Abstract
"Shoot the driver" is the paradigm of targeted cancer therapy. However, resistance to targeted inhibitors of signaling pathways is a major problem. In part the redundancy of signaling networks can bypass targeted inhibitors and thereby reduce their biological effect. In this case the driver turns out to be one of several potential messengers and is easily replaced. Cocktails of multiple targeted inhibitors are an obvious solution. This is limited, however, by the lack of potent inhibitors and may also produce increased toxicity. Therefore we explored the direct blockade of a key biological activity downstream from multiple converging oncogenic signals. Specifically, several oncogenic signaling pathways including AKT, MAPK and PIM kinase signals converge on the activation of cap-dependent translation. In cancer cells, aberrant activation of cap-dependent translation favors the increased expression of short-lived oncoproteins like c-MYC, MCL1, CYCLIN D1 and the PIM kinases. Intriguingly, cancer cells are especially sensitive to even temporary reductions in these proteins. We will discuss our findings concerning translational inhibitor therapy in cancer.
Targeted cancer therapies are designed to block selected pathways or molecules that are required for tumor cell survival. The most successful examples are inhibitors of the BCR-ABL fusion protein that drive chronic myeloid leukemia (CML). By shutting down the activity of a single molecule driving the growth of CML cells, imatinib and its successors dasatinib and nilotinib can produce complete and sustained remissions as single agents (reviewed in ref. Citation1). However, it has proven difficult to translate this success to other cancers. In metastatic melanoma, for example, the BRAF inhibitor vemurafenib produces high response rates in patients whose tumors bear the BRAF-V600E activating mutation (reviewed in ref. Citation2). Excitement generated by these results is well-deserved, as they have opened a new treatment paradigm in a disease with few available options and a grim prognosis. Unfortunately, however, resistance to the drug generally is seen within a few months. Median progression-free survival resulting from vemurafenib for treatment-naïve metastatic melanoma patients with V600E was about six months,Citation3 compared with imatinib in CML, which kept 93% of patients progression-free at five years.Citation4 The success of TKIs in CML is still the exception and not the rule in targeted cancer therapeutics.
Resistance to targeted inhibitors is an emerging problem with multiple causes and potential solutions. While mechanisms of resistance to BRAF inhibition in melanoma remain to be elucidated, they are well-described for some targets in other cancers. Drugs against the epidermal growth factor receptor (EGFR) kinase, for example, are bypassed both by activation of downstream mediators, most prominently KRAS, and by signaling through parallel pathways like the MET oncogene (reviewed in ref. Citation5). Resistance mechanisms are varied and complex in some ways, but most boil down to the same idea: evolution has provided multiple routes to the crucial endpoints that allow sustained growth and proliferation of cancer cells. Cocktails of multiple inhibitors have been proposed to prevent or thwart resistance (reviewed in ref. Citation6), and this approach appears highly promising based on some recent preclinical studies. In prostate cancer, for example, combined blockade of androgen receptor signaling and the PI3K/AKT/mTOR pathway showed potent synergy in model systems.Citation7 Unfortunately, similar approaches against many other cancers are currently limited by the availability of potent, selective inhibitors and a need to identity which combinations will be effective. Moreover, combined toxicities from simultaneous use of multiple inhibitors will pose limits on the number and intensity of drugs that can be used.
An alternative and potentially complementary approach is to directly target the downstream biological processes that are activated by signaling pathways and that cancer cells rely on. Experiences with inhibitors of EGFR, BRAF, and other signaling molecules suggest that most tumors can reliably activate parallel or downstream messengers to thwart efficacy. So inhibiting a signaling intermediate (i.e., a messenger) allows resistance if the biological effects can be achieved via an alternate route. Ultimately, tumor cells don't depend on the messenger, often a kinase, and, instead, require a downstream biological function. This opens the possibility that targeting the critical effect directly may be an effective cancer therapy and could overcome the problem of redundant messengers.
