Abstract
Increasing chronological age is the most significant risk factor for cancer. Recently, we proposed a new paradigm for understanding the role of the aging and the tumor microenvironment in cancer onset. In this model, cancer cells induce oxidative stress in adjacent stromal fibroblasts. This, in turn, causes several changes in the phenotype of the fibroblast including mitochondrial dysfunction, hydrogen peroxide production, and aerobic glycolysis, resulting in high levels of L-lactate production. L-lactate is then transferred from these glycolytic fibroblasts to adjacent epithelial cancer cells and used as “fuel” for oxidative mitochondrial metabolism. Here, we created a new pre-clinical model system to directly test this hypothesis experimentally. To synthetically generate glycolytic fibroblasts, we genetically-induced mitochondrial dysfunction by knocking down TFAM using an sh-RNA approach. TFAM is mitochondrial transcription factor A, which is important in functionally maintaining the mitochondrial respiratory chain. Interestingly, TFAM-deficient fibroblasts showed evidence of mitochondrial dysfunction and oxidative stress, with the loss of certain mitochondrial respiratory chain components, and the over-production of hydrogen peroxide and L-lactate. Thus, TFAM-deficient fibroblasts underwent metabolic reprogramming towards aerobic glycolysis. Most importantly, TFAM-deficient fibroblasts significantly promoted tumor growth, as assayed using a human breast cancer (MDA-MB-231) xenograft model. These increases in glycolytic fibroblast driven tumor growth were independent of tumor angiogenesis. Mechanistically, TFAM-deficient fibroblasts increased the mitochondrial activity of adjacent epithelial cancer cells in a co-culture system, as seen using MitoTracker. Finally, TFAM-deficient fibroblasts also showed a loss of caveolin-1 (Cav-1), a known breast cancer stromal biomarker. Loss of stromal fibroblast Cav-1 is associated with early tumor recurrence, metastasis, and treatment failure, resulting in poor clinical outcome in breast cancer patients. Thus, this new experimental model system, employing glycolytic fibroblasts, may be highly clinically relevant. These studies also have implications for understanding the role of hydrogen peroxide production in oxidative damage and “host cell aging,” in providing a permissive metabolic microenvironment for promoting and sustaining tumor growth.
Introduction
Mitochondrial oxidative stress has long been implicated in both ”normal” aging and cancer. Located within the cell's mitochondria, the inner-membrane has a large surface area containing the enzymes for energy production, via oxidative phosphorylation. During this process (OXPHOS), reactive oxygen species (ROS) are generated as by-products of the reduction of molecular oxygen to water. In addition to ROS, the mitochondrial respiratory chain can also produce nitric oxide, which is another free radical able to induce oxidative stress. ROS production by mitochondria is critical for normal cell functioning; however, aberrant ROS production can have detrimental side effects. Progressive defects in mitochondrial function during aging lead to the increased production of ROS, resulting in accumulated DNA damage.Citation1–Citation3 Ultimately, this aging-associated DNA damage also increases our susceptibility towards the onset of cancer.Citation4–Citation6 Thus, we believe that aging, mitochondrial dysfunction and cancer metabolism are all inextricably linked.
One hypothesis is that aging-associated ROS production “fertilizes” the host microenvironment, converting it to a more glycolytic state via mitochondrial dysfunction, thereby providing energy-rich nutrients (L-lactate) to “fuel” cancer cell growth.Citation7,Citation8 Thus, with increasing age, the augmented glycolytic power of the “aging host microenvironment,” could provide a more metabolically permissive “soil” for sustaining anabolic tumor cell growth (“the seeds”).Citation7,Citation8 This would mechanistically explain why increasing chronological age is the most significant risk factor for the development of human cancers. Similarly, cancer cells and chronic inflammation could accelerate aging of the host microenvironment, via increased localized production of hydrogen peroxide.Citation7,Citation8 Cancer cells, myofibroblasts, macrophages and neutrophils are thought to be the largest producers of hydrogen peroxide and hence, oxidative stress.Citation7–Citation9
Our recent work has shown that cancer-associated fibroblasts (CAFs) undergo tumor cell-initiated oxidative stress, which can, in turn, foster the metabolic and mutagenic activities of tumor cells.Citation10 More specifically, we found that stromal fibroblasts undergo ROS production and aerobic glycolysis to generate energy-rich metabolites, such as L-lactate and ketones,Citation11,Citation12 which are directly used by tumor cells to support mitochondrial oxidative phosphorylation. We have termed this new paradigm “The Reverse Warburg Effect.” Supporting a role for energy-rich metabolites in aiding cancer pathogenesis, the sole addition of L-lactate to MCF7 cancer cells dramatically increased mitochondrial mass.Citation10 Moreover, genome-wide transcriptional profiling of L-lactate or ketone-treated MCF7 cells yielded a “gene signature” that was strikingly similar to bona fide stem cells, and was predictive of recurrence, metastasis and poor overall survival in human breast cancer patients.Citation13 We also found evidence for the existence of a stromal-epithelial lactate shuttle in human tumors, analogous to the lactate shuttles that are essential for the normal physiological function of muscle tissue and brain.Citation14 Finally, intraperitoneal (i.p.) injection of L-lactate or ketones in human tumor xenograft models was sufficient to increase both tumor growth and lung metastasis.Citation15 These data support the idea that “energy transfer” or “metabolic-coupling” between the tumor stroma and epithelial cancer cells “fuels” tumor cell growth and metastasis, via oxidative mitochondrial metabolism in anabolic cancer cells.Citation16–Citation18
In addition to driving aging-associated metabolic changes, oxidative stress can significantly promote genomic instability in adjacent cancer cells, indicating that the tumor stroma can potentially increase cancer cell aggressive behavior, via a bystander effect. Clinical support for these observations includes a women's increased risk for invasive breast cancer, based on mitochondrial polymorphisms associated with alterations in mitochondrial function.Citation19 Furthermore, a recent study reported the identification of frequent frameshift mutations in the coding mononucleotide repeat of mitochondrial transcription factor A (TFAM) in colorectal primary tumors with microsatellite instability (MSI).Citation20 These mutations may lead to mitochondrial copy number reductions and mitochondrial instability in colorectal tumors, and the question arises whether these mutations are present in the tumor stroma.
