Abstract
The cisplatin-induced ATM-dependent phosphorylated (p)-ΔNp63α plays an important role in transcriptional regulation of specific genes encoding mRNAs and microRNAs (miRs) implicated in cell death, cell survival, and chemoresistance. The p-ΔNp63α-induced miR-885-3p functions as a critical regulator of MDM4, ATK1, BCL2, ATG16L2, ULK2, CASP2, and CASP3 mRNAs via pairing with their respective 'recognition' sequences. Cisplatin exposure modulated the levels of target proteins (reduced BCL2, AKT1, ATG16L2, and ULK2, while activated MDM4) in cisplatin-sensitive wild type ΔNp63α cells leading to distinct changes in cell viability. Finally, miR-885-3p modulated the cisplatin-induced TP53-dependent mitochondrial apoptosis by up regulation of MDM4 levels and down regulation of BCL2 levels in mitochondria. Altogether, our results support the notion that miR-885-3p might contribute in regulation of cell viability, apoptosis and/or autophagy in squamous cell carcinoma cells upon cisplatin exposure.
Introduction
Tumor protein (TP)-63 displays a similar modular structure and extensive homology to TP53; it encodes six distinct protein isotypes resulting from two promoters and alternative splicing.Citation1,Citation2 TP63 isotypes consist of two groups, those with the transactivation (TA) domain at the N terminus (TA-) and those without it (ΔN-).Citation1,Citation2 As a result of alternative splicing, Tp63 encodes α, β and γ isotypes of various length.Citation1,Citation2 TAp63 isotypes bind specific DNA sequences activating or repressing transcription of responsive genes that induce cell cycle arrest or cell death.Citation2–Citation8 Since ΔNp63 proteins retain the core DNA binding domain, simple competition for DNA responsive elements might prevent Tp53 or TAp63 from binding to the downstream gene promoters.Citation8–Citation12
The dominant-negative inhibitor ΔNp63α is the most abundant TP63 isotype expressed in proliferating basal layers of many epithelial tissues and is highly expressed in various human epithelial cancers (e.g., squamous cell carcinomas, SCC), supporting its role in cell proliferation and neoplastic development.Citation13,Citation14 ΔNp63α was also shown to play a more proactive role as a transcriptional regulator by activating or repressing several critical downstream gene targets.Citation9–Citation12 Moreover, ΔNp63α was found to induce nuclear accumulation and signaling of β-catenin, supporting an oncogenic role for ΔNp63 in SCC cells.Citation14 ΔNp63α overexpression was reported to increase cell proliferation and enhance tumor growth in vitro and in vivoCitation13 and also blocked UV-induced Tp53-dependent apoptosis in vivo.Citation15 However, siRNA silencing of ΔNp63α inhibited proliferation and mediated the death of SCC cells, supporting the notion that ΔNp63α functions as an anti-apoptotic factor.Citation6,Citation7,Citation16,Citation17
We previously found that cisplatin, which is well-known for a dramatic increase of tumor cell death, leads to a ATM-dependent phosphorylation of ΔNp63α.Citation18 We further found that the cisplatin-induced phosphorylated (p)-ΔNp63α transcription factor is indispensable for activation and inhibition of specific gene expression involved in cell death and survival, thereby potentially counteracting the function of the non-p-ΔNp63α anti-apoptotic factor.Citation12,Citation19,Citation20 We also previously showed the cisplatin-induced and p-ΔNp63α-dependent regulation of the expression of specific mRNAs and miRNAs implicated in tumor cell response to cisplatin treatment and likely contributing to chemosensitivity or chemoresistance.Citation19,Citation21–Citation23 miR-885-3p was found to be the most responsive to the cisplatin-induced p-ΔNp63α-dependent regulation; however, the role and function of miR-885-3p in tumor cell response to cisplatin exposure remains to be elucidated. We suggest that miR-885-3p might contribute to regulation of cell viability, apoptosis and/or autophagy.
Results
Cisplatin-induced miR-885-3p modulates the target mRNA-derived UTR-reporter activities.
The complexity of molecular mechanisms underlying the tumor cell response to cisplatin is not clear and, once in while, novel targets and biomarkers of drug resistance emerge in the literature.Citation21–Citation40 We previously suggested that tumor protein TP63, and especially its dominant-negative isotype, ΔNp63α plays a central role in the response of SCC cells to platinum chemotherapy as reviewed in references Citation18–Citation21. We also showed that the cisplatin exposure leads to the key event, the ATM-dependent phosphorylation of ΔNp63α in SCC cells, thereby triggering the series of molecular events, including activation or repression of genes implicated in cell survival and cell death via apoptosis upon DNA damage and stress.Citation19,Citation21
We further showed that p-ΔNp63α modulates the expression of both mRNAs and miRNAs through both transcriptional and post-transcriptional mechanisms.Citation19,Citation21 We have previously showed that the p-ΔNp63α transcriptional factor could play a critical role in cisplatin-induced repression and activation of specific miRNAs (e.g., miR-181a, miR-519a, miR-374a and miR-630, ref. Citation21).
In this study, we used the SCC stable clones, which have been shown to exclusively produce wild-type (wt) ΔNp63α or ΔNp63α-S385G with an altered ability to be phosphorylated by ATM kinase. The goal of this study was to examine a potential role for miR-885-3p activated by cisplatin-induced p-ΔNp63αCitation21 in regulation of cell death and survival. Using quantitative PCR (qPCR), we first showed that the cisplatin treatment, indeed, upregulated the levels of mature miR-885-3p by 2.2 + 0.2-fold in SCC-wt-ΔNp63α cells, while no significant changes were observed in SCC-ΔNp63α-S385G cells upon cisplatin exposure (). We further employed the web-based database (www.microrna.org) to identify potential miR-885-3p targets and define their respective “recognition” sequences as well as the miR-885-3p consensus sequence shown in . We found that the potential “recognition” sequences for miR-885-3p were present in the 5′-untranslated region (UTR) of Mdm4 () and in the 3′-UTR of Akt1 (), Bcl2 (), Atg16L2 (), Ulk2 (), Casp2 () and Casp3 ().
Although, we have not observed the perfect complementation between miR-885-3p “seed” sequence and “recognition” sequences in the target mRNAs, we found sufficient base pairing ranging from 8 to 11 nucleotides (), suggesting that miR-885-3p might downregulate levels of specific proteins via preventing the target mRNA translation. Reporter constructs with a luciferase sequence fused to multiple, partially complementary “recognition” sites for specific miRNAs were shown to display the repressed protein level and, in turn, reduced enzymatic activity, thereby reinforcing the idea that complementation is the key factor that decides the fate of the target message.Citation41–Citation45
To further examine the effect of miR-885-3p, its inhibitor or mimic on the specific 5′-UTR or 3′-UTR sequences fused to the luciferase reporter gene, we monitored the activity of the latter. We then employed a custom-generated Mdm4 5′-UTR luciferase enhancer construct and commercially available 3′-UTR plasmids for Akt1, Bcl2, Atg16L2, Ulk2, Casp2 and Casp3. These constructs were introduced into SCC-wt-ΔNp63α cells for 24 h along with the scrambled RNA and an inhibitor or mimic for miR-885-3p, as indicated ( and ). Resulting cells were subsequently exposed to control media or 10 µg/ml cisplatin for an additional 16 h, as indicated ( and ). Intriguingly, we found that cisplatin and miR-885-3p mimic activated the luciferase activity fused to the Mdm4 5′-UTR sequence, while miR-885-3p inhibitor dramatically reduced this activity compared with the control sample, even in spite of the presence of cisplatin in the medium ().
We further found that the cisplatin treatment and miR-885-3p mimic dramatically reduced the luciferase reporter activity fused to 3′-UTR of Akt1 (), Bcl2 (), Atg16L2 (), Ulk2 (), Casp2 () and Casp3 () compared with control conditions, while the miR-885-3p inhibitor significantly activated the luciferase reporter activity beyond the control readings, thereby counteracting the cisplatin inhibitory effect (, C and ).
