support was provided by American Heart Association Scientist Development Program Grant SDG 0730069N (to C.M.F.) and National Institutes of Health Grants R01 CA136810 (to C.M.F.).
References
- Woodgett JR. Recent advances in the protein kinase B signaling pathway. Curr Opin Cell Biol 2005; 17:150 - 157; PMID: 15780591; http://dx.doi.org/10.1016/j.ceb.2005.02.010
- Brazil DP, Yang ZZ, Hemmings BA. Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 2004; 29:233 - 242; PMID: 15130559; http://dx.doi.org/10.1016/j.tibs.2004.03.006
- Bellacosa A, Testa JR, Moore R, Larue L. A portrait of AKT kinases: human cancer and animal models depict a family with strong individualities. Cancer Biol Ther 2004; 3:268 - 275; PMID: 15034304; http://dx.doi.org/10.4161/cbt.3.3.703
- Dummler B, Tschopp O, Hynx D, Yang ZZ, Dirnhofer S, Hemmings BA. Life with a single isoform of Akt: mice lacking Akt2 and Akt3 are viable but display impaired glucose homeostasis and growth deficiencies. Mol Cell Biol 2006; 26:8042 - 8051; PMID: 16923958; http://dx.doi.org/10.1128/MCB.00722-06
- Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev 2003; 17:1352 - 1365; PMID: 12782654; http://dx.doi.org/10.1101/gad.1089403
- Franke TF. Intracellular signaling by Akt: bound to be specific. Sci Signal 2008; 1:29; PMID: 18560018; http://dx.doi.org/10.1126/scisignal.124pe29
- Gonzalez E, McGraw TE. Insulin-modulated Akt subcellular localization determines Akt isoform-specific signaling. Proc Natl Acad Sci USA 2009; 106:7004 - 7009; PMID: 19372382; http://dx.doi.org/10.1073/pnas.0901933106
- Santi SA, Lee H. The Akt isoforms are present at distinct subcellular locations. Am J Physiol Cell Physiol 2010; 298:580 - 591; PMID: 20018949; http://dx.doi.org/10.1152/ajpcell.00375.2009
- Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, et al. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci USA 2011; 108:10550 - 10555; PMID: 21670275; http://dx.doi.org/10.1073/pnas.1011665108
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science 2003; 302:1704 - 1709; PMID: 14657486; http://dx.doi.org/10.1126/science.1092053
- Merlot S, Firtel RA. Leading the way: Directional sensing through phosphatidylinositol-3-kinase and other signaling pathways. J Cell Sci 2003; 116:3471 - 3478; PMID: 12893811; http://dx.doi.org/10.1242/jcs.00703
- Devreotes P, Janetopoulos C. Eukaryotic chemotaxis: distinctions between directional sensing and polarization. J Biol Chem 2003; 278:20445 - 20448; PMID: 12672811; http://dx.doi.org/10.1074/jbc.R300010200
- Yoeli-Lerner M, Yiu GK, Rabinovitz I, Erhardt P, Jauliac S, Toker A. Akt blocks breast cancer cell motility and invasion through the transcription factor NFAT. Mol Cell 2005; 20:539 - 550; PMID: 16307918; http://dx.doi.org/10.1016/j.molcel.2005.10.033
- Smits M, Nilsson J, Mir SE, van der Stoop PM, Hulleman E, Niers JM, et al. miR-101 is downregulated in glioblastoma resulting in EZH2-induced proliferation, migration and angiogenesis. Oncotarget 2010; 1:710 - 720; PMID: 21321380
- Cha TL, Zhou BP, Xia W, Wu Y, Yang CC, Chen CT, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science 2005; 310:306 - 310; PMID: 16224021; http://dx.doi.org/10.1126/science.1118947
- Arboleda MJ, Lyons JF, Kabbinavar FF, Bray MR, Snow BE, Ayala R, et al. Overexpression of AKT2/protein kinase Bbeta leads to upregulation of beta1 integrins, increased invasion and metastasis of human breast and ovarian cancer cells. Cancer Res 2003; 63:196 - 206; PMID: 12517798
