Abstract
DNA double-strand breaks (DSBs) are arguably the most important lesions induced by ionizing radiation (IR) since unrepaired or mis-repaired DSBs can lead to chromosomal aberrations and cell death. The two major pathways to repair IR-induced DSBs are non-homologous end-joining (NHEJ) and homologous recombination (HR). Perhaps surprisingly, NHEJ represents the predominant pathway in the G1 and G2 phases of the cell cycle, but HR also contributes and repairs a subset of IR-induced DSBs in G2. Following S-phase-dependent genotoxins, HR events give rise to sister chromatid exchanges (SCEs), which can be detected cytogenetically in mitosis. Here, we describe that HR occurring in G2-irradiated cells also generates SCEs in ~50% of HR events. Since HR of IR-induced DSBs in G2 is a slow process, SCE formation in G2-irradiated cells requires several hours. During this time, irradiated S-phase cells can also reach mitosis, which has contributed to the widely held belief that SCEs form only during S phase. We describe procedures to measure SCEs exclusively in G2-irradiated cells and provide evidence that following IR cells do not need to progress through S phase in order to form SCEs.
Introduction
Sister chromatid exchange (SCE) is a natural molecular process exchanging genetic material between two identical sister chromatids. SCEs were originally discovered by McClintock and later rediscovered by Taylor et al. using plant cells labeled with H3-thymidine.Citation1,Citation2 Technical improvements by Perry and Wolff to differentially stain the sister chromatids using incorporated 5′-bromodeoxyuridine (BrdU) in combination with Hoechst 33258 led to a dramatic increase in the number of publications dealing with the mechanism of SCE formation.Citation3–Citation5 Since SCEs represent recombinogenic events arising at DNA lesions, they became a widely used endpoint in studying the mutagenic and clastogenic effects of different agents. Systematic investigations revealed that S-phase-dependent agents are generally strong inducers of SCEs, whereas ionizing radiation (IR), radiomimetic drugs or restriction enzymes are weak inducers.Citation6–Citation11 Therefore, it became a common belief in the scientific community that cells have to pass through S phase to manifest SCEs after damage induction.Citation12–Citation14 Although some investigators suggested models that involve non-homologous end-joining (NHEJ) in the generation of SCEs, the prevailing evidence suggests that homologous recombination (HR) is the underlying mechanism for SCE formation.Citation15–Citation19 As a result of the extensive research, the mechanism of formation of SCEs arising in cells after treatment with S-phase-independent clastogens such as IR was critically discussed.Citation8,Citation9,Citation14,Citation16 For example, Mühlmann-Diaz and Bedford suggested that SCEs induced after κ- or X-irradiation might represent “false” SCEs that arise from chromosomal aberrations, in particular paracentric inversions, produced in G0/G1.Citation20 Color-jumps on chromatids were also observed in cells that were treated with DSB-inducing agents in G2, but these jumps were attributed to a two-lesion exchange process.Citation21 The widely held belief that “true” SCEs are not formed in G2 was further supported by work of Wojcik et al. who observed that exponentially growing cell populations do not exhibit SCEs above background level within the first 4 h post irradiation.Citation22
Here, we present evidence that “true” SCEs are formed in G2-irradiated cells, by a mechanism based on HR. We discuss the results of our recent work in the field of DNA double-strand break (DSB) repair and explain pitfalls in the experimental setup that have to be considered when measuring SCEs which arise after irradiation in G2.
Results
A subset of ionizing radiation induced DSBs are slowly repaired by HR in G2.
HR and NHEJ represent the two major pathways to repair DSBs.Citation23–Citation25 HR has a major role in repairing replication-associated lesions by dealing with stalled or collapsed replication forks.Citation26–Citation28 In contrast, NHEJ represents the predominant pathway for repairing IR-induced DSBs in the G1 and the G2 phases of the cell cycle.Citation29–Citation31 In G2, the majority of DSBs are rapidly repaired by NHEJ within the first 2–3 h but a subset of breaks (∼15–20%) is repaired by HR with much slower kinetics.Citation32 In order to study HR in G2-irradiated cells, we analyzed asynchronously growing HeLa S3 cells and prevented irradiated S-phase cells from progressing into G2 during analysis. For this, we added aphidicolin to the cell culture medium immediately after irradiation, which efficiently blocks DNA synthesis and arrests S-phase cells, preventing them from reaching G2 and mitosis (see ).