Cisplatin-induced miR-885-3p modulates the target protein levels.
Since modulation of the UTR-fused luciferase reporter activity by miR-885-3p ultimately leads to a potential reduction of the target protein levels, we next examined the effect of cisplatin exposure on the expression levels of proteins encoded by target mRNAs. Both SCC-wt-ΔNp63α cells and SCC-ΔNp63α-S385G cells were transfected with scrambled RNA and an miR-885-3p inhibitor or mimic (), while resulting cells were incubated with control media or 10 µg/ml cisplatin as indicated (). We found that the exposure of SCC-wt-ΔNp63α cells to cisplatin reduced levels of ΔNp63α while increasing its phosphorylation (, left parts), as was previously reported in reference Citation18 and Citation21. However, SCC-ΔNp63α-S385G cells failed to display such a response (, right parts). Although the cisplatin treatment dramatically induced the MDM4 protein levels in SCC-wt-ΔNp63α cells (, left part), no changes were observed in SCC-ΔNp63α-S385G cells (, right part). While miR-885-3p mimic induced MDM4 protein levels, miR-885-3p inhibitor in a combination with cisplatin treatment significantly reduced MDM4 expression (, left part) thereby partially ”rescuing” cells from the cisplatin-induced effect.
At the same time, cisplatin treatment reduced protein levels of BCL2, AKT1, ATG16L2 and ULK2 (, parts), while CASP2 and CASP3 levels (, left parts) in SCC-wt-ΔNp63α cells were likely activated by other mechanisms. However, levels for ATG16L2 and ULK2 showed no change in SCC-ΔNp63α-S385G cells upon cisplatin exposure (, right parts) suggesting that the p-ΔNp63α and non-p-ΔNp63α-dependent regulatory mechanisms are underlying such differences. Although the levels for CASP2 and CASP3 were upregulated, the BCL2 level was downregulated in SCC-ΔNp63α-S385G cells, supporting the notion that the p-ΔNp63α-independent regulatory mechanisms could be involved in the regulation of CASP2 and CASP3 levels. Interestingly, that miR-885-3 mimic reduced all target proteins (but MDM4) compared with control samples. However, miR-885-3p inhibitor in a combination with cisplatin induced the expression of target proteins at various degrees and dramatically reduced MDM4 levels (, left parts).
Cisplatin-induced miR-885-3p modulates cell viability through its target mRNAs.
We next examined the effect of cisplatin treatment, miR-885-3p inhibitor and miR-885-3p mimic on cell viability, monitored by a mitochondrial-based MTT assay. Both SCC-wt-ΔNp63α cells () and SCC-ΔNp63α-S385G cells () were transfected with scrambled RNA or miR-885-3p mimic and then exposed to control medium (Con), while, when cells were transfected with the scrambled RNA or miR-885-3p inhibitor, they were subsequently incubated with 10 µg/ml cisplatin (CIS), as indicated ( and B). We found that the cisplatin exposure (, curve 2) and, to a lesser degree, the miR-885-3p mimic (, curve 4) dramatically decreased the number of SCC-wt-ΔNp63α cells monitored by the MTT cell viability assay, while the miR-885-3p inhibitor (, curve 3) partially “rescued” the cells from cisplatin-induced cell death as compared with control sample or cisplatin alone (, curves 1 and 2, respectively).
However, we further found that while the miR-885-3p mimic showed a similar effect on SCC-ΔNp63α-S385G cells (, curve 4), cisplatin (, curve 2) had no significant effect on MTT-measured cell viability, and the effect of miR-885-3p inhibitor (, curve 3) was even closer to control treatment of SCC-ΔNp63α-S385G cells (, curve 1), further supporting the notion that SCC-ΔNp63α-S385G cells displayed the cisplatin-resistant phenotype, while SCC-wt-ΔNp63α cells appeared to be cisplatin-sensitive.Citation18–Citation21
We further examined the potential role of specific target mRNAs regulated by miR-885-3p in the SCC cell response to cisplatin exposure. SCC-wt-ΔNp63α cells were transfected with Mdm4 siRNA and expression cassettes for Akt1, Bcl2 or Ulk2 (), while SCC-ΔNp63α-S385G cells were transfected with the Mdm4 expression cassette and siRNAs against Akt1, Bcl2 or Ulk2 (). Using the MTT-based cell viability assay, we then showed that the cisplatin treatment dramatically induced the cell death response of SCC-wt-ΔNp63α cells, while Mdm4 siRNA and expression cassettes for Akt1, Bcl2 or Ulk2 reduced the cell death response at various degrees (). At the same time, the cisplatin treatment failed to significantly induce the cell death response of SCC-ΔNp63α-S385G cells, while the Mdm4 expression cassette and siRNAs against Akt1, Bcl2 or Ulk2 partially stimulated the cell death response ().
Cisplatin-induced miR-885-3p modulates the MDM4-mediated TP53/BCL2 protein interactions.
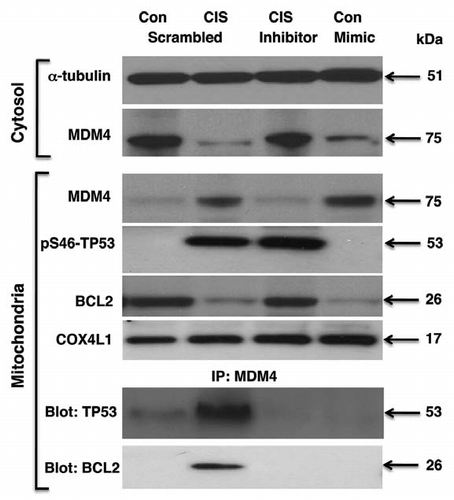
Finally, we examined the effect of cisplatin on the MDM4, which was defined by us as a potential target for miR-885-3p ( and ). Previous reports showed that MDM4 might function as a “double-faced regulator of TP53-dependent apoptosis.”Citation46–Citation49 Under unstressed conditions, MDM4 physically associates with TP53, modulating its transcriptional activity and protein stability.Citation46 After DNA damage, MDM4 is exported from the cytoplasm to the mitochondria, re-localizing the p-S46-Tp53 protein to the mitochondria.Citation46,Citation47,Citation49 This relocalization enables p-S46-Tp53 to bind to and inactivate the apoptotic inhibitor BCL2 in the mitochondria and promotes the cytochrome c release.Citation46,Citation47,Citation49 MDM4 downregulation plays an active role in cisplatin resistance by inhibiting TP53-dependent mitochondrial apoptosis.Citation46,Citation47,Citation49
SCC-wt-ΔNp63α cells were transfected with a mimic or inhibitor for miR-885-3p for 24 h and then incubated with control medium or 10 µg/ml of cisplatin for an additional 16 h. We found that cisplatin and the miR-885-3p mimic alone dramatically reduced, while the miR-885-3p inhibitor slightly induced, the MDM4 protein levels in cytoplasm as compared with control/scrambled sample (). We further found that the cisplatin treatment increased MDM4 levels and decreased BCL2 levels in mitochondria () and subsequently induced a formation of protein-protein complexes between MDM4 and p-S46-Tp53 or BCL2 (). We then observed that the miR-885-3p inhibitor blocked the migration of MDM4 into the mitochondria, while miR-885-3p mimic activated this migration as compared with the control sample (), suggesting that miR-885-3p modulates the cisplatin-induced complex formation between MDM4, p-S46-Tp53 and BCL2 and leading a TP53-dependent mitochondrial apoptosis in SCC cells as previously described in references Citation46, Citation47 and Citation50.