Mitochondrial transcription factor A (TFAM) is a nucleusencoded protein that binds and promotes the transcription of mtDNA. TFAM not only regulates mtDNA transcription and replication,Citation21 but also is known to be essential for the maintenance of mtDNA, mitochondrial biogenesis and functionCitation22 and is a component of mitochondrial nucleoids.Citation23,Citation24 Although TFAM has no known genetic relationship with any mitochondrial disease, in mice TFAM knockout causes severe mtDNA depletion and death in the embryo.Citation22 Mice having a 50% reduction in their TFAM transcript and protein levels, exhibited a 34% reduction in the mtDNA copy number, a 22% reduction in mitochondrial transcript levels, and a partial reduction in cytochrome c oxidase levels in the heart.Citation22 Although these studies have identified that aberrant TFAM regulation results in heart failure and pre-mature aging in mice, little is known about the role of TFAM in cancer development.Citation20
In the present study, we set out to determine how altered levels of TFAM in stromal fibroblasts influence tumor growth. Truncated TFAM is observed in whole tumors from colorectal cancers,Citation20 therefore, we examined if knocking-down TFAM in fibroblasts is sufficient to generate the cancer associated fibroblast phenotype.Citation10 Our results support the idea that decreased TFAM levels not only result in increased ROS, via hydrogen peroxide production, but also in the secretion of L-lactate. Interestingly, knocking-down TFAM in stromal fibroblasts was sufficient to promote tumor formation in an MDA-MB-231 xenograft system in mice. These results indicate that TFAM expression in the tumor stroma is critical to retard tumor growth.
Finally, these studies also have implications for understanding the role of hydrogen peroxide production in providing a permissive environment for tumor growth during aging, as a result of accumulated oxidative damage, metabolic reprogramming and accelerated “host aging” in the tumor stroma (reviewed in refs. Citation7 and Citation8). As such, mitochondrial oxidative stress in cancer-associated fibroblasts may be viewed as “accelerated host aging” in the tumor microenvironment.
Results
TFAM-deficient fibroblasts show a loss of Caveolin-1 and mitochondrial dysfunction.
Previous studies have demonstrated an important role for mitochondrial oxidative stress in cancer pathogenesis.Citation10,Citation25 Variants of TFAM, an important transcription factor required for mitochondrial DNA (mtDNA) replication and transcription, have recently been identified to be associated with sporadic colorectal cancer (CRC) from whole tumors with microsatellite instability (MSI).Citation20 Taken together, these studies suggest an important role for TFAM in the development of human cancers.
To directly examine the role of TFAM in cancer pathogenesis, we generated TFAM-deficient immortalized fibroblast cell lines (hTERT-BJ1), using an sh-RNA approach. illustrates the successful knock-down of TFAM in stromal fibroblasts (sh-TFAM), as compared to control fibroblasts (sh-Ctrl) by immuno-blot analysis. Next, we examined the status of the caveolin-1 (Cav-1) protein, because a loss of Cav-1 expression in the tumor stroma has previously been established as a new powerful biomarker for tumor progression.Citation26–Citation29 Interestingly, immuno-blot analysis shows that downregulation of TFAM also results in the loss of Cav-1 protein expression (). Notably, genome-wide transcriptional profiling of the tumor stroma of Cav-1 deficient breast cancer patients demonstrates a functional association with aging, mitochondrial dysfunction, glycolysis and inflammation.Citation30
TFAM expression has been shown to correlate with the activities of mitochondrial complexes I, III and IV, all of which contain mtDNA-encoded subunits.Citation31 illustrates the differences in the expression of the complexes involved in oxidative phosphorylation in hTERT sh-Ctrl and sh-TFAM fibroblasts, under both normoxic or hypoxic conditions. Under normoxic conditions, both cell lines exhibit similar expression of the 5 complexes; however, under hypoxic conditions, the expression of complexes I, II, III and IV is decreased in TFAM-deficient fibroblasts.
We examined functional mitochondrial activity under normoxic conditions and found that sh-TFAM fibroblasts show a small, but significant, reduction (∼12%; p = 0.005) in activity (); however, we could not examine this under hypoxic conditions, due to assay limitations.
TFAM-deficient fibroblasts produce more hydrogen peroxide.
Recent studies have suggested that reactive oxygen species, such as hydrogen peroxide, are involved in both tumor initiation and progression.Citation7,Citation9 Hydrogen peroxide production is sufficient to convert normal fibroblasts to activated myo-fibroblasts which, in turn, produce hydrogen peroxide themselves.Citation32,Citation33 These studies suggest that the tumor-stroma co-evolution is dependent on the secretion of hydrogen peroxide by cancer cells which then induces oxidative stress in neighboring stromal cells, such as fibroblasts, resulting in oxidative stress and increased ROS production in the tumor stroma.
Here, we determined if knocking-down TFAM in stromal fibroblasts is sufficient to induce an oxidative stress phenotype normally associated with myo-fibroblasts and cancer-associated fibroblasts. illustrates that hydrogen peroxide production is significantly increased (∼2-fold; p = 0.001) in sh-TFAM fibroblasts, as compared to sh-Ctrl fibroblasts. The ability of TFAM-deficient fibroblasts to produce excess hydrogen peroxide suggests that a loss of TFAM expression could mimic the phenotypic behavior of tumor-associated myo-fibrobasts undergoing oxidative stress.
TFAM-deficient fibroblasts undergo aerobic glycolysis and secrete L-lactate.