shows typical DSB repair kinetics for wild-type (wt) and Brca2-depleted HeLa S3 cells that were irradiated in G2 with 2 Gy of X-rays; γH2AX foci analysis was utilized as a highly sensitive assay to assess the level of DSBs.Citation33,Citation34 The Brca2-depleted cells exhibit an elevated level of unrepaired DSBs (33 vs. 23 γH2AX foci) at prolonged repair times (here determined at 8 h), showing that the repair of DSBs by HR represents a slow process. In G2-phase cells we also measured RPA foci, which form at resected DSBs and provide a measure of the DSBs at which HR is initiated ( and reviewed in ref. Citation32). The maximum number of about 27 RPA foci is formed at 2 h post IR, followed by a slow decrease within the next 6 h with kinetics similar to the slow component of DSB repair in G2. Note that the number of RPA foci in irradiated HeLa S3 cells is slightly higher than reported for primary human fibroblast, possibly reflecting their higher DNA content.Citation32 Since the γH2AX and RPA foci levels are similar for wt and Brca2-depleted cells at 2 h but significantly different at 8 h post IR, HR is a slow process. Thus, the experimental procedure to assess HR in G2 requires that cells damaged by IR remain in G2 for at least 6–8 h. This is normally warranted since the IR-induced G2 checkpoint provides additional time for repair (see and reviewed in ref. Citation35 and Citation36).
Taken together, our findings on the kinetics of IR-induced HR events suggest that it would be necessary to analyze cells at prolonged times post IR (≥6 h) in order to detect SCEs that might arise in G2-irradiated cells. Moreover, since S-phase cells can enter G2 phase and mitosis within 6 h post IR, it is further necessary to either use synchronized G2 populations or to prevent S-phase cells from entering G2 phase and mitosis.
SCEs are induced in G2 by IR.
We used exponentially growing HeLa S3 cells, irradiated them with 2 Gy, added aphidicolin to the cell culture medium immediately after IR, harvested metaphases at 6–9 h after IR and then stained the metaphases by standard Giemsa staining (for representative images see ). Caffeine was added at 6 h after irradiation to abrogate the G2 checkpoint and to enhance the number of G2 cells entering mitosis. Although caffeine is known to affect HR frequencies (reviewed in ref. Citation37) it is important to consider that caffeine in our studies was added at later times post irradiation and control experiments confirmed that the addition of caffeine 6 h post IR does not affect the SCE levels (). Unirradiated HeLa S3 cells exhibited approximately 7 SCEs; a dose of 2 Gy induced approximately 7 additional SCEs per cell, corresponding to 0.1 additional SCEs per chromosome (). Consistent with the paradigm that HR represents the underlying mechanism of SCE formation, downregulation of the HR core components Brca2 or Rad51 completely abolished the formation of IR-induced SCEs and lead to an increase in the level of unrepaired chromatid breaks and gaps ( and C). Interestingly, also the spontaneous SCE level was reduced in Brca2- and Rad51-depleted cells suggesting that spontaneous SCEs arising during the first or second S phase after adding BrdU to the cell culture medium are also formed by HR. Moreover, downregulation of the NHEJ core components Ku80 or DNA ligase IV did not affect the formation of IR-induced SCEs or the baseline SCE level.Citation32 Further support for the notion that the observed SCEs do not represent “false” SCEs arising, e.g., from paracentric inversions of cells irradiated in G1 or early S phase is provided by the analysis of the types of chromosome aberrations. In the metaphase spreads analyzed 6–9 h post IR, we only observed chromatid-type aberrations such as gaps and chromatid breaks, no chromosome-type aberrations. If cells irradiated in G1 or early S phase would have been able to reach mitosis during the repair time of 6–9 h despite the aphidicolin block, one would have expected to observe also chromosome-type aberrations such as dicentrics, rings or chromosome breaks.
Taken together, these findings provide strong evidence that the SCEs observed are indeed macroscopic endpoints of HR events of G2-irradiated cells. The level of residual chromatid breaks/gaps in Brca2-downregulated cells reflects the residual number of γH2AX foci at late repair times, showing that most of the unrepaired DSBs in Brca2-depleted cells are cytogenetically visible as chromatid breaks/gaps (). The number of 7 SCEs induced by 2 Gy supports the notion that a substantial fraction (about 50%) of the IR-induced DSBs that undergo repair by HR in G2 give rise to SCEs (compare 7 IR-induced SCEs with the loss of about 12 RPA foci in wt cells, ).