Discussion
Cisplatin is the most widely used and effective therapeutic agents for human epithelial cancers.Citation51 However, its efficiency is limited due to development of drug resistance.Citation51,Citation52 Multiple mechanisms have been implicated in cisplatin resistance: mitochondrial apoptosis, AKT1, BCL2 and caspase cascade pathways and potential involvement of autophagic pathway.Citation21–Citation38 The regulation of these signaling processes could be achieved at several levels, i.e., by transcriptional factors, protein-protein interactions and/or miRNA-dependent modulation.Citation6,Citation18,Citation21–Citation23,Citation28–Citation35,Citation40,Citation53–Citation56
miRNAs are non-coding RNAs that regulate multiple cellular processes, including cell survival and cell death.Citation41–Citation45,Citation53–Citation56 We and others recently reported that several miRNAs, including miR-181a, miR-591a, miR-374a and miR-630, could act as novel modulators of tumor cell response to cisplatin.Citation21–Citation23,Citation28 Some miRNAs (e.g., miR-181a, miR-591a, miR-374a) were downregulated, while others (e.g., miR-630 and miR-885-3p) were upregulated in SCC cells upon cisplatin exposure in a p-ΔNp63α-dependent manner.Citation21 Moreover, we previously found that the member of the RNA-induced silencing complex (RISC), DICER1, is involved in the p-ΔNp63α-dependent maturation of miRNAs.Citation21 Here, we observed that the miR-885-3p “recognition” sequences could be found in the specific mRNAs, including Mdm4, Akt1, Bcl2, Atg16L2, Ulk2, Casp2 and Casp3. Using the luciferase reporter assay, we further found that miR-885-3p, indeed, paired with the “recognition” sequences in these mRNA targets, leading to a significant modulation of the activity of the luciferase gene fused to the 5′-UTR or 3′-UTR of target mRNAs.
We then observed that cisplatin and miR-885-3p mimic reduced the activity of the luciferase gene fused to the 3′-UTR of Akt1, Bcl2, Atg16L2, Ulk2, Casp2 or Casp3, while the miR-885-3p inhibitor partially reversed the cisplatin-induced effect on these target mRNAs, potentially leading them to degradation or at least to a translation inhibition. Intriguingly, cisplatin and miR-885-3p mimic activated, while miR-885-3p inhibitor reduced, activity of the luciferase gene fused to the Mdm4 5′-UTR “recognition” sequence for miR-885-3p. Cisplatin treatment was shown to exclusively induce the Mdm4-dependent luciferase activity and MDM4 protein levels in SCC-wt-ΔNp63α cells.
Gene silencing may occur either via mRNA degradation or by preventing mRNA from being translated.Citation41–Citation45,Citation53–Citation56 Recent studies provide evidence that the mRNAs silenced by miRNAs are localized to cytoplasmic processing bodies (P-bodies) for storage or degradation.Citation57–Citation60 If there is complete complementation (perfect base pairing) between the miRNA and target mRNA sequences, the RISC members (e.g., AGO, DROSHA or DICER) can cleave the mRNA leading to direct mRNA degradation, while partial complementation (imperfect base pairing) prevents mRNA translation or speeds up mRNA deadenylation, causing the accelerated mRNA degradation.Citation57,Citation59 For partially complementary miRNAs to recognize their target mRNAs, ∼2–7 nucleotides of the miRNA consensus “seed” sequence still needed to be perfectly complementary to the target mRNA “recognition” sequence.Citation45,Citation57,Citation59 In both cases, the miRNA/mRNA pairing would ultimately lead to a reduction of specific protein levels. miRNAs are usually complementary to a 3′-UTR site of target mRNA and sometimes to a 5′-UTR site or both.Citation45,Citation57–Citation60 However, miRNA could also increase gene translation depending of the target mRNAs or even the cell proliferating stage.Citation58,Citation60
In the current study, we confirmed our previous findingsCitation19,Citation21 that the exposure of SCC-wt-ΔNp63α cells to cisplatin activated the p-ΔNp63α levels. We further found that the cisplatin treatment reduced the BCL2, AKT1, ATG16L2 and ULK2 protein levels while activating CASP2 and CASP3 in cisplatin-sensitive SCC-wt-ΔNp63α cells.Citation19,Citation21 However, levels for ATG16L2 and ULK2 showed no change in SCC-ΔNp63α-S385G cells, suggesting the p-ΔNp63α-dependent mechanism for ATGl6L2 and ULK2. Although CASP2 and CASP3 were upregulated, the BCL2 level was downregulated in SCC-ΔNp63α-S385G cells, suggesting the p-ΔNp63α independent regulatory mechanisms for these targets. We also showed that the cisplatin treatment and miR-885-3p mimic dramatically decreased the cell viability of SCC-wt-ΔNp63α cells. However, we further found that while the miR-885-3p mimic showed a similar effect on SCC-ΔNp63α-S385G cells, cisplatin treatment had no significant effect in cell viability, further supporting the notion that SCC-ΔNp63α-S385G cells display the cisplatin-resistant phenotype, while SCC-wt-ΔNp63α cells are cisplatin-sensitive.Citation19–Citation21 Moreover, we observed that the miR-885-3p inhibitor diminished the cisplatin-induced cell death response in SCC-wt-ΔNp63α cells and to a greater degree in SCC-ΔNp63α-S385G cells. We further observed Mdm4 siRNA and the Akt1, Bcl2 or Ulk2 expression cassettes reduced the cell death response of SCC-wt-ΔNp63α cells. However, the Mdm4 expression cassette and siRNAs against Akt1, Bcl2 or Ulk2 partially stimulated the cell death response of SCC-ΔNp63α-S385G cells. Finally, we showed that miR-885-3p modulated the cisplatin-induced complex formation between MDM4, p-S46-Tp53 and BCL2 in mitochondria, thereby leading to a TP53-dependent mitochondrial apoptosis in SCC cells.
Inhibition of apoptotic response of tumor cells to chemotherapy is the major reason for the failure of anticancer therapies.Citation51,Citation52 Malignant cells harbor mechanisms allowing escape from drug-induced apoptosis, and the drug-resistance phenotype can be significantly associated with resistance to cell death.Citation51,Citation52 Accumulating evidence shows that mitochondria play a key role in the tumorigenic phenotype, including the resistance to apoptosis.Citation31,Citation46,Citation47,Citation49,Citation50,Citation57,Citation61–Citation63 MDM4 was shown to activate Tp53 mitochondrial apoptosis by stably localizing at the mitochondria, where upon lethal stress conditions, MDM4 promotes the mitochondrial localization of pS46-Tp53 and binding of the latter to an anti-apoptotic factor BCL2, followed by cytochrome c release.Citation46,Citation47,Citation49
MDM4 expression was observed to correlate with cisplatin resistance of human epithelial ovarian cancers.Citation46,Citation47 Intriguingly, our data showed that miR-885-3p activated by cisplatin treatment by binding to the 5-UTR of MDM4 induced MDM4 levels and facilitated MDM4 export to mitochondria from cytoplasm, while enhancing the ability of MDM4 to bind pS46-Tp53 and BCL2. Similarly, the miR-885-3p mimic alone followed the pattern displayed by cisplatin treatment, suggesting that an miR-885-3p-dependent mechanism contributes to this p53-induced mitochondrial apoptotic pathway. However, miR-885-3p inhibitor is likely to prevent translocation of MDM4 to the mitochondria, while pS46-Tp53 seems to be abundant in the mitochondria, which is somewhat different from a popular hypothesis that MDM4 facilitates bringing pS46-Tp53 to the mitochondria.Citation46,Citation47,Citation49 One potential explanation of this effect is that, since the miR-885-3p mimic could enhance the MDM4 mRNA translation and stability, leading to MDM4 accumulation, miR-885-3p inhibitor might interfere with MDM4 expression or trafficking by a yet-unknown mechanism.