Another key attribute associated with tumor progression is the parasitic relationship between cancer cells and the surrounding tumor stroma. Cancer-associated fibroblasts undergo aerobic glycolysis and extrude lactate to “feed” adjacent cancer cells.Citation15,Citation34 Furthermore, recent studies have provided evidence of a lactate shuttle between stromal fibroblasts and epithelial cancer cells in human tumors.Citation14
As L-lactate is a critical fuel which drives and provides continued energetic support for tumor epithelial cells, we next examined the ability of TFAM-deficient fibroblasts to secrete L-Lactate. illustrates that TFAM-deficient fibroblasts secrete increased levels of L-lactate (∼2-fold; p = 0.006), relative to control fibroblasts processed in parallel.
TFAM-deficient fibroblasts promote breast cancer tumor growth in vivo, without increasing tumor angiogenesis.
Next, we used an in vivo murine xenograft model to evaluate the tumorpromoting properties of TFAM-deficient fibroblasts. Fibroblasts were co-injected with MDA-MB-231 human breast cancer cells into the flanks of immuno-deficient nude mice. After 4 weeks, the tumors were harvested and subjected to detailed analysis.
Consistent with the oxidative stress phenotype exhibited in vitro, sh-TFAM fibroblasts were able to promote tumorigenesis. shows an up to 2-fold increase in tumor growth. However, there were no observed differences in tumor vessel density, as measured using CD31 immuno-staining (). Thus, the tumor growth promoting phenotype of TFAM-deficient fibroblasts appears to independent of tumor angiogenesis.
TFAM-deficient fibroblasts increase the mitochondrial activity of adjacent epithelial cancer cells.
One hypothesis is that TFAM-deficient fibroblasts promote tumor growth by providing “fuel” in the form of L-lactate to epithelial cancer cells. This would increase mitochondrial oxidative phosphorylation in adjacent cancer cells.
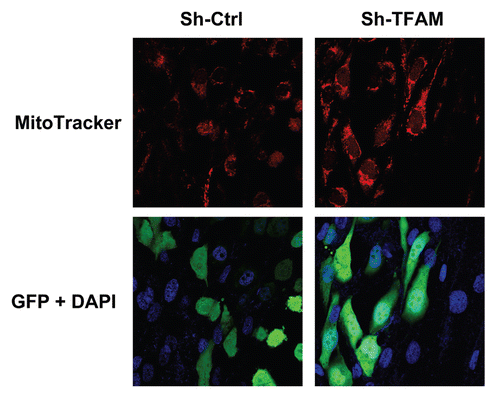
To test the validity of this hypothesis, we co-cultured sh-TFAM and sh-Ctrl fibroblasts with GFP-tagged MDA-MB-231 breast cancer cells. shows that TFAM-fibroblasts increased the mitochondrial activity of MDA-MB-231 cells, as visualized by MitoTracker staining. These findings are consistent with the idea that glycolytic fibroblasts fuel the growth of oxidative epithelial cancer cells, via mitochondrial oxidative phosphorylation.
Discussion
Mitochondria play an important role in tumor formation and cancer pathogenesis. This was first suggested in the early 1920s when Otto Warburg proposed that that tumor cells have defects in mitochondrial oxidative phosphorylation and, therefore, rely on high levels of aerobic glycolysis as the major source of ATP to fuel cellular proliferation (the “conventional” Warburg effect).Citation35–Citation40
Our new model termed the “Reverse Warburg Effect,” modifies this original notion, however, and supports an important role for functional cancer cell mitochondria and oxidative phosphorylation in tumorigenesis.Citation16–Citation18,Citation30 We have previously shown, using a co-culture model, that oxidative stress, via ROS production, is a key component of tumor initiation and development.Citation10 Furthermore, a recent study showed a direct link between mitochondrial oxidative stress and tumor pathogenesis, by targeting a powerful anti-oxidant protein (catalase, which inactivates hydrogen peroxide) specifically to mitochondria, effectively reducing tumor grade and lung metastasis in mice.Citation25 Although these studies link mitochondrial oxidative stress to tumor pathogenesis, a direct role for mitochondrial dysfunction in the tumor-stroma remained to be examined. Here, we directly demonstrated that TFAM-deficient fibroblasts with targeted mitochondrial dysfunction undergo oxidative stress, produce more hydrogen peroxide and L-lactate, and are sufficient to promote tumor growth.
Other studies have examined the role mitochondrial oxidative phosphorylation in relation to tumor cell growth (reviewed in refs. Citation41 and Citation42). For example, p32 is a key protein capable of promoting mitochondrial oxidative phosphorylation. Knock-down of p32 expression in human cancer cells shifted their metabolism from oxidative phosphorylation (OXPHOS) towards aerobic glycolysis, making these cells less tumorigenic.Citation43 This metabolic shift towards aerobic glycolysis is consistent with our TFAM knockdown data, in which we observed the increased production of a glycolytic byproduct, L-Lactate and a reduction in OXPHOSassociated proteins.
Another study by Weinberg et al.Citation4 examined the effects of TFAM loss in a mouse model designed to assess the effects of mitochondrial metabolism and ROS generation in K-rasmediated tumorigenicity. They found that CRE-mediated excision of TFAM from epithelial tumor cells in TFAM-floxed mice, resulted in fewer lesions per unit area and significant reductions in tumor growth.Citation44
Thus, a metabolic shift towards aerobic glycolysis in epithelial cancer cells inhibits tumor growth, due to defective mitochondrial function. However, aerobic glycolysis in cancer-associated fibroblasts has just the opposite effect. Our current TFAM knock-down data directly shows that glycolytic fibroblasts stimulate tumor growth via a paracrine effect, likely due to the secretion of L-lactate into the tumor microenvironment.