Several inherent properties of signaling pathways are relevant to targeted therapies, their side effects and mechanisms of resistance.Citation8 For example, signaling pathways frequently converge on key activities. As explained above, this redundancy can produce resistance to targeted agents. Often redundant pathways are induced via feedback mechanisms, which provide robust signals and can also bypass selective inhibitors. Signals also diverge, however, and seemingly parallel pathways therefore also produce pleiotropic and non-overlapping effects. It is possible not all activities are required by cancer cells. Hence, an upstream block may produce toxicities, in part, by blocking activities that are not strictly required by tumor cells as much as by normal tissues. Finally, unlike metabolic pathways, where a limiting substrate is passed down, signaling cascades amplify signals toward a key activity, and the initial signal or message is both energetically “cheap” and infinitely recurrent. Accordingly, it is conceivable that direct block of the downstream effects provide an alternative or complementary approach to targeting upstream signaling molecules.
Multiple oncogenic signals, including the PI3K, MAPK/ERK and PIM kinase pathways converge on the activation of cap-dependent translation, the process by which most capped mRNAs are translated into proteins (reviewed in ref. Citation9). Signaling pathways control the availability of the cap-binding protein eIF4E that is the limiting component of the multimeric translation complex eIF4F, which also includes scaffolds (eIF4G) and RNA helicase activities (eIF4A).Citation10–Citation12 The complex ultimately mediates loading of mRNAs onto ribosomes. Availability of the eIF4E factor is especially important for mRNAs with long and structured 5′ UTRs. These include, in particular, short-lived cell cycle regulators and oncoproteins. Hence, regulation of eIF4E via upstream signals provides an immediate level of expression control that directly controls levels of proteins, including c-MYC, cyclin D1, BCL2, MCL1 and PIM1.Citation13–Citation17 Cancer cells require continuous expression of these proteins. For example, even brief loss of MYC expression produces widespread cell death in several cancers but only produces reversible cell cycle arrest in normal tissues.Citation18 Hence, the increased requirement for the continuous translation of oncoproteins in cancer cells may provide a therapeutic window for inhibitors of capdependent translation.
We recently investigated the therapeutic potential of directly blocking capdependent translation in non-Hodgkin lymphoma (NHL).Citation15,Citation16,Citation19 Rapalogs, inhibitors of mTORC1, the upstream activator of cap-dependent translation, have been extensively studied in NHL in clinical trials (reviewed in ref. Citation20). However, their activity has, overall, been modest, and our results implicate mTORC1-independent activation of translation by PIM kinases as one mechanism of rapalog resistance in NHLs.Citation16 The PIM family kinases are active upon expression and do not require activating modifications. They have been known for some time to be able to promote phosphorylation of 4E-BP1 in a manner resistant to rapamycin.Citation21,Citation22 We now report expression of PIM1 and/or PIM2 in more than 60% of diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL) and more than 75% of mantle cell and small lymphocytic lymphomas. Our study found PIM expression (either PIM1 or PIM2) was associated with worse time to event and overall survival in FL, while another recent report points to PIM2 as a driver of aggressive disease in activated B-cell type DLBCL.Citation23 In addition, two recent studies of chromosomal translocations mediated by activation-induced deaminase (AID) identified PIM1 as a frequent target.Citation24,Citation25 In sum, expression of PIM kinases is common in NHLs, may be associated with a more aggressive clinical course and exemplifies how the redundancy of messaging molecules can bypass the clinical activity of selective signaling inhibitors.
In experimental systems, we found that direct blockade of cap-dependent translation was highly effective against lymphomas with redundant PI3K/AKT and PIM signals.Citation16 Briefly, using both a constitutively active mutant 4E-BP1 allele that blocks eIF4E activity and a small molecule inhibitor of the eIF4A helicase, silvestrol, we were able to completely restore rapalog sensitivity in lymphomas engineered to express PIM2 kinase activity. Mechanistically, we found that silvestrol dramatically reduced the translation of critical oncoproteins, including c-MYC, cyclin D1 and MCL1. Interestingly, silvestrol also blocked the translation of both PIM kinases themselves. Moreover, consistent with prior reports, silvestrol treatment at an effective dose was well-tolerated in animals, and we observed no frank toxicity.Citation19 Hence, blocking cap-dependent translation disrupts upstream signaling molecules, the PIM kinases and also key oncoproteins commonly considered “undruggable” oncoproteins, including c-MYC.