The original model proposed by Fabiola Moretti's research team consists of the following steps: (1) MDM4 stably localizes at the mitochondria, where it (2) binds BCL2, (3) facilitates mitochondrial localization of pS46-TP53, (4) promotes binding between pS46-Tp53 and BCL2 and (5) induces release of cytochrome C, leading to apoptosis. The specific mechanism by which pS46-Tp53 is exported from cytoplasm to mitochondria is very likely to be more complex and might include multiple pathways induced by stress (e.g., cisplatin exposure) as described in references Citation61–Citation64.
Furthermore, CASP2 and CASP3 were predominantly activated in skin cancers and oral SCC upon cisplatin exposure.Citation27,Citation30,Citation31 Cisplatin was shown to induce the TP53-mediated CASP2 activation and the mitochondrial release of apoptosis-inducing factor (AIF) in renal tubular epithelial cells.Citation31 However, the AIF silencing markedly inhibited cisplatin-induced cell death.Citation31 Although, the CASP2 silencing significantly prevented AIF translocation from mitochondria to cytoplasm, Tp53 overexpression not only resulted in CASP2 activation, but also in AIF mitochondrial release, while Tp53 silencing prevented both events.Citation31
AKT1 gene amplification and overexpression in human lung cancer cells is associated with an acquired cisplatin resistance.Citation12,Citation24,Citation25 The AKT1 forced expression was sufficient to render the cells to become drug resistant through the mammalian target of the rapamycin (mTOR) signaling pathway.Citation25 However, AKT1 inhibition reversed the resistant cells to be sensitive to the drug.Citation25 In addition, the concomitant inhibition of AKT and autophagy was found to be required for an efficient cisplatin-induced apoptosis of metastatic skin carcinoma.Citation35 Counteracting the autophagic process using ATG5 inhibitors along with AKT inhibitors dramatically enhanced the cytotoxicity of cisplatin, thus revealing a key role for autophagy in chemoresistance.Citation35 While forced expression of ΔNp63α was shown leading to a AKT1 transcriptional upregulation,Citation12 the cisplatin-mediated and p-ΔNp63α-dependent miR-885-3p reduced AKT1 levels in SCC cells.
Autophagy was shown to appear at early stages after cisplatin exposure, as indicated by induction of LC3-I, conversion of LC3-I to LC3-II protein, upregulation of BECN1 and ATG5 and the appearance of cisplatin-induced punctuated staining of LC3-II.Citation33–Citation35 However, autophagy inhibitors reduced the formation of autophagosomes but induced apoptosis after 2–4 h of cisplatin treatment, as indicated by CASP3/7 and -6 activation, nuclear fragmentation and cell death. At later stages of cisplatin injury, apoptosis was also found to be associated with autophagy, as autophagic inhibitors and siRNAs to Becn1 and Atg5 enhanced activation of caspases and apoptosis.Citation33–Citation35
ULK1 and ULK2 are functionally redundant protein kinases required to mediate/initiate autophagy under nutrient-deprived conditions.Citation65–Citation69 There is strong genetic evidence that ULK1 is an essential component of the autophagic signaling pathway; the ability of ULK2 to compensate for the loss of ULK1 function is cell type-specific.Citation65 Both ULK1 and ULK2 are transcriptional targets of Tp53 and are upregulated upon DNA damage, suggesting that ULK1 and ULK2 may mediate tumor suppression activity and contribute to the efficacy of chemotherapeutic drugs.Citation66–Citation70 Accumulating evidence along with our present data suggest that cisplatin treatment unveils a complex plethora of molecular events leading to pro- and anti-survival responses, and potential outcome for tumor cell fate needs to be assessed on the personalized basis.Citation16,Citation21–Citation23,Citation28,Citation35–Citation38,Citation52,Citation53 Emerging novel targets and biomarkers, including miRNAs implicated in tumor cell resistance to platinum agents could provide new venues to counteract drug resistance and improve chemotherapeutic strategies.Citation16,Citation21–Citation23,Citation28,Citation35–Citation38,Citation52,Citation53
Materials and Methods
Cells, reagents and transfections.
The SCC cell line JHU-029 (with wt-TP53, TP63 is amplified, and ΔNp63α is overexpressed, ref. Citation71) was isolated from primary tissue at the Department of Otolaryngology/Head and Neck Surgery (JHMI). Cells were obtained from the Head and Neck Cancer Research Division Tissue Bank at the JHMI. The cell line was authenticated by a short tandem repeat profiling analysis using the AmpFISTR Identifiler PCR Amplification Lit (Applied Biosystems) at the JHMI Fragment Analysis Facility. The stable SCC cell lines expressing wt-ΔNp63α or ΔNp63α-S385G were generated using Flp-In technology as described in reference Citation19 and Citation21. Cells were maintained in RPMI medium 1640 and 10% fetal bovine serum and incubated with control medium or 10 µg/ml cisplatin (Sigma) for the indicated time periods. Cells were lysed with 50 mM Tris, pH 7.5, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 0.5% Brij-50, 1 mM PMSF, 0.5 mM NaF, 0.1 mM Na3VO4, 2x complete protease inhibitor cocktail, sonicated for 10 sec intervals and spun for 30 min at 15,000x g. Supernatants were analyzed by immunoblotting and immunoprecipitation, and the levels of target proteins were normalized against the β-actin level as previously described in references Citation19 and Citation21. Blots were scanned and quantified by Image Quant software version 3.3 (Molecular Dynamics). Values were expressed as percentage of a control sample (defined as 1).
In some experiments, cells were transfected with siRNAs against the following targets: Akt1 (H00000207-R08), Bcl2 (H00000596-R01), Ulk2 (H00009706-R02) (all obtained from Novus Biologicals) and Mdm4 (Santa Cruz Biotechnology, sc-37448). Cells were grown in 24-well plates at ∼35–50% confluency and then transfected with 10 pmole siRNA and 1 µl RNAiMAX Lipofectamine in 100 µl of transfection solution with Opti-MEM I (Invitrogen) performed for 48 h according to the manufacturer's instructions. Cells were also transfected with the following pCMV6-Myc-tagged expression cassettes for: Bcl2 (RC21055), Akt1 (RC201850), Ulk2 (RC206010) and Mdm4 (RC209620), all obtained from Origene Technology. Cells were grown in 24-well plates at ∼60–70% confluency and then transfected for 48 h with 1 µg of plasmids using a preincubated transfection solution (200 µl) and 300 µl of Opti-MEM I to each well in the presence of 1.5 µl of Lipofectamine 2000 (Invitrogen).
Isolation of mitochondrial/cytoplasmic fractions.
1–2 × 106 cells were resuspended in hypotonic lysis buffer (10 mM HEPES pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA) with protease inhibitors (Sigma). After resuspension, 0.6% Triton X-100 (final concentration) was added, and the nuclei were pelleted by centrifugation at 2,500–3,000x g for 10 min at 4°C, as described in reference Citation72. Mitochondria were isolated from the supernatant (cytoplamic fraction) by differential centrifugation at the 12,000x g for 20 min. Pellets were subjected to a mild digitonin (Sigma) treatment to remove the outer membrane with mitochondria-bound cytoplasmic polyribosomes.Citation72 After that, mitochondria were purified by a centrifugation in a sucrose density gradient (25–50% w/w), yielding a homogenous band with buoyant density 1.160–1.170 g/ml. Purified mitochondria were pelleted at the 10,000x g for 15 min at 4°C. Mitochondrial lysates were obtained by 1% Triton X-100 as previously described in reference Citation72.
Validation of miRNA expression by qPCR.
To validate the differential expression of miRNAs, we isolated total small RNAs using a miRVana miRNA isolation kit (Applied Biosystems, AM1560). We then used the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, #4374966) to produce single-stranded cDNA probes. Next, we performed a qPCR using the TaqMan MicroRNA Assay Kit TaqMan® U47 (#4380911) and TaqMan® Gene Expression Master Mix, 1-Pack (#4369016), both obtained from Applied Biosystems.Citation21 For mature miRNAs, we also used the following: hsa-miR-885-3p (#002372). The reaction conditions were 50°C for 2 min, 95°C for 10 min, 40 cycles of 95°C for 15 sec and 60°C for 1 min with a sample volume of 20 ml. Expression was normalized as described in reference Citation21.
