Abstract
Targeting DNA repair with poly(ADP-ribose) polymerase (PARP) inhibitors has shown a broad range of anti-tumor activity in patients with advanced malignancies with and without BRCA deficiency. It remains unclear what role p53 plays in response to PARP inhibition in BRCA-proficient cancer cells treated with DNA damaging agents. Using gene expression microarray analysis, we find that DNA damage response (DDR) pathways elicited by veliparib (ABT-888), a PARP inhibitor, plus topotecan comprise the G1/S checkpoint, ATM, and p53 signaling pathways in p53-wildtype cancer cell lines and BRCA1, BRCA2 and ATR pathway in p53-mutant lines. In contrast, topotecan alone induces the G1/S checkpoint pathway in p53-wildtype lines and not in p53-mutant cells. These responses are coupled with G2/G1 checkpoint effectors p21CDKN1A upregulation, and Chk1 and Chk2 activation. The drug combination enhances G2 cell cycle arrest, apoptosis and a marked increase in cell death relative to topotecan alone in p53-wildtype and p53-mutant or -null cells. We also show that the checkpoint kinase inhibitor UCN-01 abolishes the G2 arrest induced by the veliparib and topotecan combination and further increases cell death in both p53-wildtype and -mutant cells. Collectively, PARP inhibition by veliparib enhances DDR and cell death in BRCA-proficient cancer cells in a p53-dependent and -independent fashion. Abrogating the cell-cycle arrest induced by PARP inhibition plus chemotherapeutics may be a strategy in the treatment of BRCA-proficient cancer.
Introduction
Poly(ADP-ribose) polymerases (PARPs) are nuclear proteins that bind to DNA strand breaks.Citation1,Citation2 Upon binding, the activated PARPs cleave NAD+ into nicotinamide and ADP-ribose that is polymerized onto nuclear proteins including PARP-1 itself (auto-ADP-ribosylation), DNA-binding proteins and histones.Citation3 Poly(ADP-ribosyl)ation causes chromatin relaxation and functions as a scaffold that facilitates the recruitment and assembly of the DNA repair proteins.Citation4 Inhibition of PARP activity prevents the recruitment of XRCC1 and the repair of DNA single strand breaks (SSBs) by base excision repair.Citation5 The accumulation of SSBs produces DNA double-strand breaks (DSBs) during DNA replication.Citation6,Citation7 In addition, PARP-1 is involved in the repair of DSBs through non-homologous end joining.Citation8,Citation9 The loss of function of BRCA1 or BRCA2, which is implicated in DSB repair through homologous recombination (HR),Citation10 sensitizes cells to PARP inhibition through a mechanism of synthetic lethality.Citation11–Citation13 Recent clinical studies have shown that clinical activity of PARP inhibitors in combination with chemotherapy is not limited to the tumors with BRCA deficiency.Citation14–Citation16
The cellular responses to DNA damage include DNA repair, transcriptional alteration, DNA damage checkpoint activation and apoptosis.Citation17 p53 is a nuclear transcription factor encoded by the Tp53 gene, which is mutated in more than 50% of human tumors.Citation18 p53 plays important roles in the cellular responses to DNA damage, regulation of cell cycle and genomic stability.Citation19 p53 also participates in the processes of base excision repair and nucleotide excision repair,Citation20 and wild-type p53 downregulates Rad51 expression in response to DSBs.Citation21 It also controls the entry of cells into mitosis when they enter G2 with damaged DNA.Citation22
Previous studies have focused on the roles of PARPs in SSB or DSB repairs, and recently on DNA repair defects such as BRCA deficiency as well as loss of function of other proteins with roles in DSB repair.Citation13 What role p53 may play in response to PARP inhibition in BRCA-proficient cancer cells treated with DNA damaging agents remains unclear. Veliparib (ABT-888) is a potent small molecule PARP inhibitor, which was developed by the Abbott Laboratories and is in clinical trials.Citation23–Citation26 In the present study, we use cDNA microarray analyses to identify and delineate the molecular pathways implicated in the responses to veliparib plus topotecan compared with topotecan alone in cells with various p53 status. We find that PARP inhibition markedly enhances the cellular DNA damage responses by alteration of multiple DNA damage response pathways and the death of cancer cells in a p53-dependent and -independent manner. The alteration and activation of crucial cell cycle-related genes across the identified pathways in association with DNA damage responses have been validated and are discussed.
Results
PARP inhibition enhances DNA damage responses via multiple damage response pathways in p53-dependent and -independent fashion.