Further evidence for “true” SCE formation in G2-irradiated cells.
Using FACS analysis, we showed that HeLa S3 cells treated with aphidicolin are blocked in S phase and cannot progress through G2 and subsequently reach mitosis (). Therefore, only cells that are in G2 at the time of irradiation can enter mitosis. However, to address the possibility that a few cells might pass from late S to G2 phase despite the replication block and affect the SCE levels in the metaphases analyzed, we performed additional experiments using EdU as a thymidine analogue, which was added at the time of irradiation. In these experiments, we differentiated S-phase-irradiated from G2-phase-irradiated cells by their EdU signal on metaphase spreads (). Cells that were in S phase at the time of irradiation were able to incorporate EdU whereas cells in G2 were not. If aphidicolin was added after EdU labeling, only EdU-negative metaphases were observed, confirming that no S-phase cells progressed through G2 and into mitosis. In experiments without aphidicolin, we observed EdU-negative as well as EdU-positive metaphases, indicating that S-phase as well as G2-phase cells can enter mitosis. Metaphases analyzed at 6–9 h post irradiation mainly represented EdU-negative cells while metaphases collected at 9–12 h post IR were predominantly positive for EdU. Counting SCEs in EdU-negative as well as in EdU-positive metaphases on the same slide revealed that the SCE level of EdU-positive cells (S-phase-irradiated) is slightly higher than that of EdU-negative cells (G2-irradiated) (). This suggests that either EdU potentiates IR-induced SCEs or that SCE formation is more efficient in S than in G2. Importantly, however, EdU-negative G2-irradiated cells show clearly elevated SCE levels compared with unirradiated cells, consolidating our contention that SCEs arise in G2-irradiated cells.
To provide further evidence for our notion that DSBs induced by IR lead to SCEs in G2 we used the alkylating agent methyl methanesulfonate (MMS). MMS generates DNA damage that causes stalled or collapsed replication forks but is not expected to produce DSBs in G2-phase cells. Consistent with this, 0.5 mM MMS causes γH2AX foci in S-phase but not in G2-phase cells.Citation38 Thus, using 0.5 mM MMS we expected that only cells passing through S phase and not cells treated in G2 phase would show elevated SCE levels. Supporting this is the observation that only EdU-positive but not EdU-negative metaphases exhibit elevated SCE levels ().
Conclusion
HR is a highly conserved mechanism repairing DSBs at stalled or collapsed replication forks. In vertebrate cells, HR relies on the existence of sister chromatids and is, hence, only active in the S and G2 phases of the cell cycle. Perhaps surprisingly, HR plays only a minor role in the repair of IR-induced DSBs in G2, whereas NHEJ is the major repair mechanism.Citation32 Nevertheless, a subset of IR-induced DSBs in G2 is repaired by HR with slow kinetics. Consistent with this finding, we observed that SCEs arise in G2-irradiated cells by a process dependent on Brca2 and Rad51, strongly suggesting that SCEs arising in G2 are macroscopic endpoints of HR. Thus, G2-irradiated cells do not need to progress through S phase in order to form SCEs.
Figures and Tables
Figure 1 (A) Cell cycle analysis of irradiated HeLa S3 cells at different time points after 2 Gy IR. Cells were pulse-labeled with BrdU (10 µM) for 1 h directly before irradiation to mark all S-phase cells and aphidicolin (3 µg/ml) was added immediately after irradiation. Without aphidicolin, S-phase cells progressed to G2 and the subsequent G1 phase during repair incubation. If aphidicolin was added, all BrdU-positive cells stayed in S during the entire incubation period. The BrdU-negative G2 population, that constitutes the cells of interest, slowly decreased up to 12 h post IR. By adding 1 mM caffeine at 6 h, BrdU-negative G2 cells enter mitosis more efficiently without affecting the S-phase block. (B) γH2AX foci analysis of wt and Brca2-depleted HeLa S3 cells after 2 Gy IR. The siRNA sequences and concentrations used are reported in reference Citation32. Only cells irradiated in G2 were analyzed. Note that the induction value likely represents an underestimation since significant repair can occur within the first 15 min post IR.Citation39,Citation40 In the first hours post IR the repair in wt and Brca2-depleted cells is similar. At 8 h post IR the Brca2-depleted cells exhibit significantly elevated foci level compared to wt cells. (C) Single-stranded DNA formation in wt and Brca2-depleted HeLa S3 cells measured by the formation of RPA foci after 2 Gy IR in G2-phase cells. Brca2-depleted cells show a constant level of RPA foci between 2 and 8 h post IR whereas wt cells lose RPA foci between 2 and 8 h similar to the loss of γH2AX foci, indicating that γH2AX foci at prolonged repair times represent resected DSBs which are repaired by HR.