Antibodies.
We used antibodies against β-actin (Sigma), ΔNp63 (EMD/Calbiochem, anti-p40, PC373, residues 5–17 epitope), BCL2 (Abcam, clone 124, ab694), ULK2 (Abcam, ab81021), CASP2 (Abnova, clone 4i13, ab18029), AKT1 (Abcam, 9A4, ab89402), α-tubulin (Abcam, clone AA13, #ab28037), COX4L1 (Abcam, mAbcam59426-mitochondrial loading control, #ab59426), CASP3 (Abnova Corporation, H00000836-D01P), ATG16L2 (Novus Biologicals, NBP1-21331), MDM4 (Abnova Corporation, PAB8758) and p-S46-TP53 (Cell Signaling Technology, phospho-Ser46, 2521S). A custom rabbit polyclonal antibody against a phosphorylated peptide encompassing the ΔNp63α protein sequence (ATM motif, NKLPSVpS-QLINPQQ, residues 379–392) was prepared and purified against the phosphorylated peptide vs. non-phosphorylated peptide.Citation18
5′-UTR Luciferase reporter assay.
The pGL3-MDM4 5′-UTR construct was generated by us as follows: the sequence encompassing the 5′-UTR (−156 to +144 base pairs) was amplified from the cDNA template using the following primers, sense, (−156) 5′-GAT AGG TAC CGA GCT CGG GAG GCC GGA AGT TGC GGC TT-3′ (−121) and antisense, (+125) 5′-CTT ACT TAG ATC GCA GAT CTC CAC CAC AAT ATA CCA TAT-3′ (+158) containing the Sac1 and BglII restriction sites, respectively. cDNA template was prepared from 1 µg of total RNA isolated from human lung cancer cells (A549, CCL-185, American Tissue Culture Collection) using TRIzol reagent (Invitrogen) followed by reverse transcription with the aid of 10 pmol random hexamer and the High Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Invitrogen, #4374966). The PCR amplification was carried-out at (95°C, 4 min; for 30 cycles at 95°C for 30 sec; at 60°C for 1 min; at 72°C for 1 min) and at 72°C for 7 min.
Resulting 300 base pair PCR product was cloned first into the pCR2.1 TOPO TA cloning vector (Invitrogen) and then into SacI and BglII sites of the pGL3-Basic vector (Promega), thereby producing the pGL3-MDM4 5′-UTR plasmid. At all steps, sequence was verified by sequencing.
For the 5′-UTR reporter assays, cells grown in 96-well plates were transfected with the 30 nM 885-3p miRNA hairpin precursors (Ambion, PM12458) or inhibitors (Ambion, AM12458), 0.15 µg pGL3 enhancer luciferase vector and 0.02 µg Renilla vector (pRL-TK) using Lipofectamine-2000. Forty-eight h after transfection and cisplatin treatment, luciferase activity was measured by the Dual-Glo Luciferase Assay (Promega).
3′-UTR luciferase reporter assay.
We used the 3′-UTR luciferase reporter plasmids for ULK2 (#S811113), ATG16L2 (#S800799), AKT1 (#S812411), CASP2 (#S811761), CASP3 (#S809759) and MDM4 (#S812965) were obtained from SwitchGear Genomics, and the BCL2 3′-UTR construct (#SC222289) was purchased from Origene Technologies. For the 3′-UTR-mediated luciferase activity assay, cells grown in a 96-well plate were transfected for 24 h with the control (empty) pLightSwitch_3UTR vector (#S890005), respectively using Fugene HD reagent (Roche). In addition, cells were transfected with the selected 3′-UTR plasmids (Switchgear Genomics or Origene Technologies). The LightSwitch Luciferase Assay Reagent (SwitchGear Genomics) enabled us to measure luciferase reporter signal according to the manufacturer's protocol. For the 3′-UTR assays, cells were also transfected with 30 nM of the mimic (PM12458) or inhibitor (AM12458) for miRNA-885-3p obtained from Ambion/Applied Biosystems. Each experiment was performed independently at least three times and in triplicate. At 24 h, cells were also treated with 10 µg/ml cisplatin or control medium for an additional 24 h. The RenSP luciferase activity was measured at 480 nm using a luminometer.
Cell viability assay.
Cells were plated at 20–30% confluence in each well of a 6-well plate. Growth was assayed at 0–120 h by MTT cell viability assay (American Tissue Culture Collection) as described in reference Citation12. Absorbance readings were taken using a SpectraMax M2e Microplate fluorescence reader (Molecular Devices) at 570- and 650-nm wavelengths. All samples were run in triplicate.
Statistical analysis.
Statistical analysis was performed using the unpaired Student t-test. p-values of < 0.05 were considered to indicate statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| UTR | = | untranslated region |
| TP | = | tumor protein |
| SCC | = | squamous cell carcinoma |
| wt | = | wild-type |
| TA | = | transactivation |
| RISC | = | RNA-induced silencing complex |
Figures and Tables
Figure 1 Cisplatin induced miR-885-3p expression through p-ΔNp63α. (A) qPCR analysis of the miR-885-3p levels in SCC-wt-ΔNp63α or SCC-ΔNp63α-S385G cells. Cells were incubated with control medium or 10 µg/ml cisplatin for 16 h. miRNA levels were monitored by qPCR. Data were normalized against the glyceraldehyde-3-phospho dehydrogenase levels and plotted as relative units (RU) with measurements obtained from wt-ΔNp63α cells treated with control medium set as 1. Experiments were performed in triplicate with +SD as indicated (p < 0.01). (B–I) Representation of the consensus miR-885-3p sequence (B) and “recognition” sequences in the 5′-UTR of Mdm4 (C), 3′-UTR of Akt1 (D), 3′-UTR of Bcl2 (E), 3′-UTR of Atg16L2 (F), 3′-UTR of Ulk2 (G), 3′-UTR of Casp2 (H) and 3′-UTR of Casp3 (I). The miR-885-3p consensus sequence and 5′-UTR/3′-UTR “recognition” sequences were obtained from the www.microrna.org web database. The capital letters indicate the nucleotides in the target mRNA sequences that are complementary to the nucleotides in the miR-885-3p consensus sequence.
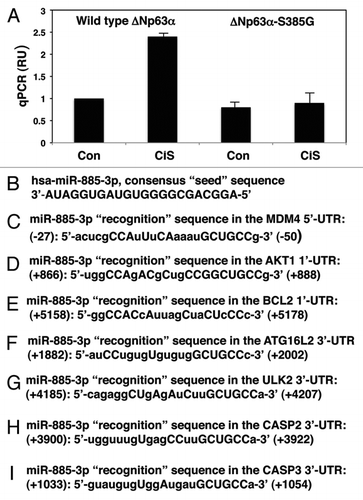
Figure 2 Cisplatin exposure modulated the miR-885-3p-mediated effect on specific target mRNAs. SCC-wt-ΔNp63α cells were transfected for 24 h with the luciferase reporter plasmids with 5′-UTR of Mdm4 (A), 3′-UTR of Akt1 (B) and 3′-UTR of Bcl2 (C). Each UTR sequence was found to contain the specific “recognition” sequences, which potentially could form double-stranded complexes with the miR-885-3p consensus sequence. Cells were also transfected for 24 h with the scrambled RNA and an inhibitor or mimic for miR-885-3p. Resulting cells were further incubated with control medium or 10 µg/ml cisplatin for an additional 16 h, as indicated. The luciferase activity was measured at 480 nm using a luminometer. Each experiment was performed independently at least three times and in triplicate with +SD as indicated (p < 0.01).