To identify transcripts significantly changed by treatments in the pair of HCT-116 p53+/+ and p53−/− cells, we compared gene expression profiles between treatments with topotecan alone and veliparib plus topotecan, and vehicle control by Affymetrix MAS 5.0 Statistical Comparison Analysis. p21CDKN1A and BTG2 transcripts relevant to DNA damage response were increased by topotecan in p53+/+ cells (Table S1A). More transcripts, in contrast, were significantly upregulated by the combination treatment. Those included PA26, DDB2, Bax, FasR and MDM2 in addition to p21CDKN1A and BTG2 (Table S1B). In p53−/− cells, no changes were detected in the context of DNA damage response by topotecan alone (Table S1C), whereas veliparib plus topotecan treatment induced RAD51, a critical DSB repair gene, and CDC2 as well as CDC6 (Table S1D).Citation27 Therefore, the veliparib and topotecan combination induces more significant gene expression alterations relevant to DNA damage response than topotecan alone, and those alterations are both dependent and independent of p53.
The functional class scoring analysis as described in the Materials and Methods was performed to identify global DNA damage responses to veliparib plus topotecan or topotecan alone vs. vehicle control in cancer cells with endogenous wild-type and mutant p53. In p53-wild-type cells, G1/S checkpoint pathway alterations were detected following exposure to topotecan alone ( and S2A). ELAC1, p21CDKN1A, ATR and CDK2 were the top altered transcripts in this pathway. In response to veliparib plus topotecan, changes included the ATM and p53 signaling pathways plus the G1/S checkpoint pathway ( and S2B). The top differentially expressed genes included p21CDKN1A, SMAD3, CDK2, CCNA1, MDM2 and RBBP8. No pathway effect relevant to DNA damage response was observed in p53-mutant lines when exposed to topotecan alone (Table S2C). The BRCA1, BRCA2 and ATR pathway, by contrast, was induced by the two drug combination, in which RAD50, ATR, GAS2 and FANCF were differentially expressed ( and S2D).
PARP inhibition enhances cell cycle arrest in G2 induced by topotecan in p53-wild-type and p53-mutant or -null cells.
To investigate whether the cell cycle arrest is one of the major cellular responses to treatments, we performed the cell cycle analysis by flow cytometry. Cells accumulated in G2 phase were increased in response to topotecan, and this response was further augmented in all cell lines by concurrent treatment with veliparib and topotecan (). Of note, veliparib alone at 1 µM had little effect on the cell cycle. The fact that cells were predominantly arrested in G2 phase rather than M phase was substantiated by a depletion or a near depletion in phosphorylated histone 3 (pH3), a mitosis marker ().
Next, we examined cell cycle checkpoint effectors implicated in DNA damage response such as p21CDKN1A, and phosphorylation of the checkpoint kinase 1 (Chk1) at Ser345 and of checkpoint kinase 2 (Chk2) at Thr68. Chk1 and Chk2 are critical in the molecular pathways of G1/S checkpoint, p53 and ATM signaling as well as in the BRCA1, BRCA2 and ATR pathway. The veliparib and topotecan combination enhanced expression of p21CDKN1A protein in p53-wild-type HCT-116 p53+/+ and MCF-7 cells (). The combination also enhanced activation of Chk1 in HCT116 p53+/+, MCF-7 and p53−/− cells, and Chk2 in HT-29, MDA-231 and p53−/− cells (). The key G2 or G1 phase checkpoint protein p21CDKN1A as a member of p53 and ATM signaling pathways is increased by topotecan alone and enhanced by the veliparib and topotecan combination in cells with wild-type p53. The activation of Chk1 and/or Chk2 in the ATM signaling and BRCA1, BRCA2 and ATR pathways is enhanced by veliparib in cells with wild-type, mutant or null p53 gene.
PARP inhibition augments cancer cell apoptosis and death induced by topotecan in p53-wild-type and p53-mutant or -null cells.