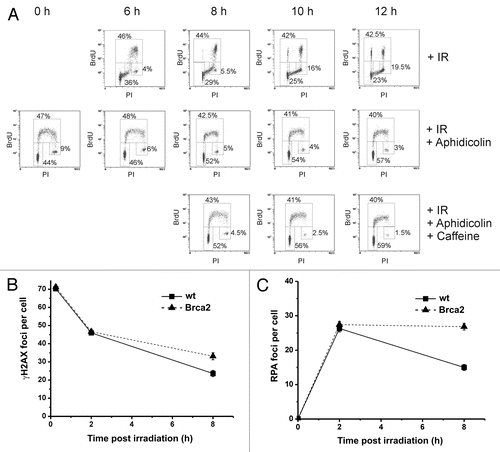
Figure 2 (A) Representative images of metaphase spreads after standard fluorescence plus Giemsa (FPG) staining. HeLa S3 cells were grown for 48 h (about two rounds of replication) in medium containing 10 µM BrdU. Cells were then irradiated with 2 Gy and aphidicolin (3 µg/ml) was added immediately. 1 mM caffeine (to abrogate the G2 checkpoint) and 200 ng/ml colcemid (to arrest cells in mitosis) were added 6 h later and metaphases were harvested at 9 h after IR. The mean number of chromosomes in HeLa S3 cells varied between 68 and 72, and all data were normalized to 70 chromosomes per cell. Untreated (left parts) and Brca2-siRNA-treated cells are shown (right parts); SCEs are marked by white arrows, chromatid breaks/gaps by black arrows. The 4 insets show examples of a chromosome with a chromatid break/gap (the two upper insets), an SCE (the lower inset on the left) or a chromatid break/gap together with an SCE (the chromosome in the lower inset on the right). (B) SCE analysis of control, Brca2- and Rad51-siRNA-treated HeLa S3 cells. siRNA knock-down of Brca2 and Rad51 results in a complete abolishment of IR-induced SCE formation compared to control cells. Caffeine added at 6 h post IR does not affect the SCE levels. (C) Analysis of chromatid breaks and gaps in control and Brca2-siRNA-treated HeLa S3 cells. Brca2-downregulated cells exhibit an elevated level of chromatid breaks and gaps compared to control cells. We have also measured chromatid breaks and gaps in HeLa S3 cells that were not pre-labeled with BrdU and have obtained similar results. Thus, under our experimental conditions, the radiosensitizing agent BrdU does not significantly affect the breakage level in G2-irradiated cells.Citation41
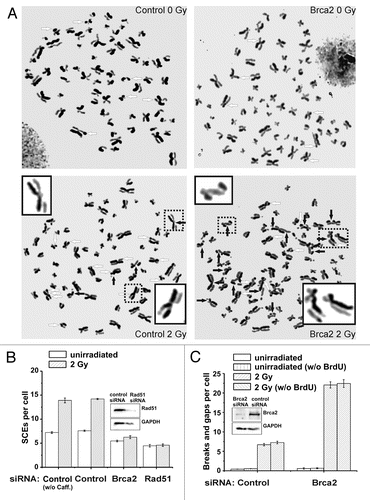
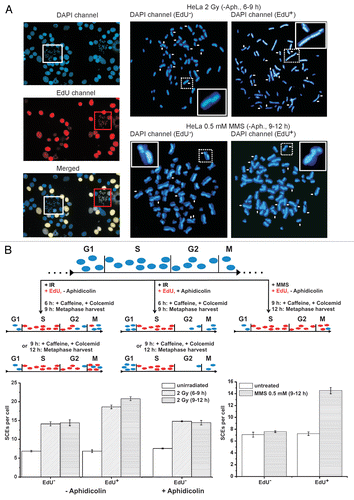
Figure 3 (A) Representative images of DAPI (blue) and EdU stained (red) metaphases after 2 Gy IR or 0.5 mM MMS for 0.5 h. HeLa S3 cells were grown for 48 h in medium containing 10 µM BrdU and SCEs (marked by white arrows) were visualized by DAPI instead of FPG staining; 10 µM EdU was added immediately after irradiation or MMS treatment and served to identify cells in S phase at the time of irradiation or MMS treatment. EdU-positive cells exhibit a bright, pan-chromosomal staining (red). Caffeine (1 mM) and colcemid (200 ng/ml) were added at 6 or 9 h post treatment and metaphases were harvested 3 h later. (B) SCE analysis of cells collected during two different time intervals after 2 Gy IR (6–9 h and 9–12 h) or between 9 and 12 h after 0.5 mM MMS. Unirradiated cells were collected between 3–6 h post EdU labeling in order to collect EdU-positive as well as EdU-negative metaphases on the same slide. The different experimental procedures are schematically described in the upper part of the part. For the SCE analysis after IR (lower left part), cells were either investigated without aphidicolin treatment or aphidicolin (3 µg/ml) was added shortly after EdU labeling. In experiments without aphidicolin, SCEs were scored in EdU-negative (G2-irradiated) as well as in EdU-positive cells (S-phase-irradiated) on the same slide; at 6–9 h, about 72% of the analyzed metaphases were EdU negative; at 9–12 h, this proportion decreased to about 25%. In experiments using aphidicolin, only EdU-negative cells could reach mitosis. EdU-negative metaphases exhibit ∼7 IR-induced SCEs (i.e., a total of ∼14 SCEs), EdU-positive cells a slightly higher level. For the SCE analysis after MMS (lower right part), aphidicolin was avoided to allow S-phase cells to enter mitosis. Metaphases were harvested between 9 and 12 h post MMS treatment. Only EdU-positive cells (in S during MMS treatment) exhibit an elevated level of SCEs; EdU-negative metaphases (cells in G2 during MMS treatment) show background SCE levels.