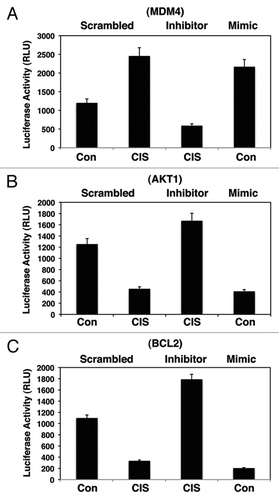
Figure 3 Cisplatin exposure modulated the miR-885-3p-mediated effect on specific target mRNAs. SCC-wt-ΔNp63α cells were transfected for 24 h with the luciferase reporter plasmids with 3′-UTR of Atg16L2 (A), 3′-UTR of Ulk2 (B), 3′-UTR of Casp2 (C) and 3′-UTR of Casp3 (D). Each UTR sequence was found to contain the specific “recognition” sequences, which potentially could form double-stranded complexes with the miR-885-3p consensus sequence. Cells were also transfected for 24 h with the scrambled RNA and an inhibitor or mimic for miR-885-3p. Resulting cells were further incubated with control medium or 10 µg/ml cisplatin for an additional 16 h, as indicated. The luciferase activity was measured at 480 nm using a luminometer. Each experiment was performed independently at least three times and in triplicate with +SD as indicated (p < 0.01).
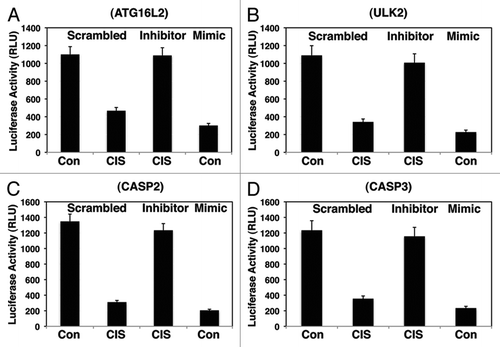
Figure 4 Cisplatin induced the p-ΔNp63α mediated modulation of target protein levels through miR-885-3p. SCC-wt-ΔNp63α cells (left parts) and SCC-ΔNp63α-S385G cells (right parts) were transfected for 24 h with the scrambled RNA and an inhibitor or mimic for miR-885-3p. Resulting cells were further incubated with control medium or 10 µg/ml cisplatin for an additional 16 h, as indicated. Protein expression was tested in total lysates using the indicated antibodies, while loading was monitored by anti-β-actin antibody.
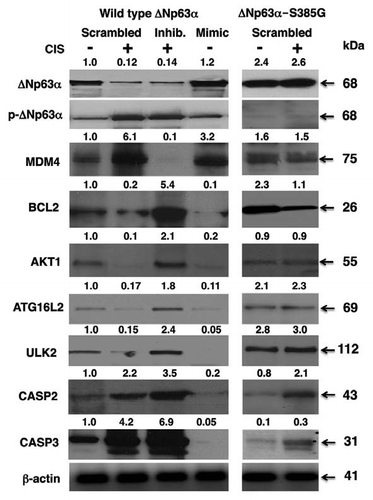
Figure 5 Cisplatin modulated cell viability via p-ΔNp63α/miR-885-3p axis. SCC-wt-ΔNp63α cells (A) and SCC-ΔNp63α-S385G cells (B) were transfected with the miR-885-3p inhibitor (curve 3) or miR-885-3p mimic (curve 4) for 24 h. Cells were then exposed to control medium (Con, curves 1 and 4) or 10 µg/ml cisplatin (CIS, curves 2 and 3) for an additional 0–120 h. Cell survival was monitored at 24, 48, 72, 96 and 120 h by measuring the mitochondrial activity by MTT reagent. Experiments were performed in triplicate with +SD as indicated (p < 0.05). In parallel experiments, SCC-wt-ΔNp63α cells were transfected with Mdm4 siRNA, pCMV6-Akt1, pCMV6-Bcl2 and pCMV6-Ulk2 (C), while SCC-ΔNp63α-S385G cells were transfected with pCMV6-Mdm4, siRNA against Akt1, Bcl2 or Ulk2 (D) for 24 h. Cells then were exposed to control medium or 10 µg/ml cisplatin for an additional 48 h. Cell viability was monitored at 72 h by measuring the mitochondrial activity by MTT reagent. Experiments were performed in triplicate with + SD, as indicated (p < 0.05).
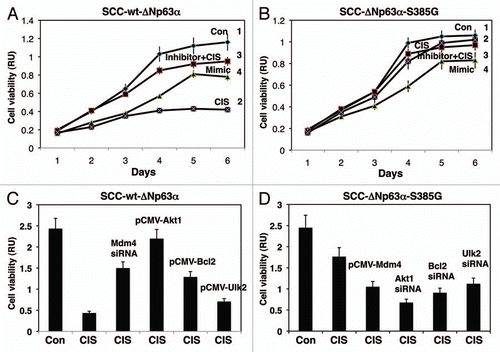
Figure 6 Cisplatin induced miR-885-3p-modulated MDM4/p-S46-TP53/BCL2 protein interactions. Wt-ΔNp63α cells were transfected with scrambled RNA and an inhibitor or mimic for miR-885-3p for 24 h. Resulting cells were exposed to control medium and 10 µg/ml of cisplatin for an additional 16 h. Protein levels for MDM4, BCL2 and p-S46-TP53 in cytosol (cyto) and mitochondria were examined by immunoblotting with the indicated antibodies. Protein loading levels were tested with anti-α-tubulin and anti-cytochrome c oxidase (COX4L1) antibodies for cytosol (cyto) and mitochondria, respectively. Mitochondrial lysates were immunoprecipitated (IP) with antibody to MDM4 and then blotted with antibodies to TP53 and BCL2.
Acknowledgments
This work is partly supported by the R01 grant DE13561 from NCI/NIDCR (E.A.R.) and by the R01 grant EY018636-01 from NEI (E.A.R.).