We also sought to examine whether this combination approach leads to the enhancement of subsequent cancer cell death and apoptosis. PARP cleavage, which is indicative of cells undergoing apoptosis, was detectable 96 h after exposure to topotecan alone and to the two drug combination (). PARP inhibition markedly enhanced topotecan-induced cancer cell death (measured as the reduced clonogenic cell survival) in p53-wild-type, as well as p53-null or -mutant cancer cells (all p ≤ 0.05, unpaired two-sided t-test; ). Noticeably, there were comparable enhancement of cell death by veliparib in HCT116 p53+/+ and p53−/− cells (45.8% vs. 43.0%; ). In addition, varying levels of enhancement of cell death were observed in p53-mutant MDA-231 and HT-29 cells. In all cases, G2 arrest by veliparib, topotecan and veliparib plus topotecan was associated with the death of cancer cells (Spearman's correlation coefficient = 0.70, p = 0.004; ). These data indicate that veliparib augments the death of cancer cells induced by topotecan. G2 arrest, despite not being causal for the cell death, reflects the collective DNA damage effects induced by the drug treatments. p53 deficiency alone does not appear to specifically confer more sensitivity or resistance to treatment with a PARP inhibitor plus topotecan or with topotecan alone ().
UCN-01 abrogates G2 arrest induced by veliparib plus topotecan in p53-wild-type and p53-mutant cells.
UCN-01, a cell cycle checkpoint kinase inhibitor, has been shown to abrogate the cell cycle checkpoints induced by DNA damaging drugs.Citation28,Citation29 To study whether UCN-01 abolishes the cell cycle G2 arrest induced by drug treatments and further enhances cell death, we performed cell cycle analysis in cells treated with veliparib plus topotecan in the presence and absence of UCN-01. UCN-01 at low dose (100 nM) nearly abrogated G2 arrest induced by veliparib plus topotecan in p53-wild-type HCT-116 and p53-mutant HT-29 cells relative to the cells treated without UCN-01 (). Importantly, UCN-01 further increased cell death (). Together, these results show that UCN-01 abolishes the cell cycle arrest induced by PARP inhibition plus topotecan and further increases cell death in both p53-wild-type and -mutant cells.
Discussion
Our findings demonstrate that PARP inhibition by the small molecular PARP inhibitor veliparib enhances the effects of a DNA damaging agent in BRCA-proficient cancer cells. In p53+/+ HCT116 cells, a set of p53 regulated gene transcripts such as p21CDKN1A, BTG2, PA26, DDB2, Bax, FasR and MDM2 were induced by veliparib plus topotecan, relative to p21CDKN1A and BTG2 by topotecan alone (Table S1B vs. S1A).Citation22,Citation30–Citation32 These are genes implicated in the regulation of cell cycle, DNA damage responses and apoptosis. In contrast, expression of RAD51 and CDC2 as well as CDC6 was selectively and significantly increased by the combination treatment in p53−/− cells. Increased expression of Rad51 and formation of Rad51 foci after DNA damage has been observed in HCT116 p53−/− cells compared with HCT116 p53+/+ cells.Citation21 Wild-type p53 may repress Rad51 transcription by binding to the p53 response element in the Rad51 promoter and inhibit Rad51 foci formation in response to DSBs. CDC2 and CDC6 are important in the cell cycle checkpoints, which are utilized by p53−/− cells to increase DNA damage responses (Table S1D). Our gene expression microarray results reveal that cellular DNA damage responses to veliparib plus topotecan tend to be centered on the genes regulated by p53 in p53+/+ cells, whereas the response primarily involves the genes related to the DNA repair and cell cycle in p53−/− cells. Although the set of genes in response to DNA damage are substantially different, similar levels of enhancement of cell death were observed in the pair of HCT116 p53+/+ and p53−/− cells (). p53 status alone does not appear to play a critical role in the enhanced cell death by the PARP inhibitor in combination with topotecan. In addition, more gene expression alterations relevant to DNA damage response are induced by veliparib in combination with topotecan than topotecan alone.
The combination treatment also widens the cellular DNA damage responses in cancer cells with endogenous wild-type as well as mutant p53 gene. Veliparib plus topotecan elicits p53, ATM signaling and G1/S checkpoint pathways relative to the latter pathway by topotecan alone (Table S2A and S2B). The three pathways comprise an array of highly coordinated genes with interrelated functions on the cell cycle checkpoints, DNA damage response signaling, DNA repair and apoptosis. In p53-mutant cells, the BRCA1, BRCA2 and ATR pathway is induced by the drug combination but not by topotecan alone. This pathway consists of genes with functions related primarily to the DNA repair and DNA damage response signaling. For instance, RAD50 is a critical component of the MRE11-RAD50-NBS1 (MRN) complex, which is involved in sensing and localizing to the DSB sites, leading to the activation of DNA damage response.Citation13 Moreover, PARP inhibition augments expression or activation levels of several critical genes related to DNA damage response and cell cycle checkpoint (e.g., p21, Chk1 and/or Chk2). In essence, more genes and pathways responded to veliparib plus topotecan compared with topotecan alone in an interrelated DNA damage response network that is both dependent and independent of p53 (Tables S2A–D, and ). As a result of the increase in DNA damage responses to the combination treatment, cell death is substantially enhanced in both p53-wildtype and p53-mutant or -null cells (). Additionally, the two-drug combination elicits other molecular pathways related to apoptosis and stress response such as Fas signaling and TNF/stress related signaling as well as stress induction of HSP regulation in p53-wild-type cells (Table S2B).