Acknowledgements
We thank Bernd Kaina, Andrzej Wojcik and Larry Thompson for valuable comments on the manuscript. The M.L. laboratory is supported by the Deutsche Forschungsgemeinschaft (Lo 677/4) and the Bundesministerium für Bildung und Forschung (02S8135, 02S8355 and 03NUK001C).
References
- McClintock B. The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behavior of ring-shaped chromosomes. Genetics 1938; 23:315 - 376
- Taylor JH, Woods PS, Hughes WL. The organization and duplication of chromosomes as revealed by autoradiographic studies using tritium-labeled thymidinee. Proc Natl Acad Sci USA 1957; 43:122 - 128
- Perry P, Wolff S. New giemsa method for the differential staining of sister chromatids. Nature 1974; 251:156 - 158
- Kaina B. Mechanisms and consequences of methylating agent-induced SCEs and chromosomal aberrations: a long road traveled and still a far way to go. Cytogenet Genome Res 2004; 104:77 - 86
- Wilson DM III, Thompson LH. Molecular mechanisms of sister-chromatid exchange. Mutat Res 2007; 616:11 - 23
- Au WW, O'Neill JP, Wang W, Luippold HE, Preston RJ. Induction of chromosome aberrations and specific locus mutation but not sister chromatid exchanges in Chinese hamster ovary cells by neocarzinostatin. Teratog Carcinog Mutagen 1984; 4:515 - 522
- Kaina B, Aurich O. Dependency of the yield of sister-chromatid exchanges induced by alkylating agents on fixation time. Possible involvement of secondary lesions in sister-chromatid exchange induction. Mutat Res 1985; 149:451 - 461
- Littlefield LG, Colyer SP, Sayer AM, DuFrain RJ. Sister-chromatid exchanges in human lymphocytes exposed during G0 to four classes of DNA-damaging chemicals. Mutat Res 1979; 67:259 - 269
- Littlefield LG, Colyer SP, Joiner EE, DuFrain RJ. Sister chromatid exchanges in human lymphocytes exposed to ionizing radiation during G0. Radiat Res 1979; 78:514 - 521
- Morgan WF, Crossen PE. X irradiation and sister chromatid exchange in cultured human lymphocytes. Environ Mutagen 1980; 2:149 - 155
- Perry P, Evans HJ. Cytological detection of mutagen-carcinogen exposure by sister chromatid exchange. Nature 1975; 258:121 - 125
- Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 2001; 307:1235 - 1245
- Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res 2003; 532:103 - 115
- Wolff S, Bodycote J, Painter RB. Sister chromatid exchanges induced in Chinese hamster cells by UV irradiation of different stages of the cell cycle: the necessity for cells to pass through S. Mutat Res 1974; 25:73 - 81
- Ishii Y, Bender MA. Effects of inhibitors of DNA synthesis on spontaneous and ultraviolet light-induced sister-chromatid exchanges in Chinese hamster cells. Mutat Res 1980; 79:19 - 32
- Painter RB. A replication model for sister-chromatid exchange. Mutat Res 1980; 70:337 - 341
- Dronkert ML, Beverloo HB, Johnson RD, Hoeijmakers JH, Jasin M, Kanaar R. Mouse RAD54 affects DNA double-strand break repair and sister chromatid exchange. Mol Cell Biol 2000; 20:3147 - 3156