References
- Yang A, Kaghad M, Wang Y, Gillett E, Fleming M, Dotsch V, et al. p63, a p53 homologue at 3q27–29, encodes multiple products with transactivating, deathinducing and dominant-negative activities. Mol Cell 1998; 2:305 - 316; PMID: 9774969; http://dx.doi.org/10.1016/S1097-2765(00)80275-0
- Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006; 13:962 - 972; PMID: 16601753; http://dx.doi.org/10.1038/sj.cdd.4401914
- Trink B, Osada M, Ratovitski E, Sidransky D. p63 transcriptional regulation of epithelial integrity and cancer. Cell Cycle 2007; 6:240 - 245; PMID: 17297296; http://dx.doi.org/10.4161/cc.6.3.3803
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene 2008; 27:6507 - 6521; PMID: 18955976; http://dx.doi.org/10.1038/onc.2008.315
- Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, et al. TAp63 and ΔNp63 in cancer and epidermal development. Cell Cycle 2007; 6:274 - 285; PMID: 17264681; http://dx.doi.org/10.4161/cc.6.3.3797
- Flores ER, Tsai K, Crowley D, Sengupta S, Yang A, McKeon F, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature 2002; 416:560 - 564; PMID: 11932750; http://dx.doi.org/10.1038/416560a
- Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, et al. TAp63α induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J 2005; 24:2458 - 2471; PMID: 15944736; http://dx.doi.org/10.1038/sj.emboj.7600708
- Martin AG, Trama J, Crighton D, Ryan KM, Fearnhead HO. Activation of p73 and induction of Noxa by DNA damage requires NFkappaB. Aging 2009; 1:335 - 349; PMID: 20195489
- Shimada A, Kato S, Enjo K, Osada M, Ikawa Y, Kohno K, et al. Transcriptional activities of p53 and its homologue p51/p63: similarities and differences. Cancer Res 1999; 59:2781 - 2786; PMID: 10383130
- King KE, Ponnamperuma RM, Yamashita T, Tokino T, Lee LA, Young MF, et al. ΔNp63α functions as both positive and a negative transcriptional regulator and blocks in vitro differentiation of murine keratinocytes. Oncogene 2003; 22:3635 - 3644; PMID: 12789272; http://dx.doi.org/10.1038/sj.onc.1206536
- Papoutsaki M, Moretti F, Lanza M, Marinari B, Sartorelli V, Guerrini L, et al. A p38-dependent pathway regulates ΔNp63 DNA binding to p53-dependent promoters in UV-induced apoptosis of keratinocytes. Oncogene 2005; 24:6970 - 6975; PMID: 16007154; http://dx.doi.org/10.1038/sj.onc.1208835
- Sen T, Sen N, Brait M, Begum S, Chatterjee A, Hoque MO, et al. ΔNp63α confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res 2011; 71:1167 - 1176; PMID: 21266360; http://dx.doi.org/10.1158/0008-5472.CAN-10-1481
- Hibi K, Trink B, Patturajan M, Westra W, Caballero O, Hill D, et al. AIS is an oncogene amplified in squamous cell carcinoma. Proc Natl Acad Sci USA 2000; 97:5462 - 5467; PMID: 10805802; http://dx.doi.org/10.1073/pnas.97.10.5462
- Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, et al. ΔNp63 induces β-catenin nuclear accumulation and signaling. Cancer Cell 2002; 1:369 - 379; PMID: 12086851; http://dx.doi.org/10.1016/S1535-6108(02)00057-0
- Liefer KM, Koster M, Wang X, Yang A, McKeon F, Roop D. Downregulation of p63 is required for epidermal UV-B-induced apoptosis. Cancer Res 2000; 60:4016 - 4020; PMID: 10945600
- Rocco JW, Leong CO, Kuperwasser N, DeYoung MP, Ellisen LW. p63 mediates survival in squamous cell carcinoma by suppression of p73-dependent apoptosis. Cancer Cell 2006; 9:45 - 56; PMID: 16413471; http://dx.doi.org/10.1016/j.ccr.2005.12.013
- Cheng W, Jacobs WB, Zhang JJ, Moro A, Park JH, Kushida M, et al. ΔNp63 plays an anti-apoptotic role in ventral bladder development. Development 2006; 133:4783 - 4792; PMID: 17079275; http://dx.doi.org/10.1242/dev.02621
- Huang Y, Sen T, Nagpal J, Upadhyay S, Trink B, Ratovitski EA, et al. ATM kinase is a master switch for the ΔNp63α phosphorylation/degradation in human head and neck squamous cell carcinoma cells upon DNA damage. Cell Cycle 2008; 7:2846 - 2855; PMID: 18769144; http://dx.doi.org/10.4161/cc.7.18.6627
- Huang Y, Chuang AY, Romano RA, Liégeois NJ, Sinha S, Trink B, et al. Phospho-ΔNp63α/NF-Y protein complex transcriptionally regulates DDIT3 expression in squamous cell carcinoma cells upon cisplatin exposure. Cell Cycle 2010; 9:328 - 338; PMID: 20023394; http://dx.doi.org/10.4161/cc.9.2.10432
- Huang Y, Ratovitski EA. Phospho-ΔNp63α/Rpn13-dependent regulation of LKB1 degradation modulates autophagy in cancer cells. Aging 2010; 2:959 - 968; PMID: 21191146
- Huang Y, Chuang A, Hao H, Talbot C, Sen T, Trink B, et al. Phospho-ΔNp63α is a key regulator of the cisplatin-induced microRNAome in cancer cells. Cell Death Differ 2011; 18:1220 - 1230; PMID: 21274007; http://dx.doi.org/10.1038/cdd.2010.188
- Ory B, Ramsey MR, Wilson C, Vadysirisack DD, Forster N, Rocco JW, et al. A microRNA-dependent program controls p53-independent survival and chemosensitivity in human and murine squamous cell carcinoma. J Clin Invest 2011; 121:809 - 820; PMID: 21293058; http://dx.doi.org/10.1172/JCI43897
- Ory B, Ellisen LW. A microRNA-dependent circuit controlling p63/p73 homeostasis: p53 family cross talk meets therapeutic opportunity. Oncotarget 2011; 2:259 - 264; PMID: 21436470
- Lee MW, Kim DS, Min NY, Kim HT. Akt1 inhibition by RNA interference sensitizes human non-small cell lung cancer cells to cisplatin. Int J Cancer 2008; 122:2380 - 2384; PMID: 18224693; http://dx.doi.org/10.1002/ijc.23371
- Liu LZ, Zhou XD, Qian G, Shi X, Fang J, Jiang BH. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res 2007; 67:6325 - 6332; PMID: 17616691; http://dx.doi.org/10.1158/0008-5472.CAN-06-4261
- Floros KV, Thomadaki H, Lallas G, Katsaros N, Talieri M, Scorilas A. Cisplatin-induced apoptosis in HL-60 human promyelocytic leukemia cells: differential expression of BCL2 and novel apoptosis-related gene BCL2L12. Ann NY Acad Sci 2003; 1010:153 - 158; PMID: 15033711; http://dx.doi.org/10.1196/annals.1299.025
- Xu JH, Wang AX, Huang HZ, Wang JG, Pan CB, Zhang B. Survivin shRNA induces caspase-3-dependent apoptosis and enhances cisplatin sensitivity in squamous cell carcinoma of the tongue. Oncol Res 2010; 18:377 - 385; PMID: 20441052; http://dx.doi.org/10.3727/096504010X12644422320663
- Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res 2010; 70:1793 - 1803; PMID: 20145152; http://dx.doi.org/10.1158/0008-5472.CAN-09-3112
- Yang C, Kaushal V, Haun RS, Seth R, Shah SV, Kaushal GP. Transcriptional activation of caspase-6 and -7 genes by cisplatin-induced p53 and its functional significance in cisplatin nephrotoxicity. Cell Death Differ 2008; 15:530 - 544; PMID: 18064040; http://dx.doi.org/10.1038/sj.cdd.4402287
- Zhu J, Yang Y, Wu J. Bcl-2 cleavages at two adjacent sites by different caspases promote cisplatin-induced apoptosis. Cell Res 2007; 17:441 - 448; PMID: 17452997
- Seth R, Yang C, Kaushal V, Shah SV, Kaushal GP. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem 2005; 280:31230 - 31239; PMID: 15983031; http://dx.doi.org/10.1074/jbc.M503305200
- Michaud WA, Nichols AC, Mroz EA, Faquin WC, Clark JR, Begum S, et al. Bcl-2 blocks cisplatin-induced apoptosis and predicts poor outcome following chemoradiation treatment in advanced oropharyngeal squamous cell carcinoma. Clin Cancer Res 2009; 15:1645 - 1654; PMID: 19240170; http://dx.doi.org/10.1158/1078-0432.CCR-08-2581