Subsequently, we focused on validating the critical genes of the cell cycle checkpoints in the identified molecular pathways. G2 arrest in cancer cells with wild-type p53 gene was associated with the upregulation of p21CDKN1A, a crucial effector of the p53 signaling pathway as well as the activation of Chk1 and/or Chk2.Citation28,Citation33,Citation34 The G2 and/or S arrest(s) in those cells with mutant or deficient p53 gene were primarily associated with the activation of Chk1 and/or Chk2. The two cell cycle checkpoint kinases are members of the ATM signaling and BRCA1, BRCA2 and ATR pathways (Table S1B and S1D). Our results and published data suggest that p53-regulated expression of p21CDKN1A and activation of Chk1/or Chk2 are associated with G2 arrest in p53-wild-type cells.Citation17–Citation19 Chk1 and/or Chk2 are largely associated with G2 or S arrest in p53-mutant or -null cells.Citation35
Importantly, we demonstrate that the G2 cell cycle arrest, as a collective effect of DNA damage and DNA damage response to veliparib, topotecan and veliparib plus topotecan, is associated with the death of cancer cells. The G2 arrest allows the cells to repair the damaged DNA as a protective measure for cancer cells as well as for normal human fibroblasts and epithelial cells,Citation36 and, therefore, G2 arrest limits the treatment efficacy of DNA damaging drugs and ionizing radiation.Citation28,Citation37 UCN-01 at low dose (100 nM) nearly abrogated G2 arrest induced by veliparib plus topotecan in HCT-116 p53+/+ and p53-mutant HT-29 cells, which led to a further increase in cell death (). Thus, abrogation of the cell cycle arrest, which is a conserved protective means of cells, further sensitizes the cancer cells to the veliparib and topotecan combination treatment.
In summary, our findings using a gene expression profiling approach delineate the cellular responses to the combination of a PARP inhibitor and a DNA damaging agent in p53-proficient and -deficient, and BRCA-proficient cancer cells. The results increase our understanding of how PARP inhibition augments the efficacy of DNA damaging agents. They may have implications for the design of treatment strategies by pharmacological PARP inhibition using small molecule PARP inhibitors in combination with additional agents that specifically target the key members of cell cycle checkpoints in the treatment of BRCA-proficient cancer.
Materials and Methods
Cells, drugs and drug treatment.
Veliparib and topotecan were obtained from the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute. Veliparib was dissolved in DMSO and reconstituted to 10 mM with H2O; aliquots of the drug were stored frozen at −20°C. Topotecan at 1 mM in H2O was stored at −20°C until use. UCN-01 (7-hydroxystaurosporine) was purchased from Calbiochem; aliquots at 1 mM in DMSO were stored at −20°C until use. p53-wild-type human HCT-116 (p53+/+; BRCA2-mutant but HR proficient) colon carcinoma cellsCitation38 and its p53-null (p53−/−) isogenic cells were kindly provided by Dr. Bert Vogelstein at Johns Hopkins Sydney Kimmel Comprehensive Cancer Center.Citation22 Breast cancer cell lines MCF-7 (p53-wild type and BRCA-wild type) and MDA-MB-231 (MDA-231; p53mutant and BRCA-wild type) and colorectal cancer cell line HT-29 (p53-mutant and BRCA-wild type) were obtained from ATCC.Citation39 Exponentially growing cells in 10% fetal bovine serum RPMI at 60–70% confluence were treated with 10 nM topotecan alone, 1 µM veliparib alone, or in combination for 24 h. The treated cells were subjected to gene expression microarray, protein gel blotting, cell survival or cell cycle analysis.
RNA extraction, microarray hybridization and analysis.
Total RNA extraction and cRNA synthesis, microarray hybridization have been described in detail previously in reference Citation40. All data were uploaded into the microarray statistical package BRB-ArrayTools version 3.6.0 for filtering and statistical analysis (http://linus.nci.nih.gov). All analyses were performed on data from log2-transformed median-normalized expression values. To perform analysis, the numbers of genes were reduced by filtering out genes with less than 50% of expression data and those with less than a 2.0-fold change in either direction from the gene's median value. A set of 8,905 genes remaining representing 171 BioCarta pathways was used for further data analyses.