Silvestrol worked dramatically better than inhibition of the upstream kinases. Briefly, we tested the SuperGen Inc. PIM kinase inhibitors SGI-1776 and SGI-1773 side by side with silvestrol in a panel of these PIM-expressing human NHL cell lines. Notably, SGI-1776 is the only PIM inhibitor that has entered clinical trials, although these had to be discontinued due to cardiac toxicity of the compound (SuperGen press release, 2010). In any case, silvestrol showed in vitro potency at IC50 of less than 10 nM in all cases, and the PIM kinase inhibitors were 100 to 1,000 times less active. These results highlight some problems associated with the “inhibitor cocktail” approach and indicate a complementary strategy that includes direct blockade of a key biological activity, in this instance, cap-dependent translation of oncoproteins.
Silvestrol is not the only means to block cap-dependent translation, and others are reviewed in reference Citation9. These include antisense oligonucleotides against eIF4E and peptide inhibitors of eIF4F complex formation, though neither has entered clinical trials. Silvestrol is the most well-studied of several compounds that emerged from library screens for ability to disrupt the function of the eIF4F subunit eIF4A, an RNA helicase required for its ability to promote mRNA translation. A plant-derived flavagline, silvestrol has shown activity against a variety of tumor types and can be given to mice at high enough concentrations for antitumor activity without major toxicity. The drug shows activity as a single agent against human breast and prostate cancer cell lines in xenograft experiments in nude mice.Citation26 This produced mild transient impairment of hepatic synthetic function but no toxicities producing morbidity or mortality. In genetically defined murine tumor models, silvestrol showed potent synergy with chemotherapy when used against tumors bearing translational activation due to loss of Pten or overexpression of eIf4e.Citation19 Originally isolated from Aglaia silvestris, silvestrol has a complex structure that has proved difficult to chemically synthesize in quantity. For this reason, the parent compound is not an ideal clinical drug candidate. Efforts are underway by Drs. Pelletier (McGill) and Porco (Boston University) to develop analogs with more efficient synthesis profiles and that retain its biochemical properties. In sum, cap-dependent translation is a promising drug target alternate to mTORC1 and upstream kinase inhibitors.
Outlook
Shooting the driver may not be the only option in targeted therapy. Our study is a successful example of blocking cap-dependent translation in cancer as an alternate approach to targeting the upstream kinases.Citation16 However, many questions remain.
What about toxicity of blocking translation? Cap-dependent translation is a fundamental biological process in cancer and normal cells, and it seems surprising that its transient inactivation is tolerated in vivo. Temporary blockade of cap-dependent translation, however, affects primarily ephemeral oncoproteins, including c-MYC, cyclinD, MCL1 and the PIM kinases. Data on the transient inactivation of c-MYC indicate selective effects on cancer cells,Citation18 but exactly why cancer cells are more sensitive to these effects than normal regenerative tissues is not clear.
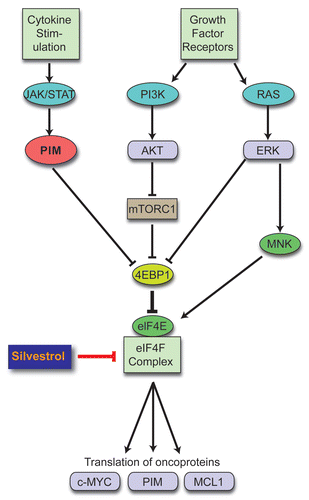
How broadly relevant is blocking translation in cancer? Translation is a key output of signaling pathways, including RAS, PI3K/AKT and PIM, that are activated in most (if not all) cancers (). We have limited data, however, on the effects of blocking the translational output of these pathways in cancer. Besides our study in lymphoma, Cencic et al.l reported activity against human breast and prostate cancer cell lines both in vitro and in vivo.Citation26
What are optimal combination strategies? Our data indicate that combination of silvestrol with rapalogs may produce a one-two punch by blocking both mTORC-dependent and -independent activation of translation. Other studies on silvestrolCitation19 or rapamycinCitation11 indicate potent synergy of translation inihibition with DNA damaging agents. Clearly, further work is needed to integrate a new therapeutic approach with existing concepts.