Both p32 and TFAM are important for the proper function of OXPHOS and engineered defects in OXPHOS shifts cancer cells towards aerobic glycolysis. An interesting difference between these previous studies and our new findings is that p32 and TFAM were knocked-down in epithelial cancer cells, which resulted in decreased tumor growth in vivo. However, in our current study, TFAM was knocked-down in stromal fibroblasts and promoted tumorigenesis in vivo. This important compartment-specific distinction shows that cancer-associated fibroblasts, via a glycolytic shift, can provide the necessary “fuel” (L-lactate) for epithelial cancer cell OXPHOS.
In addition to the metabolic shift which occurs in cancerassociated fibroblasts, increased oxidative stress has also been shown to be a driving force in tumor-stroma co-evolution. Studies have indicated that progressive defects in mitochondrial function lead to the increased production of reactive oxygen species (ROS) during aging, such as hydrogen peroxide, resulting in accumulated DNA damage, ultimately leading to increased cancer risk.Citation1–Citation6
Oxidative stress, via hydrogen peroxide production, is sufficient to convert normal fibroblasts to activated myo-fibroblasts which then produce hydrogen peroxide themselves.Citation32,Citation33 Based on our current understanding of the tumor-stroma co-evolution, it is oxidative stress from the fibroblast which drives tumor progression.Citation10 In this study, we show that TFAM knock-down fibroblasts produce ∼2-fold more hydrogen peroxide, which we believe is promoting tumor growth. This hypothesis is directly supported by a recent study which showed that mitochondrial-directed catalase (MCAT), a powerful anti-oxidant protein which inactivates hydrogen peroxide, was effective in reducing both tumor grade and nearly eliminating lung metastasis in mice.Citation25
Taken together, our data support a role for defective OXPHOS in cancer pathogenesis. However, unlike most other published studies which suggest a defect in OXPHOS in epithelial cancer cells, our results support a role for defective OXPHOS in the tumor stroma, and not the epithelial cancer cells themselves. We directly show that altering OXPHOS in the stromal fibroblast compartment results in increased L-lactate production and that these fibroblasts are able to promote tumorgenesis in vivo.
A loss of stromal caveolin-1 (Cav-1) protein expression is a powerful biomarker that predicts early tumor recurrence, metastasis and poor clinical outcome in breast cancer patients.Citation27–Citation30,Citation45,Citation46 These findings have now been validated in several independent breast cancer cohorts,Citation47–Citation49 and have also been extended to prostate cancer, as well.Citation46,Citation50 Mechanistically, a loss of stromal Cav-1 in cancer-associated fibroblasts is a functional marker for oxidative stress, mitochondrial dysfunction and aerobic glycolysis in the tumor microenvironment.Citation10,Citation12 Genome-wide transcriptional profiling of the tumor stroma from Cav-1 deficient breast cancer patients shows a strict association with aging, mitochondrial dysfunction, glycolysis and inflammation.Citation30 Caveolin-1 deficient fibroblasts also show direct evidence of mitochondrial dysfunction and are more glycolytic (secondary to increased autophagy and mitophagy), with increased ROS production.Citation10,Citation12,Citation51,Citation52 Most importantly, Cav-1 deficient fibroblasts also promote an up to 4-fold increase in tumor growth, in breast cancer xenograft models.Citation53 Suppression of this fibroblast-driven tumor-promoting phenotype can be accomplished by overexpression of SOD2 in Cav-1 deficient fibroblasts,Citation53 which functions to reduce mitochondrial oxidative stress. In accordance with these previous findings, here we demonstrate that TFAM-deficient fibroblasts show evidence of mitochondrial oxidative stress, with a dramatic loss of Cav-1 protein expression. Thus, a loss of Cav-1 is sufficient to drive mitochondrial oxidative stress,Citation10,Citation53 and mitochondrial oxidative stress drives a loss of Cav-1 (this report), providing a “feed-forward” mechanism for ROS production and aerobic glycolysis in tumor microenvironment. Finally, loss of stromal Cav-1 in fibroblasts is mediated by autophagic degradation via lysosomes, which is induced by oxidative stress.Citation51,Citation54,Citation55 As such, loss of stromal Cav-1 can be prevented with anti-oxidants (N-acetylcysteine; NAC) or autophagy inhibitors (chloroquine), both of which have significant antitumor activity.Citation51,Citation54,Citation55
In an accompanying report, we also provide in vivo functional evidence for mitochondrial dysfunction in the tumor stroma and hyper-activation of oxidative mitochondrial metabolism in epithelial cancer cells, in human breast cancer patients.Citation56 More specifically, we demonstrate that the tumor stroma shows an absence of functional mitochondrial activity, while epithelial cancer cells have even more mitochondrial activity than normal adjacent epithelial cells.Citation56 This has broad implications for cancer diagnosis and for targeted therapeutic interventions, aimed at reducing fuel production in the tumor stroma and/or reducing fuel consumption in epithelial cancer cells.Citation56 Both of these strategies would eliminate energy transfer (from the tumor stroma to epithelial cancer cells), effectively “cutting off the fuel supply” for tumor growth and metastasis.Citation56
In summary, we believe that new cancer therapies should be developed to target the metabolic inter-dependencies between epithelial cancer cells and their surrounding stromal microenvironment. Our molecular genetic data directly supports the idea that cancer chemo-prevention and treatment should focus on the development of new powerful anti-oxidants, to minimize oxidative stress, L-lactate production and tumor stroma co-evolution. In accordance with this notion, the anti-oxidant N-acetyl-cysteine (NAC) has antitumor properties, behaves as an anti-inflammatory (due to NFκB inhibition), inhibits L-lactate production,Citation57,Citation58 and increases lifespan by 25% in animal models (reviewed in refs. Citation7, Citation8 and Citation34). Interestingly, lower levels of tumor-associated lactate were observed in breast cancer patients with long-term survival (greater than 5 years survival), as compared with patients who died of breast cancer recurrence and metastasis (less than 5 years of survival).Citation59
Materials and Methods
Generation of TFAM knock-down fibroblasts.