- Kaushal GP, Kaushal V, Herzog C, Yang C. Autophagy delays apoptosis in renal tubular epithelial cells in cisplatin cytotoxicity. Autophagy 2008; 4:710 - 712; PMID: 18497570
- Yang C, Kaushal V, Shah SV, Kaushal GP. Autophagy is associated with apoptosis in cisplatin injury to renal tubular epithelial cells. Am J Physiol Renal Physiol 2008; 294:777 - 787; PMID: 18256309; http://dx.doi.org/10.1152/ajprenal.00590.2007
- Claerhout S, Verschooten L, Van Kelst S, De Vos R, Proby C, Agostinis P, et al. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int J Cancer 2010; 127:2790 - 2803; PMID: 21351258; http://dx.doi.org/10.1002/ijc.25300
- Allen KE, Weiss GJ. Resistance may not be futile: microRNA biomarkers for chemoresistance and potential therapeutics. Mol Cancer Ther 2010; 9:3126 - 3136; PMID: 20940321; http://dx.doi.org/10.1158/1535-7163.MCT-10-0397
- Al-Ejeh F, Kumar R, Wiegmans A, Lakhani SR, Brown MP, Khanna KK. Harnessing the complexity of DNA-damage response pathways to improve cancer treatment outcomes. Oncogene 2010; 29:6085 - 6098; PMID: 20818418; http://dx.doi.org/10.1038/onc.2010.407
- Steelman LS, Martelli AM, Nicoletti F, McCubrey JA. Exploiting p53 status to enhance effectiveness of chemotherapy by lowering associated toxicity. Oncotarget 2011; 2:109 - 112; PMID: 21487160
- Hölzel M, Burger K, Mühl B, Orban M, Kellner M, Eick D. The tumor suppressor p53 connects ribosome biogenesis to cell cycle control: a double-edged sword. Oncotarget 2010; 1:43 - 47; PMID: 21293052
- Noonan EJ, Place RF, Basak S, Pookot D, Li LC. miR-449a causes Rb-dependent cell cycle arrest and senescence in prostate cancer cells. Oncotarget 2010; 1:349 - 358; PMID: 20948989
- Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA 2003; 100:9779 - 9784; PMID: 12902540; http://dx.doi.org/10.1073/pnas.1630797100
- Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev 2004; 18:504 - 511; PMID: 15014042; http://dx.doi.org/10.1101/gad.1184404
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem 2010; 79:351 - 379; PMID: 20533884; http://dx.doi.org/10.1146/annurev-biochem-060308-103103
- Jopling CL, Norman KL, Sarnow P. Positive and negative modulation of viral and cellular mRNAs by liver-specific microRNA miR-122. Cold Spring Harb Symp Quant Biol 2006; 71:369 - 376; PMID: 17381319; http://dx.doi.org/10.1101/sqb.2006.71.022
- Moretti F, Thermann R, Hentze MW. Mechanism of translational regulation by miR-2 from sites in the 5′ untranslated region or the open reading frame. RNA 2010; 16:2493 - 2502; PMID: 20966199; http://dx.doi.org/10.1261/rna.2384610
- Mancini F, Di Conza G, Pellegrino M, Rinaldo C, Prodosmo A, Giglio S, et al. MDM4 (MDMX) localizes at the mitochondria and facilitates the p53-mediated intrinsic-apoptotic pathway. EMBO J 2009; 28:1926 - 1939; PMID: 19521340; http://dx.doi.org/10.1038/emboj.2009.154
- Mancini F, Moretti F. Mitochondrial MDM4 (MDMX): an unpredicted role in the p53-mediated intrinsic apoptotic pathway. Cell Cycle 2009; 8:3854 - 3859; PMID: 19887911; http://dx.doi.org/10.4161/cc.8.23.10089
- Heminger K, Markey M, Mpagi M, Berberich SJ. Alterations in gene expression and sensitivity to genotoxic stress following HdmX or Hdm2 knockdown in human tumor cells harboring wild-type p53. Aging 2009; 1:89 - 108; PMID: 19946469
- Giglio S, Mancini F, Pellegrino M, Di Conza G, Puxeddu E, Sacchi A, et al. Regulation of MDM4 (MDMX) function by p76 (MDM2): a new facet in the control of p53 activity. Oncogene 2010; 29:5935 - 5945; PMID: 20697359; http://dx.doi.org/10.1038/onc.2010.324
- Galluzzi L, Morselli E, Kepp O, Vitale I, Pinti M, Kroemer G. Mitochondrial Liaisons of p53. Antioxid Redox Signal 2011; 15:1691 - 1714; PMID: 20712408; http://dx.doi.org/10.1089/ars.2010.3504
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 584; PMID: 17625587; http://dx.doi.org/10.1038/nrc2167
- Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin?. Cell Cycle 2008; 7:1353 - 1359; PMID: 18418074; http://dx.doi.org/10.4161/cc.7.10.5930
- Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell 2010; 40:205 - 215; PMID: 20965416; http://dx.doi.org/10.1016/j.molcel.2010.09.027
- Kumarswamy R, Volkmann I, Thum T. Regulation and function of miRNA-21 in health and disease. RNA Biol 2011; In press PMID: 21712654
- Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol 2008; 5:115 - 119; PMID: 19182522; http://dx.doi.org/10.4161/rna.5.3.6558
- Nohata N, Sone Y, Hanazawa T, Fuse M, Kikkawa N, Yoshino H. miR-1 as a tumor suppressive microRNA targeting TAGLN2 in head and neck squamous cell carcinoma. Oncotarget 2011; 2:29 - 42; PMID: 21378409
- Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol 2009; 6:65 - 72; PMID: 19106625; http://dx.doi.org/10.4161/rna.6.1.7534
- Hammell CM. The microRNA-argonaute complex: a platform for mRNA modulation. RNA Biol 2008; 5:123 - 127; PMID: 18698153; http://dx.doi.org/10.4161/rna.5.3.6570
- Chan SP, Slack FJ. microRNA-mediated silencing inside P-bodies. RNA Biol 2006; 3:97 - 100; PMID: 17179742; http://dx.doi.org/10.4161/rna.3.3.3499
- Vasudevan S, Tong Y, Steitz JA. Cell cycle control of microRNA-mediated translation regulation. Cell Cycle 2008; 7:1545 - 1549; PMID: 18469529; http://dx.doi.org/10.4161/cc.7.11.6018
- Trinh DL, Elwi AN, Kim SW. Direct interaction between p53 and Tid1 proteins affects p53 mitochondrial localization and apoptosis. Oncotarget 2010; 1:396 - 404; PMID: 21311096
- Ahn BY, Trinh DL, Zajchowski LD, Lee B, Elwi AN, Kim SW. Tid1 is a new regulator of p53 mitochondrial translocation and apoptosis in cancer. Oncogene 2010; 29:1155 - 1166; PMID: 19935715; http://dx.doi.org/10.1038/onc.2009.413
- Altieri DC. Mitochondrial compartmentalized protein folding and tumor cell survival. Oncotarget 2011; 2:347 - 351; PMID: 21555789
- Meulmeester E, Pereg Y, Shiloh Y, Jochemsen AG. ATM-mediated phosphorylations inhibit Mdmx/Mdm2 stabilization by HAUSP in favor of p53 activation. Cell Cycle 2005; 4:1166 - 1170; PMID: 16082221; http://dx.doi.org/10.4161/cc.4.9.1981
- Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol 2010; 22:132 - 139; PMID: 20056399; http://dx.doi.org/10.1016/j.ceb.2009.12.004
- Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res 2010; 20:748 - 762; PMID: 20548331; http://dx.doi.org/10.1038/cr.2010.82
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456 - 461; PMID: 21205641; http://dx.doi.org/10.1126/science.1196371
- Lee EJ, Tournier C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 2011; 7:689 - 695; PMID: 21460635; http://dx.doi.org/10.4161/auto.7.7.15450
- Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ 2011; In press PMID: 21475306; http://dx.doi.org/10.1038/cdd.2011.33
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA, Kroemer G. Autophagy regulation by p53. Curr Opin Cell Biol 2010; 22:181 - 185; PMID: 20044243; http://dx.doi.org/10.1016/j.ceb.2009.12.001
- Rocco JW, Li D, Liggett WH Jr, Duan L, Saunders JK Jr, Sidransky D, et al. p16INK4A adenovirus-mediated gene therapy for human head and neck squamous cell cancer. Clin Cancer Res 1998; 4:1697 - 1704; PMID: 9676844
- Shaposhnikov JD, Ratovitski EA. Polyadenylated RNA molecules and polyribosomes in tumors of chemical and viral origin. Int J Cancer 1978; 21:395 - 399; PMID: 631936; http://dx.doi.org/10.1002/ijc.2910210322