- Wang J, Wan W, Sun R, Liu Y, Sun X, Ma D, et al. Reduction of Akt2 expression inhibits chemotaxis signal transduction in human breast cancer cells. Cell Signal 2008; 20:1025 - 1034; PMID: 18353613; http://dx.doi.org/10.1016/j.cellsig.2007.12.023
- Zhang B, Gu F, She C, Guo H, Li W, Niu R, et al. Reduction of Akt2 inhibits migration and invasion of glioma cells. Int J Cancer 2009; 125:585 - 595; PMID: 19330838; http://dx.doi.org/10.1002/ijc.24314
- Chin YR, Toker A. Akt2 regulates expression of the actin-bundling protein palladin. FEBS Lett 2010; 584:4769 - 4774; PMID: 21050850; http://dx.doi.org/10.1016/j.febslet.2010.10.056
- Cheng GZ, Zhang W, Wang LH. Regulation of cancer cell survival, migration and invasion by Twist: AKT2 comes to interplay. Cancer Res 2008; 68:957 - 960; PMID: 18281467; http://dx.doi.org/10.1158/0008-5472.CAN-07-5067
- Chin YR, Toker A. The actin-bundling protein palladin is an Akt1-specific substrate that regulates breast cancer cell migration. Mol Cell 2010; 38:333 - 344; PMID: 20471940; http://dx.doi.org/10.1016/j.molcel.2010.02.031
- Du K, Tsichlis PN. Regulation of the Akt kinase by interacting proteins. Oncogene 2005; 24:7401 - 7409; PMID: 16288287; http://dx.doi.org/10.1038/sj.onc.1209099
- Yoeli-Lerner M, Toker A. Akt/PKB signaling in cancer: a function in cell motility and invasion. Cell Cycle 2006; 5:603 - 605; PMID: 16582622; http://dx.doi.org/10.4161/cc.5.6.2561
- Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem 2006; 281:36443 - 36453; PMID: 17012749; http://dx.doi.org/10.1074/jbc.M600788200
- Higuchi M, Masuyama N, Fukui Y, Suzuki A, Gotoh Y. Akt mediates Rac/Cdc42-regulated cell motility in growth factor-stimulated cells and in invasive PTEN knockout cells. Curr Biol 2001; 11:1958 - 1962; PMID: 11747822; http://dx.doi.org/10.1016/S0960-9822(01)00599-1
- Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science 2000; 287:1037 - 1040; PMID: 10669415; http://dx.doi.org/10.1126/science.287.5455.1037
- Ho YP, Kuo CW, Hsu YT, Huang YS, Yew LP, Huang WF, et al. β-Actin is a downstream effector of the PI3K/AKT signaling pathway in myeloma cells. Mol Cell Biochem 2011; 348:129 - 139; PMID: 21069433; http://dx.doi.org/10.1007/s11010-010-0647-7
- Vandermoere F, El Yazidi-Belkoura I, Demont Y, Slomianny C, Antol J, Lemoine J, et al. Proteomics exploration reveals that actin is a signaling target of the kinase Akt. Mol Cell Proteomics 2007; 6:114 - 124; PMID: 17088265; http://dx.doi.org/10.1074/mcp.M600335-MCP200
- Poon JS, Eves R, Mak AS. Both lipid- and protein-phosphatase activities of PTEN contribute to the p53-PTEN anti-invasion pathway. Cell Cycle 2010; 9:4450 - 4454; PMID: 21084866; http://dx.doi.org/10.4161/cc.9.22.13936
- Liang J, Slingerland JM. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003; 2:339 - 345; PMID: 12851486; http://dx.doi.org/10.4161/cc.2.4.433
- Héron-Milhavet L, Franckhauser C, Rana V, Berthenet C, Fisher D, Hemmings BA, et al. Only Akt1 is required for proliferation, while Akt2 promotes cell cycle exit through p21 binding. Mol Cell Biol 2006; 26:8267 - 8280; PMID: 16982699; http://dx.doi.org/10.1128/MCB.00201-06
- Yun SJ, Tucker DF, Kim EK, Kim MS, Do KH, Ha JM, et al. Differential regulation of Akt/protein kinase B isoforms during cell cycle progression. FEBS Lett 2009; 583:685 - 690; PMID: 19166849; http://dx.doi.org/10.1016/j.febslet.2009.01.005
- Shackelford RE, Kaufmann WK, Paules RS. Oxidative stress and cell cycle checkpoint function. Free Radic Biol Med 2000; 28:1387 - 1404; PMID: 10924858; http://dx.doi.org/10.1016/S0891-5849(00)00224-0