Pathways that had more genes differentially expressed between topotecan and vehicle treatment, and between veliparib plus topotecan and vehicle treatment were identified by paired t-test in p53-wild-type (MCF-7 and HCT-116) and mutant p53 lines (MDA-MB-231 and HT-29), respectively. These were performed using a functional class scoring analysis as described previously in BRB-ArrayTools.Citation40–Citation42 Functional class scoring is a more powerful method of identifying differentially expressed gene classes than the more common over-representation analysis or annotation of gene lists based on individually analyzed genes.Citation40,Citation43 The pathways differentially expressed between the differences in phenotypic classes by treatments with a p value less than 0.05 for LS or KS statistic were reported.
To identify significantly changed transcripts in the pair of HCT-116 p53+/+ and p53−/−, each probe set on the drug-treated array (11 probes per probe-set) was compared with the corresponding vehicle-treated one using Wilcoxon's signed rank test. It was defined as statistically significant if p < 0.003 for increase and p > 0.997 for decrease in expression by Affymetrix MAS 5.0 Statistical Comparison Analysis in GCOS.Citation40
Protein gel blots.
Equal numbers of cells were lysed in Laemmli sample buffer and subjected to electrophoresis.Citation44 Nitrocellulose filters were probed with antibodies to p21 in 1:200 dilution (clone DO7, Santa Cruz Inc.), to phospho-Chk1 (Ser345), phospho-Chk2 (Thr68), phosphorylated histone H3 and cleaved PARP in 1:1,000 dilutions (Cell Signaling Technology), respectively. β-actin was probed as the protein loading control (antibody dilution 1:5,000; Sigma). Reactive proteins were viewed using enhanced chemiluminescence (Pierce Chemical Co.).
Cell cycle analysis.
Cell cycle analyses were performed as described previously in reference Citation45. Briefly, after harvesting and fixation in 70% ethanol, the cells were treated with RNase (5 µg/ml) and stained with propidium iodide (50 µg/ml). Approximately 20,000 stained cells were analyzed for DNA content by fluorescence-activated cell sorting (FACS) in a FACScan (SOBR model, Becton Dickinson Instrument). The cells in G1, S or G2/M fraction of the cell cycle were quantified with CellQuest (Becton Dickinson).
Clonogenic cell survival assay.
After 24 h of drug treatment as indicated, cells were washed and plated into 6-well plates (each condition in triplicate) for 10 to 14 d. After fixation and staining with 0.1% crystal violet (Sigma-Aldrich), colonies consisting of >50 cells were counted. The relative death fraction of the cells was calculated as [(average colonies of drug treated - vehicletreated)/vehicle-treated] × 100.
Statistical analysis.
The unpaired t-test was used to evaluate the statistical differences in cell death between topotecan and combination treatment in all five lines as well as the differences in cell death between treatments with UCN-01 and without UCN-01 after veliparib plus topotecan treatment in HCT-116 and HT29 cells. The Spearman's rank correlation test was utilized to assess the association between G2 fraction (ratio of drug-treated/vehicle-treated) and % death of cancer cells by veliparib, topotecan and veliparib/topotecan combination treatment in five cell lines. In all analyses, p values were two-sided.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 Treatment effect on cell cycle and key cell cycle checkpoint proteins and kinases. (A) Cell cycle effects by treatments examined by flow cytometry. Cells were exposed to 1 µM veliparib, 10 nM topotecan alone or in combination for 24 h. Numbers above G1, S and G2 peaks indicate the percentage of cells in each phase of the cell cycle. Shown are the representative data of three independent experiments. (B) Protein gel blotting analyses of p21CDKN1A expression, activation of Chk1 and Chk2, and histone H3 phosphorylation (pH 3) after treatments for 24 h. Shown are the representative results of at least two independent experiments. ABT, veliparib; TPT, topotecan.