While challenges remain, our study provides proof of concept that direct inhibition of a key output of multiple signaling pathways provides a conceptual and therapeutically feasible alternative to targeting multiple signaling molecules.
Figures and Tables
Figure 1 Converging pathways. Multiple oncogenic signals activate cap-dependent translation. Our study shows how targeting cap-dependent translation can bypass multiple upstream signals simultaneously and knocks down expression of short-lived translationally regulated oncoproteins. Our strategy represents an alternative or possibly complementary approach to cocktails of multiple targeted inhibitors in cancer therapy.
Acknowledgments
This work is supported by grants from the NCI (R01-CA142798-01), and a P30 supplemental award (H.G.W.), the Leukemia Research Foundation (H.G.W.), the Louis V. Gerstner Foundation (H.G.W.), the WLBH Foundation (H.G.W.), the Society of MSKCC (H.G.W.), the Starr Cancer Consortium grant I4-A410 (H.G.W.), the Charles A. Dana Foundation (J.H.S.), the Lymphoma Research Foundation (J.H.S.), the ASCO Cancer Foundation (J.H.S.), the MSKCC Translational-Integrative Medicine Research Fund (J.H.S.) and the Lacher Foundation (J.H.S.).
References
- Kantarjian HM, Baccarani M, Jabbour E, Saglio G, Cortes JE. Second-generation tyrosine kinase inhibitors: the future of frontline CML therapy. Clin Cancer Res 2011; 17:1674 - 1683; PMID: 21307148; http://dx.doi.org/10.1158/1078-0432.CCR-10-2922
- Vultur A, Villanueva J, Herlyn M. Targeting BRAF in advanced melanoma: a first step toward manageable disease. Clin Cancer Res 2011; 17:1658 - 1663; PMID: 21447722; http://dx.doi.org/10.1158/1078-0432.CCR-10-0174
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 2011; 364:2507 - 2516; PMID: 21639808; http://dx.doi.org/10.1056/NEJMoa1103782
- Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355:2408 - 2417; PMID: 17151364; http://dx.doi.org/10.1056/NEJMoa062867
- Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res 2008; 14:2895 - 2899; PMID: 18483355; http://dx.doi.org/10.1158/1078-0432.CCR-07-2248
- Sawyers CL. Cancer: mixing cocktails. Nature 2007; 449:993 - 996; PMID: 17960228; http://dx.doi.org/10.1038/449993a
- Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011; 19:575 - 586; PMID: 21575859; http://dx.doi.org/10.1016/j.ccr.2011.04.008
- Alberghina L, Hofer T, Vanoni M. Molecular networks and system-level properties. J Biotechnol 2009; 144:224 - 233; PMID: 19616593; http://dx.doi.org/10.1016/j.jbiotec.2009.07.009
- Blagden SP, Willis AE. The biological and therapeutic relevance of mRNA translation in cancer. Nature reviews. Nat Rev Clin Oncol 2011; 8:280 - 291
- Lazaris-Karatzas A, Montine KS, Sonenberg N. Malignant transformation by a eukaryotic initiation factor subunit that binds to mRNA 5′ cap. Nature 1990; 345:544 - 547; PMID: 2348862; http://dx.doi.org/10.1038/345544a0
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428:332 - 337; PMID: 15029198; http://dx.doi.org/10.1038/nature02369
- Livingstone M, Atas E, Meller A, Sonenberg N. Mechanisms governing the control of mRNA translation. Phys Biol 2010; 7:21001; PMID: 20463379; http://dx.doi.org/10.1088/1478-3975/7/2/021001