- Shin JS, Hong SW, Lee SL, Kim TH, Park IC, An SK, et al. Serum starvation induces G1 arrest through suppression of Skp2-CDK2 and CDK4 in SK-OV-3 cells. Int J Oncol 2008; 32:435 - 439; PMID: 18202766
- Sen CK, Roy S. Redox signals in wound healing. Biochim Biophys Acta 2008; 1780:1348 - 1361
- Roy S, Khanna S, Nallu K, Hunt TK, Sen CK. Dermal wound healing is subject to redox control. Mol Ther 2006; 13:211 - 220; PMID: 16126008; http://dx.doi.org/10.1016/j.ymthe.2005.07.684
- Steiling H, Munz B, Werner S, Brauchle M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp Cell Res 1999; 247:484 - 494; PMID: 10066376; http://dx.doi.org/10.1006/excr.1998.4366
- Héron-Milhavet L, Mamaeva D, Rochat A, Lamb NJ, Fernandez A. Akt2 is implicated in skeletal muscle differentiation and specifically binds Prohibitin2/REA. J Cell Physiol 2008; 214:158 - 165; PMID: 17565718; http://dx.doi.org/10.1002/jcp.21177
- Ardite E, Barbera JA, Roca J, Fernandez-Checa JC. Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NFkappaB activation. Am J Pathol 2004; 165:719 - 728; PMID: 15331397; http://dx.doi.org/10.1016/S0002-9440(10)63335-4
- Hansen JM, Klass M, Harris C, Csete M. A reducing redox environment promotes C2C12 myogenesis: implications for regeneration in aged muscle. Cell Biol Int 2007; 31:546 - 553; PMID: 17241791; http://dx.doi.org/10.1016/j.cellbi.2006.11.027
- Lyons CN, Leary SC, Moyes CD. Bioenergetic remodeling during cellular differentiation: changes in cytochrome c oxidase regulation do not affect the metabolic phenotype. Biochem Cell Biol 2004; 82:391 - 399; PMID: 15181473; http://dx.doi.org/10.1139/o04-040
- Muslin AJ. Akt2: a critical regulator of cardiomyocyte survival and metabolism. Pediatr Cardiol 2011; 32:317 - 322; PMID: 21279637; http://dx.doi.org/10.1007/s00246-010-9879-2
- DeBosch B, Sambandam N, Weinheimer C, Courtois M, Muslin AJ. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J Biol Chem 2006; 281:32841 - 32851; PMID: 16950770; http://dx.doi.org/10.1074/jbc.M513087200
- Robin E, Guzy RD, Loor G, Iwase H, Waypa GB, Marks JD, et al. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. J Biol Chem 2007; 282:19133 - 19143; PMID: 17488710; http://dx.doi.org/10.1074/jbc.M701917200
- Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol 2004; 24:816 - 823; PMID: 14976002; http://dx.doi.org/10.1161/01.ATV.0000122852.22604.78
- Maritim AC, Sanders RA, Watkins JB 3rd. Diabetes, oxidative stress and antioxidants: a review. J Biochem Mol Toxicol 2003; 17:24 - 38; PMID: 12616644; http://dx.doi.org/10.1002/jbt.10058
- Brozinick JT Jr, Roberts BR, Dohm GL. Defective signaling through Akt-2 and -3 but not Akt-1 in insulin-resistant human skeletal muscle: potential role in insulin resistance. Diabetes 2003; 52:935 - 941; PMID: 12663464; http://dx.doi.org/10.2337/diabetes.52.4.935
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB 3rd, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science 2001; 292:1728 - 1731; PMID: 11387480; http://dx.doi.org/10.1126/science.292.5522.1728
- Dummler B, Hemmings BA. Physiological roles of PKB/Akt isoforms in development and disease. Biochem Soc Trans 2007; 35:231 - 235; PMID: 17371246; http://dx.doi.org/10.1042/BST0350231
- George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, et al. A family with severe insulin resistance and diabetes due to a mutation in AKT2. Science 2004; 304:1325 - 1328; PMID: 15166380; http://dx.doi.org/10.1126/science.1096706