All cell lines used in these experiments were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum in a 37°C humidified atmosphere containing 5% CO2 until confluent, unless otherwise noted. Human immortalized fibroblasts (hTERT-BJ1) were used to generate both shRNA TFAM and shRNA control cell lines. shRNA TFAM (sc-38053-V) and shRNA control (sc-108080) lenti-viral particles were purchased from Santa Cruz Biotechnologies Inc. For the generation of stable cell lines, hTERT-BJ1 fibroblasts (75,000 cells/well) were plated in 12-well dishes in growth media. After 24 hours, the media was removed and replaced with 500 µl DMEM + 5% FBS, 12 µl of shRNA virus and 5 µg/ml polybrene for TFAM shRNA (sh-TFAM) and control (sh-Ctrl). Twenty-four hours post-transduction, the media containing virus was removed and replaced with DMEM growth media and the cells were selected with puromycin for five days.
Immunoblot analysis.
Fibroblasts were harvested in lysis buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM n-octyl-glucoside), containing protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Sigma) and centrifuged at 13,000x g for 15 min at 4°C to remove insoluble debris. Protein concentrations were analyzed using the BCA reagent (Pierce). Cell lysates were then separated by SDS-PAGE (12% acrylamide) and transferred to nitrocellulose. Subsequent wash buffers contained 10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20, which was supplemented with 1% bovine serum albumin and 4% nonfat dry milk (Carnation) for the blocking solution and 1% bovine serum albumin for the antibody diluent. After blocking for 1 h, membranes were washed with TBST and then incubated for 1 h with the primary antibody which was followed by TBST washes. The membranes were probed with Mitoprofile total OXPHOS cocktail (MS601, Mitosciences); TFAM (H00007019-B01P, Abnova), Caveolin-1 (BD-2297, BD-Biosciences) and anti-β-actin antibody (A5441, Sigma). Horseradish peroxidase-conjugated secondary antibodies [anti-mouse, 1:5,000 dilution (Pierce) or anti-rabbit 1:5,000 (BD Biosciences/Pharmingen)] were used to visualize bound primary antibodies, with the Supersignal chemiluminescence substrate (Pierce, Rockford).
Mitochondrial activity in hTERT fibroblasts.
To measure mitochondrial activity, cells were stained with MitoTracker Orange (CMTMRos; M7510, Invitrogen, Inc.), whose accumulation in the mitochondria is dependent upon their membrane potential. Briefly, sh-Crtl fibroblasts or sh-TFAM fibroblasts were cultured for 4 days in DMEM with 10% NuSerum. After 4 days, the cells were then incubated with pre-warmed MitoTracker staining solution (diluted in serum-free DMEM to a final concentration of 25 nM) for 10 min at 37°C. All subsequent steps were performed in the dark. Cells were washed in PBS, harvested and re-suspended in 300 µL of Binding Buffer (BD Biosciences). Cells were then analyzed by FACS using a PE Texas Red signal detector, with an excitation wavelength of 496 nm and emission of 615 nm (to detect MitoTracker). Data analysis was performed using FlowJo 8.8 software.
Hydrogen peroxide assay.
Sh-TFAM or sh-Ctrl fibroblasts were seeded (100,000 per well) in 12-well plates in 1 ml of complete media in quadruplicate. Two days later, new growth media was added and after 4 h, hydrogen peroxide production was analyzed using Pentafluorobenzenesulfonyl fluorescein (PFBSF; 10005983, Caymen Chemical, Inc.). Briefly, cells were washed 3x with PBS followed by a 45 min incubation (37°C) of 500 ul phenol-red free Opti-MEM containing 5 µM PFBSF. Following incubation, cells were washed with PBS (3x), then trypsinized in phenol red free tyrpsin, collected, spun (1,000x rpm) and resuspended in 300 µl of 1x FACS buffer (556454; BD Bioscience, Inc.) and quantified by FACS using the BD LSRII (BD Biosciences, Inc.). 1x FACS buffer is composed of 0.2 µm sterile filtered 0.01 M Hepes (pH 7.4), 0.14 M NaCl and 2.5 mM CaCl2. The data were analyzed with the FlowJo software (Tree Star, Inc.).
L-lactate assay.
Sh-TFAM or sh-Ctrl fibroblasts were seeded (100,000 per well) in 12-well plates in 1 ml of complete media. Two days after, the media was changed to phenol-red free Opti-MEM containing 2% FBS. After 24 h, the media of each well was collected for measuring L-lactate concentration using the EnzyChrom™ L-Lactate Assay Kit (ECLC-100, BioAssay Systems, Inc.). After removing the media, cells were tyrpsinized (0.025%), spun-down and resuspended in 1 mL media for quantification. Cells were counted in 4–6 fields using a 40x objective lens and a haemocytometer. The amount of L-lactate in the media was normalized to total cell count.
Pre-clinical studies in mouse xenografts.
All animals were housed and maintained in a barrier facility at the Kimmel Cancer Center at Thomas Jefferson University under National Institutes of Health (NIH) guidelines. Mice were kept on a 12-hour light/dark cycle with ad libitum access to food and water. Animal protocols used for this study were pre-approved by the Institutional Animal Care and Use Committee (IACUC). Briefly, MDA-MB-231-GFP human breast cancer cells (1 million cells) were admixed with sh-Ctrl or sh-TFAM fibroblasts (300,000 cells) in 100 µl of sterile PBS, were injected into the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 4–6 weeks of age). Mice were then sacrificed at 4-weeks post-injection; tumors were dissected to determine their weight and size using calipers. Tumor volume was calculated using the formula (X2Y)/2, where X and Y are the short and long dimensions, respectively, of the tumor.
Quantitation of tumor angiogenesis.