- Rhoads RE, Joshi-Barve S, Rinker-Schaeffer C. Mechanism of action and regulation of protein synthesis initiation factor 4E: effects on mRNA discrimination, cellular growth rate and oncogenesis. Prog Nucleic Acid Res Mol Biol 1993; 46:183 - 219; PMID: 8234784; http://dx.doi.org/10.1016/S0079-6603(08)61022-3
- Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis 2003; 20:265 - 273; PMID: 12741684; http://dx.doi.org/10.1023/A:1022943419011
- Wendel HG, Silva RL, Malina A, Mills JR, Zhu H, Ueda T, et al. Dissecting eIF4E action in tumorigenesis. Genes Dev 2007; 21:3232 - 3237; PMID: 18055695; http://dx.doi.org/10.1101/gad.1604407
- Schatz JH, Oricchio E, Wolfe AL, Jiang M, Linkov I, Maragulia J, et al. Targeting cap-dependent translation blocks converging survival signals by AKT and PIM kinases in lymphoma. J Exp Med 2011; 208:1799 - 1807; PMID: 21859846; http://dx.doi.org/10.1084/jem.20110846
- Hoover DS, Wingett DG, Zhang J, Reeves R, Magnuson NS. Pim-1 protein expression is regulated by its 5′-untranslated region and translation initiation factor elF-4E. Cell Growth Differ 1997; 8:1371 - 1380; PMID: 9419425
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, et al. Modelling Myc inhibition as a cancer therapy. Nature 2008; 455:679 - 683; PMID: 18716624; http://dx.doi.org/10.1038/nature07260
- Bordeleau ME, Robert F, Gerard B, Lindqvist L, Chen SM, Wendel HG, et al. Therapeutic suppression of translation initiation modulates chemosensitivity in a mouse lymphoma model. J Clin Invest 2008; 118:2651 - 2660; PMID: 18551192
- Schatz JH. Targeting the PI3K/AKT/mTOR Pathway in Non-Hodgkin's Lymphoma: Results, Biology and Development Strategies. Curr Oncol Rep 2011; 13:398 - 406; PMID: 21755275; http://dx.doi.org/10.1007/s11912-011-0187-7
- Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev 2003; 17:1841 - 1854; PMID: 12869584; http://dx.doi.org/10.1101/gad.1105003
- Hammerman PS, Fox CJ, Birnbaum MJ, Thompson CB. Pim and Akt oncogenes are independent regulators of hematopoietic cell growth and survival. Blood 2005; 105:4477 - 4483; PMID: 15705789; http://dx.doi.org/10.1182/blood-2004-09-3706
- Gómez-Abad C, Pisonero H, Blanco-Aparicio C, Roncador G, Gonzalez-Menchen A, Martinez-Climent JA, et al. PIM2 inhibition as a rational therapeutic approach in B-cell lymphoma. Blood 2011; In press PMID: 21937691; http://dx.doi.org/10.1182/blood-2011-03-344374
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide Translocation Sequencing Reveals Mechanisms of Chromosome Breaks and Rearrangements in B Cells. Cell 2011; 147:107 - 119; PMID: 21962511; http://dx.doi.org/10.1016/j.cell.2011.07.049
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-Capture Sequencing Reveals the Extent and Nature of Chromosomal Rearrangements in B Lymphocytes. Cell 2011; 147:95 - 106; PMID: 21962510; http://dx.doi.org/10.1016/j.cell.2011.07.048
- Cencic R, Carrier M, Galicia-Vazquez G, Bordeleau ME, Sukarieh R, Bourdeau A, et al. Antitumor activity and mechanism of action of the cyclopenta[b] benzofuran, silvestrol. PLoS ONE 2009; 4:5223; PMID: 19401772; http://dx.doi.org/10.1371/journal.pone.0005223