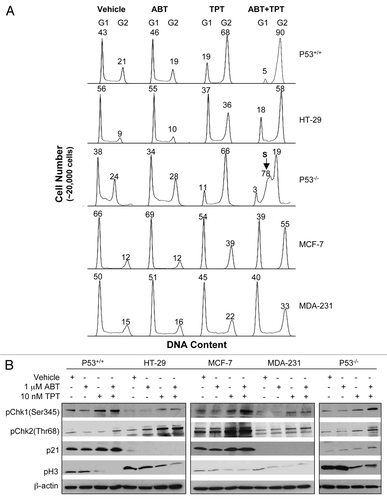
Figure 2 Enhancing effects of veliparib on apoptosis and cell death induced by topotecan. (A) Cleaved PARP detected by protein gel blotting 96 h after treatments. (B) Relative % changes in cell death in response to veliparib (gray bar), topotecan alone (white bar) or in combination (black bar) assessed by clonogenic assays. Shown are the representative data of two independent experiments. (C) Relationship between the cell death and G2 arrest (ratio of drug-treated/vehicle-treated) in response to veliparib, topotecan or in combination in HCT-116 p53+/+ (dot), HT-29 (diamond), MCF-7 (square), MDA-MB-231 (triangle), and p53−/− (star). ABT, veliparib; cl, cleaved; CC, correlation coefficient; TPT, topotecan.
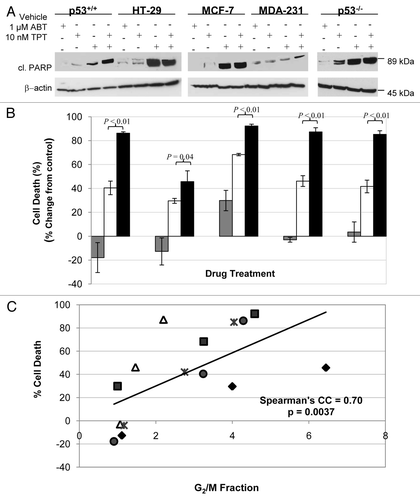
Figure 3 Effect of UCN-01 on the cell cycle distribution and cell death in cells treated with veliparib plus topotecan. After 24 h exposure to veliparib plus topotecan, the cultures were washed, and UCN-01 (100 nM) or vehicle was added for an additional 18 h. (A) Cell cycle effects of veliparib in combination with topotecan in the presence and absence of UCN-01. The numbers above G1 and G2 peaks indicate the percentage of cells in each phase of the cell cycle. (B) Percent change, relative to vehicle treatment without UCN-01, of cell death by veliparib plus topotecan without UCN-01 (gray bar), and with UCN-01(black bar) assessed by clonogenic assays. Shown are the representative data of two independent experiments. ABT, ABT-888; TPT, topotecan.
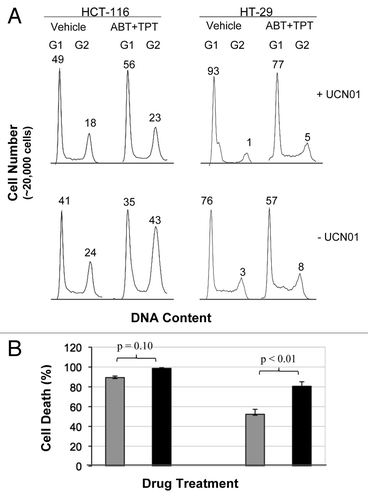
Table 1 The differentially expressed genes in the cell cycle
Table 2 The differentially expressed genes in role of BRCA1, BRCA2 and ATR in cancer susceptibility pathwayTable Footnote* by veliparib plus topotecan in p53-mutant lines
Additional material
Download Zip (595 KB)Acknowledgments
The HCT116 p53-null isogenic cells were kindly provided by Dr. Bert Vogelstein at the Johns Hopkins Sydney Kimmel Comprehensive Cancer Center, Baltimore, MD. We thank Barbara Taylor at the Flow Cytometry Core, Center for Cancer Research, National Cancer Institute for her assistance in cell cycle analysis. This work was supported by the Division of Cancer Treatment and Diagnosis and the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
References
- Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis and disease. Am J Hum Genet 1999; 64:1282 - 1288; PMID: 10205258; http://dx.doi.org/10.1086/302389
- Yélamos J, Schreiber V, Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med 2008; 14:169 - 178; PMID: 18353725; http://dx.doi.org/10.1016/j.molmed.2008.02.003
- Rouleau M, Aubin RA, Poirier GG. Poly(ADP-ribosyl) ated chromatin domains: access granted. J Cell Sci 2004; 117:815 - 825; PMID: 14963022; http://dx.doi.org/10.1242/jcs.01080
- Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 2000; 275:40974 - 40980; PMID: 11016934; http://dx.doi.org/10.1074/jbc.M006520200
- Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, et al. DNA polymerasebeta-mediated long patch base excision repair. Poly(ADP-ribose)polymerase-1 stimulates strand displacement DNA synthesis. J Biol Chem 2001; 276:32411 - 32414; PMID: 11440997; http://dx.doi.org/10.1074/jbc.C100292200
- Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004; 23:3872 - 3882; PMID: 15021907; http://dx.doi.org/10.1038/sj.onc.1207491
- Liu X, Shi Y, Guan R, Donawho C, Luo Y, Palma J, et al. Potentiation of temozolomide cytotoxicity by poly(ADP)ribose polymerase inhibitor ABT-888 requires a conversion of single-stranded DNA damages to double-stranded DNA breaks. Mol Cancer Res 2008; 6:1621 - 1629; PMID: 18922977