Immuno-histochemical staining for CD31 was performed on frozen tumor sections using a 3-step biotin-streptavidin-horseradish peroxidase method. Frozen tissue sections (6 µm) were fixed in 2% paraformaldehyde in PBS for 10 min and washed with PBS. After blocking with 10% rabbit serum, the sections were incubated overnight at 4°C with rat anti-mouse CD31 antibody (BS Biosciences) at a dilution of 1:200, followed by biotinylated rabbit anti-rat IgG (1:200) antibody and streptavidin-HRP. Immuno-reactivity was revealed with 3,3′-diaminobenzidine. For quantitation of vessels, CD31-positive vessels were enumerated in 8 fields within the central area of each tumor using a 20x objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated using Image J.
Mitochondrial activity staining in MDA/fibroblast co-cultures.
Fibroblasts (hTERT; sh-TFAM vs. sh-Ctrl cells) and epithelial cancer cells [MDA-MB-231-GFP(+)] were plated onto glass coverslips in 12-well plates. After 3 days in DMEM with 10% NuSerum, the co-cultures were stained for mitochondrial activity, as previously described in references Citation55 and Citation60. Briefly, MitoTracker Orange (CMTMRos M7510, Invitrogen, Inc.) was dissolved in DMSO to prepare a 1 mM solution, immediately before use. The 1 mM stock solution was diluted in serum-free DMEM to a final concentration of 25 nM. Cells were then incubated with pre-warmed MitoTracker staining solution for 10 minutes at 37°C. Then, cells were washed in PBS, fixed with 2% PFA, incubated with the 4,6-diamidino-2-phenylindole (DAPI; D3571, Invitrogen, Inc.) nuclear stain and mounted with Prolong Gold Anti-fade mounting reagent (P36930, Invitrogen, Inc.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Abbreviations
| TFAM | = | transcription factor A, mitochondrial |
| Cav-1 | = | caveolin-1 |
| CAFs | = | cancer associated fibroblasts |
| ROS | = | reactive oxygen species |
| OXPHOS | = | oxidative phosphorylation |
Figures and Tables
Figure 1 TFAM-deficient fibroblasts show a loss of Caveolin-1 protein expression. We generated TFAM-deficient immortalized fibroblast cell lines (hTERT-BJ1), using an sh-RNA approach. Note the successful knock-down of TFAM in stromal fibroblasts (sh-TFAM), as compared to control fibroblasts (sh-Ctrl), as seen by immuno-blot analysis. We also examined the status of the caveolin-1 (Cav-1) protein, because a loss of Cav-1 expression in the tumor stroma is a biomarker for tumor progression. Immuno-blot analysis shows that downregulation of TFAM also results in the loss of Cav-1 protein expression. Blotting for β-actin was performed in parallel as a control for equal protein loading.
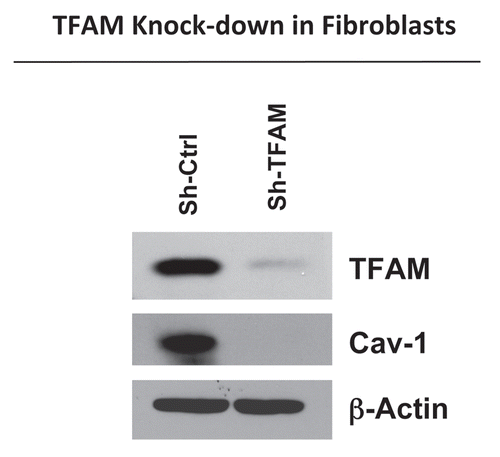
Figure 2 TFAM-deficient fibroblasts show defects in oxidative phosphorylation. To assess the status of the mitochondrial respiratory chain in fibroblasts, a battery of antibodies directed against mitochondrial complex components (I–V) were examined. Under normoxic conditions, no changes were observed between sh-TFAM and sh-Ctrl fibroblasts. However, under hypoxic conditions, note the decrease in Complexes I–IV. Immunoblotting with β-actin is shown as a control for equal loading.
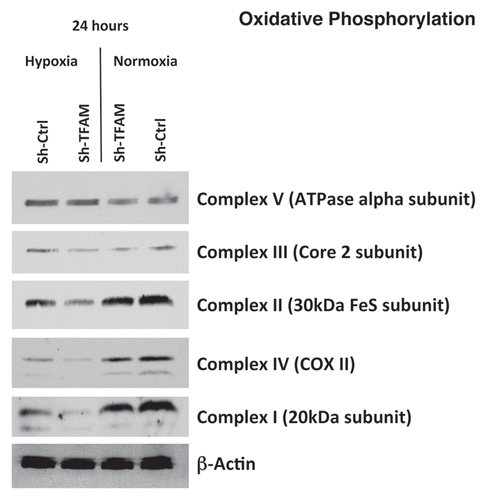
Figure 3 TFAM-deficient fibroblasts show a decrease in functional mitochondrial activity. We examined functional mitochondrial activity under normoxic conditions and found that sh-TFAM fibroblasts show a small, but significant, reduction (∼12%; p = 0.005) in activity.
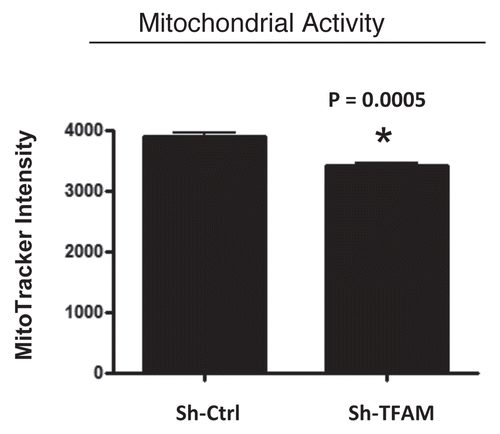
Figure 4 TFAM-deficient fibroblasts produce more hydrogen peroxide. Hydrogen peroxide production is significantly increased (∼2-fold; p = 0.001) in sh-TFAM fibroblasts, as compared to sh-Ctrl fibroblasts. The ability of TFAM-deficient fibroblasts to produce excess hydrogen peroxide suggests that a loss of TFAM expression could mimic the phenotypic behavior of tumor-associated myo-fibrobasts undergoing oxidative stress.