- Smiraldo PG, Gruver AM, Osborn JC, Pittman DL. Extensive chromosomal instability in Rad51d-deficient mouse cells. Cancer Res 2005; 65:2089 - 2096
- Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol 1999; 19:5166 - 5169
- Muhlmann-Diaz MC, Bedford JS. Comparison of gamma-ray-induced chromosome ring and inversion frequencies. Radiat Res 1995; 143:175 - 180
- Harvey AN, Savage JR. Investigating the nature of chromatid breaks produced by restriction endonucleases. Int J Radiat Biol 1997; 71:21 - 28
- Wojcik A, Bruckmann E, Obe G. Insights into the mechanisms of sister chromatid exchange formation. Cytogenet Genome Res 2004; 104:304 - 309
- Jeggo P, Lobrich M. Radiation-induced DNA damage responses. Radiat Prot Dosimetry 2006; 122:124 - 127
- Lobrich M, Jeggo PA. The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 2007; 7:861 - 869
- Iliakis G, Wang H, Perrault AR, Boecker W, Rosidi B, Windhofer F, et al. Mechanisms of DNA double strand break repair and chromosome aberration formation. Cytogenet Genome Res 2004; 104:14 - 20
- Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, et al. The structure-specific endonuclease Mus81 contributes to replication restart by generating double-strand DNA breaks. Nat Struct Mol Biol 2007; 14:1096 - 1104
- Roseaulin L, Yamada Y, Tsutsui Y, Russell P, Iwasaki H, Arcangioli B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J 2008; 27:1378 - 1387
- Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J 2006; 25:1764 - 1774
- Delacote F, Lopez BS. Importance of the cell cycle phase for the choice of the appropriate DSB repair pathway, for genome stability maintenance: The trans-S double-strand break repair model. Cell Cycle 2008; 7:33 - 38
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008; 7:2902 - 2906
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706 - 5715
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, et al. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009; 28:3413 - 3427
- Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 2010; 9:662 - 669
- Grudzenski S, Raths A, Conrad S, Rube CE, Lobrich M. Inducible response required for repair of low-dose radiation damage in human fibroblasts. Proc Natl Acad Sci USA 2010; 107:14205 - 14210
- Deckbar D, Birraux J, Krempler A, Tchouandong L, Beucher A, Walker S, et al. Chromosome breakage after G2 checkpoint release. J Cell Biol 2007; 176:749 - 755
- Krempler A, Deckbar D, Jeggo PA, Lobrich M. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle 2007; 6:1682 - 1686
- Wang H, Boecker W, Wang X, Guan J, Thompson LH, Nickoloff JA, Iliakis G. Caffeine inhibits homology-directed repair of I-SceI-indueced DNA double-strand breaks. Oncogene 2004; 23:824 - 834
- Nikolova T, Ensminger M, Lobrich M, Kaina B. Homologous recombination protects mammalian cells from replication-associated DNA double-strand breaks arising in response to methyl methanesulfonate. DNA Repair (Amst) 2010; 9:1050 - 1063
- Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. A double-strand break repair defect in ATM-deficient cells contributes to radiosensitivity. Cancer Research 2004; 64:500 - 508
- Kegel P, Riballo E, Kuhne M, Jeggo PA, Lobrich M. X-irradiation of cells on glass slides has a dose doubling impact. DNA Repair (Amst) 2007; 6:1692 - 1697
- Iliakis G, Kurtzman S, Pantelias G, Okayasu R. Mechanism of radiosensitization by halogenated pyrimidines: effect of BrdU on radiation induction of DNA and chromosome damage and its correlation with cell killing. Radiat Res 1989; 119:286 - 304