- Veuger SJ, Curtin NJ, Smith GC, Durkacz BW. Effects of novel inhibitors of poly(ADP-ribose) polymerase-1 and the DNA-dependent protein kinase on enzyme activities and DNA repair. Oncogene 2004; 23:7322 - 7329; PMID: 15286704; http://dx.doi.org/10.1038/sj.onc.1207984
- Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J 2006; 25:1305 - 1314; PMID: 16498404; http://dx.doi.org/10.1038/sj.emboj.7601015
- Hawtin RE, Stockett DE, Wong OK, Lundin C, Helleday T, Fox JA. Homologous recombination repair is essential for repair of vosaroxin-induced DNA double-strand breaks. Oncotarget 2010; 1:606 - 619; PMID: 21317456
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 921; PMID: 15829967; http://dx.doi.org/10.1038/nature03445
- Boss DS, Beijnen JH, Schellens JH. Inducing Synthetic Lethality using PARP Inhibitors. Curr Clin Pharmacol 2010; 5:192 - 195; PMID: 20406170; http://dx.doi.org/10.2174/157488410791498798
- Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle 2011; 10:1192 - 1199; PMID: 21487248; http://dx.doi.org/10.4161/cc.10.8.15273
- Plummer R, Woll P, Fyfe D, Boddy AV, Griffin M, Hewitt P, et al. A phase I and pharmacokinetic study of ixabepilone in combination with Carboplatin in patients with advanced solid malignancies. Clin Cancer Res 2008; 14:8288 - 8294; PMID: 19088046; http://dx.doi.org/10.1158/1078-0432.CCR-08-0471
- Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res 2008; 14:7917 - 7923; PMID: 19047122; http://dx.doi.org/10.1158/1078-0432.CCR-08-1223
- Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol 2011; 12:852 - 861; PMID: 21862407; http://dx.doi.org/10.1016/S1470-2045(11)70214-5
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39 - 85; PMID: 15189136; http://dx.doi.org/10.1146/annurev.biochem.73.011303.073723
- Joerger AC, Fersht AR. Structure-function-rescue: the diverse nature of common p53 cancer mutants. Oncogene 2007; 26:2226 - 2242; PMID: 17401432; http://dx.doi.org/10.1038/sj.onc.1210291
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 310; PMID: 11099028; http://dx.doi.org/10.1038/35042675
- Smith ML, Seo YR. p53 regulation of DNA excision repair pathways. Mutagenesis 2002; 17:149 - 156; PMID: 11880544; http://dx.doi.org/10.1093/mutage/17.2.149
- Arias-Lopez C, Lazaro-Trueba I, Kerr P, Lord CJ, Dexter T, Iravani M, et al. p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep 2006; 7:219 - 224; PMID: 16322760; http://dx.doi.org/10.1038/sj.embor.7400587
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 1501; PMID: 9822382; http://dx.doi.org/10.1126/science.282.5393.1497
- Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res 2007; 13:2728 - 2737; PMID: 17473206; http://dx.doi.org/10.1158/1078-0432.CCR-06-3039
- Kummar S, Kinders R, Gutierrez ME, Rubinstein L, Parchment RE, Phillips LR, et al. Phase 0 clinical trial of the poly (ADP-ribose) polymerase inhibitor ABT-888 in patients with advanced malignancies. J Clin Oncol 2009; 27:2705 - 2711; PMID: 19364967; http://dx.doi.org/10.1200/JCO.2008.19.7681
- Yang SX, Kummar S, Steinberg SM, Murgo AJ, Gutierrez M, Rubinstein L, et al. Immunohistochemical detection of poly(ADP-ribose) polymerase inhibition by ABT-888 in patients with refractory solid tumors and lymphomas. Cancer Biol Ther 2009; 8:2004 - 2009; PMID: 19823047; http://dx.doi.org/10.4161/cbt.8.21.9917
- Kummar S, Chen A, Ji J, Zhang Y, Reid JM, Ames M, et al. Phase I Study of PARP Inhibitor ABT-888 in Combination with Topotecan in Adults with Refractory Solid Tumors and Lymphomas. Cancer Res 2011; 71:5626 - 5634; PMID: 21795476; http://dx.doi.org/10.1158/0008-5472.CAN-11-1227
- Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M, et al. Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 2005; 25:3737 - 3751; PMID: 15831478; http://dx.doi.org/10.1128/MCB.25.9.3737-51.2005
- Yu Q, La Rose J, Zhang H, Takemura H, Kohn KW, Pommier Y. UCN-01 inhibits p53 upregulation and abrogates gamma-radiation-induced G(2)-M checkpoint independently of p53 by targeting both of the checkpoint kinases, Chk2 and Chk1. Cancer Res 2002; 62:5743 - 5748; PMID: 12384533
- Furuta T, Hayward RL, Meng LH, Takemura H, Aune GJ, Bonner WM, et al. p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene 2006; 25:2839 - 2849; PMID: 16407843; http://dx.doi.org/10.1038/sj.onc.1209313
- Rouault JP, Falette N, Guehenneux F, Guillot C, Rimokh R, Wang Q, et al. Identification of BTG2, an antiproliferative p53-dependent component of the DNA damage cellular response pathway. Nat Genet 1996; 14:482 - 486; PMID: 8944033; http://dx.doi.org/10.1038/ng1296-482
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, et al. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999; 18:127 - 137; PMID: 9926927; http://dx.doi.org/10.1038/sj.onc.1202274
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004; 303:1010 - 1014; PMID: 14963330; http://dx.doi.org/10.1126/science.1092734
- Fan J, Wray J, Meng X, Shen Z. BCCIP is required for the nuclear localization of the p21 protein. Cell Cycle 2009; 8:3019 - 3024; PMID: 19713748; http://dx.doi.org/10.4161/cc.8.18.9622
- Eymin B, Claverie P, Salon C, Leduc C, Col E, Brambilla E, et al. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol Cell Biol 2006; 26:4339 - 4350; PMID: 16705183; http://dx.doi.org/10.1128/MCB.02240-05
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 439; PMID: 11100718; http://dx.doi.org/10.1038/35044005
- Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011; 2:222 - 233; PMID: 21447859
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene 2001; 20:1803 - 1815; PMID: 11313928; http://dx.doi.org/10.1038/sj.onc.1204252
- Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med 2009; 1:315 - 322; PMID: 20049735; http://dx.doi.org/10.1002/emmm.200900041
- Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S, et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther 2006; 5:2606 - 2612; PMID: 17088437; http://dx.doi.org/10.1158/1535-7163.MCT-06-0433
- Yang SX, Simon RM, Tan AR, Nguyen D, Swain SM. Gene expression patterns and profile changes pre- and post-erlotinib treatment in patients with metastatic breast cancer. Clin Cancer Res 2005; 11:6226 - 6232; PMID: 16144925; http://dx.doi.org/10.1158/1078-0432.CCR-05-0270
- Pavlidis P, Qin J, Arango V, Mann JJ, Sibille E. Using the gene ontology for microarray data mining: a comparison of methods and application to age effects in human prefrontal cortex. Neurochem Res 2004; 29:1213 - 1222; PMID: 15176478; http://dx.doi.org/10.1023/B:NERE.0000023608.29741.45
- Yang SX, Steinberg SM, Nguyen D, Wu TD, Modrusan Z, Swain SM. Gene expression profile and angiogenic marker correlates with response to neoadjuvant bevacizumab followed by bevacizumab plus chemotherapy in breast cancer. Clin Cancer Res 2008; 14:5893 - 5899; PMID: 18794102; http://dx.doi.org/10.1158/1078-0432.CCR-07-4762
- Simon R. Analysis of DNA microarray expression data. Best Pract Res Clin Haematol 2009; 22:271 - 282; PMID: 19698933; http://dx.doi.org/10.1016/j.beha.2009.07.001
- Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, et al. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest 1999; 104:263 - 269; PMID: 10430607; http://dx.doi.org/10.1172/JCI6863
- Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J Biol Chem 2003; 278:28045 - 28051; PMID: 12732644; http://dx.doi.org/10.1074/jbc.M300554200