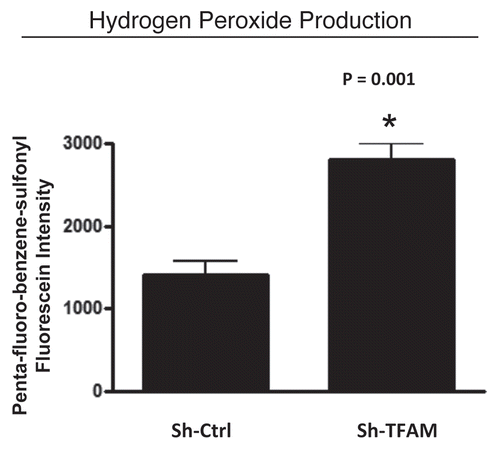
Figure 5 TFAM-deficient fibroblasts secrete elevated levels of L-lactate, consistent with increased aerobic glycolysis. L-lactate is a critical fuel which provides continued energetic support for tumor epithelial cells. Note that TFAM-deficient fibroblast secrete increased levels of L-lactate (∼2-fold; p = 0.006), relative to control fibroblasts processed in parallel.
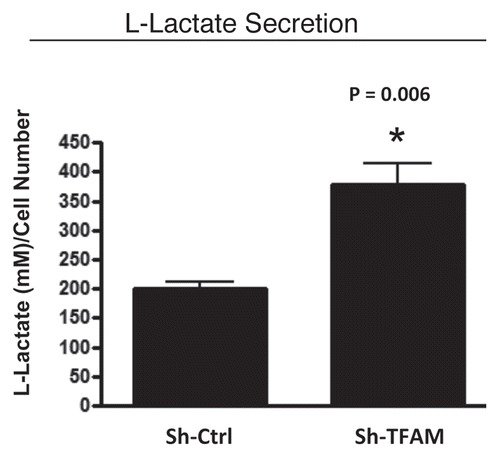
Figure 6 TFAM-deficient fibroblasts promote tumor growth. We used an in vivo murine xenograft model to evaluate the tumor-promoting properties of TFAM-deficient fibroblasts. Fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of immuno-deficient nude mice. After 4 weeks, the tumors were harvested and subjected to detailed analysis. sh-TFAM fibroblasts were able to promote tumorigenesis, with an up to 2-fold increase in tumor growth. This represents an ∼1.6-fold increase (p = 0.03) in tumor weight and an ∼2.1-fold increase (p = 0.006) in tumor volume.
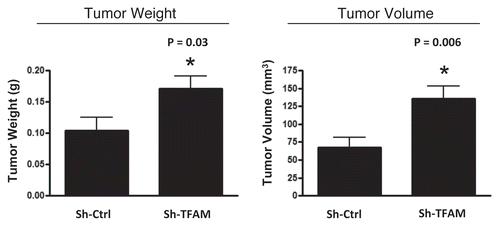
Figure 7 TFAM-deficient fibroblasts do not increase tumor angiogenesis. We used an in vivo murine xenograft model to evaluate the tumor-promoting properties of TFAM-deficient fibroblasts. Fibroblasts were co-injected with MDA-MB-231 breast cancer cells into the flanks of immuno-deficient nude mice. After 4 weeks, the tumors were harvested and subjected to detailed analysis. There were no observed differences in tumor vessel density density (number of vessels per field), as measured using CD31 immuno-staining. As such, the tumor growth promoting activity of TFAM-deficient fibroblasts is independent of tumor angiogenesis.
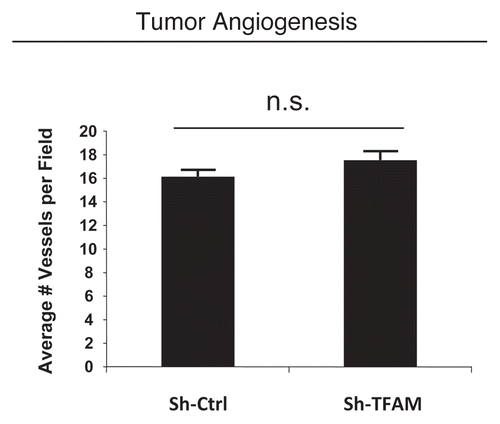
Figure 8 TFAM-deficient fibroblasts increase mitochondrial activity in adjacent cancer epithelial cells. We co-cultured sh-TFAM and sh-Ctrl fibroblasts with GFP-tagged MDA-MB-231 breast cancer cells. Then, we visualized mitochondrial activity in these co-cultures using MitoTracker. Note that TFAM-fibroblasts specifically increase the mitochondrial activity of adjacent MDA-MB-231 cells. MitoTracker (red); epithelial cancer cells/GFP (green); nuclei/DAP I (blue). See online version for color version of the figure.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
References
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005; 308:1909 - 1911
- Treiber N, Maity P, Singh K, Kohn M, Keist AF, Ferchiu F, et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell 2011; 10:239 - 254
- Woo DK, Shadel GS. Mitochondrial stress signals revise an old aging theory. Cell 2011; 144:11 - 12
- Blagosklonny MV. Increasing healthy lifespan by suppressing aging in our lifetime: preliminary proposal. Cell Cycle 2010; 9:4788 - 4794
- Blagosklonny MV, Campisi J, Sinclair DA. Aging: past, present and future. Aging (Albany NY) 2009; 1:1 - 5
- Gil Del Valle L. Oxidative stress in aging: Theoretical outcomes and clinical evidences in humans. Biomed Pharmacother 2010; In Press
- Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasisThe seed and soil also needs “fertilizer”. Cell Cycle 2011; 10:2440 - 2449
- Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, Sotgia F. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle 2011; 10:2059 - 2063
- Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: Implications for PET imaging of human tumors. Cell Cycle 2011; 10:2504 - 2520
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010; 9:2201 - 2219
- Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011; 10:1271 - 1286
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 1783
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 3514
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, Lisanti MP. Stromalepithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 2011; 43:1045 - 1051
- Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res 2011; 13:213
- Pavlides S, Vera I, Gandara R, Sneddon S, Pestell R, Mercier I, et al. Warburg Meets Autophagy: Cancer Associated Fibroblasts Accelerate Tumor Growth and Metastasis Via Oxidative Stress, Mitophagy and Aerobic Glycolysis. Antioxid Redox Signal 12012; In press
- Bai RK, Leal SM, Covarrubias D, Liu A, Wong LJ. Mitochondrial genetic background modifies breast cancer risk. Cancer Res 2007; 67:4687 - 4694
- Guo J, Zheng L, Liu W, Wang X, Wang Z, Wang Z, et al. Frequent truncating mutation of TFAM induces mitochondrial DNA depletion and apoptotic resistance in microsatellite-unstable colorectal cancer. Cancer Res 2011; 71:2978 - 2987
- Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta 2002; 1576:1 - 14
- Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, et al. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat Genet 1998; 18:231 - 236
- Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, et al. Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 2003; 31:1640 - 1645
- Wang Y, Bogenhagen DF. Human mitochondrial DNA nucleoids are linked to protein folding machinery and metabolic enzymes at the mitochondrial inner membrane. J Biol Chem 2006; 281:25791 - 25802
- Goh J, Enns L, Fatemie S, Hopkins H, Morton J, Pettan-Brewer C, Ladiges W. Mitochondrial targeted catalase suppresses invasive breast cancer in mice. BMC Cancer 2011; 11:191
- Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, et al. Caveolin-1-/- null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol 2009; 174:746 - 761
- Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8:1071 - 1079
- Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther 2010; 10:135 - 143
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 2034
- Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle 2011; 10:1794 - 1809
- Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001; 88:529 - 535
- Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med 2010; 2:211 - 230
- Waghray M, Cui Z, Horowitz JC, Subramanian IM, Martinez FJ, Toews GB, Thannickal VJ. Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. Faseb J 2005; 19:854 - 856
- Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther 2010; 10:537 - 542
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11:325 - 337
- Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol 2005; 70:363 - 374
- Warburg O. On respiratory impairment in cancer cells. Science 1956; 124:269 - 270
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 314
- Warburg O, Gawehn K, Geissler AW, Kayser D, Lorenz S. Experiments on Anaerobiosis of Cancer Cells. Klin Wochenschr 1965; 43:289 - 293
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 1033
- Dang CV. p32 (C1QBP) and cancer cell metabolism: is the Warburg effect a lot of hot air?. Mol Cell Biol 2010; 30:1300 - 1302
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy and fiction. Biochem Biophys Res Commun 2004; 313:459 - 465
- Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol 2010; 30:1303 - 1318
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Krasmediated tumorigenicity. Proc Natl Acad Sci USA 2010; 107:8788 - 8793
- Ghajar CM, Meier R, Bissell MJ. Quis custodiet ipsos custodies: who watches the watchmen?. Am J Pathol 2009; 174:1996 - 1999
- Furuta S, Ghajar CM, Bissell MJ. Caveolin-1: Would-be Achilles' heel of tumor microenvironment?. Cell Cycle 2011; 10:3431
- Koo JS, Park S, Kim SI, Lee S, Park BW. The impact of caveolin protein expression in tumor stroma on prognosis of breast cancer. Tumour Biol 2011; 32:787 - 799
- Qian N, Ueno T, Kawaguchi-Sakita N, Kawashima M, Yoshida N, Mikami Y, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci 2011; 102:1590 - 1596
- Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol 2009; 174:2035 - 2043
- Di Vizio D, Morello M, Sotgia F, Pestell RG, Freeman MR, Lisanti MP. An absence of stromal caveolin-1 is associated with advanced prostate cancer, metastatic disease and epithelial Akt activation. Cell Cycle 2009; 8:2420 - 2424
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533
- Bosch M, Mari M, Herms A, Fernandez A, Fajardo A, Kassan A, et al. Caveolin-1 deficiency causes cholesterol-dependent mitochondrial dysfunction and apoptotic susceptibility. Curr Biol 2011; 21:681 - 686
- Trimmer C, Sotgia F, Whitaker-Menezes D, Balliet RM, Eaton G, Martinez-Outschoorn UE, et al. Caveolin-1 and mitochondrial SOD2 (MnSOD) function as tumor suppressors in the stromal microenvironment: a new genetically tractable model for human cancer associated fibroblasts. Cancer Biol Ther 2011; 11:383 - 394
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartmentspecific oncogenesis. Cell Cycle 2010; 9:3534 - 3551
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle 2011; 10:4047 - 4064
- Watt G, Jongsakul K, Ruangvirayuth R. A pilot study of N-acetylcysteine as adjunctive therapy for severe malaria. Qjm 2002; 95:285 - 290
- Leelarungrayub D, Khansuwan R, Pothongsunun P, Klaphajone J. N-Acetylcysteine Supplementation Controls Total Antioxidant Capacity, Creatine Kinase, Lactate and Tumor Necrotic Factor-Alpha against Oxidative Stress Induced by Graded Exercise in Sedentary Men. Oxid Med Cell Longev 2011; 2011:329643
- Cao MD, Sitter B, Bathen TF, Bofin A, Lonning PE, Lundgren S, Gribbestad IS. Predicting long-term survival and treatment response in breast cancer patients receiving neoadjuvant chemotherapy by MR metabolic profiling. NMR Biomed 2011; In Press
- Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010; 9:2412 - 2422