Abstract
Complement 1q-Binding Protein (C1qbp) is a mitochondrial protein reported to be upregulated in cancer. However, whether C1qbp plays a tumor suppressive or tumorigenic role in the progression of cancer is controversial. Moreover, the exact effects of C1qbp on cell proliferation, migration, and death/survival have not been definitely proven. To this end, we comprehensively examined the effects of C1qbp on mitochondrial-dependent cell death, proliferation, and migration in both normal and breast cancer cells using genetic gain- and loss-of-function approaches. In normal fibroblasts, overexpression of C1qbp protected the cells against staurosporine-induce apoptosis, increased proliferation, decreased cellular ATP, and increased cell migration in a wound-healing assay. In contrast, the opposite effects were observed in fibroblasts depleted of C1qbp by RNA interference. C1qbp expression was found to be markedly elevated in 4 different human breast cancer cell lines as well as in ductal and adenocarcinoma tumors from breast cancer patients. Stable knockdown of C1qbp by shRNA in the aggressive MDA-MB-231 breast cancer cell line greatly reduced cell proliferation, increased ATP levels, and decreased cell migration compared to control shRNA-transfected cells. Moreover, C1qbp knockdown elicited a significant increase in doxorubicin-induced apoptosis in the MDA-MB-231 cells. Finally, C1qbp upregulation was not restricted to breast cancer cells and tumors, as levels of C1qbp were also found to be significantly elevated in both human lung and colon cancer cell lines and carcinomas. Together, these results establish a pro-tumor, rather than anti-tumor, role for C1qbp, and indicate that C1qbp could serve as a molecular target for cancer therapeutics.
Introduction
Complement 1q binding protein (C1qbp, gC1qr, p32, HAPB1 or SF2p32), was first described as a globular protein capable of interacting with complement subcomponent 1q.Citation1 As a result, C1qbp was originally thought to act as plasmalemmal receptor for C1q and thus play a critical role in the inflammatory response.Citation2,Citation3 However, several studies, including our own, have demonstrated that C1qbp exclusively localizes to the mitochondrial matrix in variety of cell types,Citation4–Citation8 and indeed, C1qbp possesses a canonical N-terminal mitochondrial localization sequence that targets it to this organelle.Citation5 Despite this, the data ascribing a specific function(s) to mitochondrially localized C1qbp is still limited, especially with regards to the role C1qbp may play in pathogenesis of diseases, such as cancer, where alterations in mitochondrial phenotype appear to contribute.Citation8–Citation10
C1qbp has been reported to be upregulated in human cancers,Citation11–Citation14 which would tend to suggest that C1qbp plays a role in tumorigenesis. Indeed, the degree to which C1qbp is elevated in breast and prostate cancers is inversely correlated with the prognosis.Citation13,Citation14 Consistent with this, inhibition of C1qbp reduced proliferation of breast and prostate cancer cells,Citation8,Citation14 and we have recently reported that C1qbp suppresses mitochondrial permeability transition and cell death in fibroblasts.Citation4 However, recent studies have also reported that induction of mitochondrial-dependent apoptosis in cancer cells by the pro-death Bcl2 family member Hrk,Citation15 the tumor suppressor p14ARF,Citation16 and the cytotoxic drug cisplatinCitation16 requires C1qbp. In contrast, C1qbp overexpression drove mitochondrial-dependent death in fibroblasts.Citation17 Together, these data would suggest that C1qbp is part of the mitochondrial death machinery and, as such, would be tumor suppressive.
Because of these conflicting reports regarding the function of mitochondrial C1qbp and its potential role in the progression of cancer, the purpose of the present study was to comprehensively examine the effects of C1qbp on mitochondrial-dependent cell death, cell proliferation and cell migration using both genetic gain- and loss-of-function approaches. Here, we have established that C1qbp is sufficient to protect both normal and breast cancer cells against mitochondrial-driven death and to promote both cell proliferation and migration. We also found marked upregulation in human breast, lung and colon cancer cell lines and tumors. Together, these data suggest that C1qbp could a potential molecular target for anticancer drug therapy.
Results
C1qbp inhibits staurosporine-induced apoptosis.
We have previously reported that C1qbp can attenuate oxidative stress-induced mitochondrial permeability transition and necrotic cell death.Citation4 Here, we wanted to examine whether C1qbp could also inhibit the more canonical mitochondrial-dependent apoptotic pathway. Consequently, we infected MEFs with viruses expressing either β-galactosidase (β-gal, control) or Myc-tagged C1qbp () and treated them with staurosporine, which induces mitochondrial-dependent apoptosis in a Bax/Bak-dependent manner.Citation18 Exposure to staurosporine caused a dose-dependent increase in apoptotic cell death in β-gal-expressing MEFs, as determined by cleavage of caspase-3 and PARP () and TUNEL staining (). However, MEFs overexpressing C1qbp exhibited markedly less caspase-3 and PARP cleavage as well as TUNEL staining than control cells ( and B). We also conducted RNA interference experiments, where we knocked down C1qbp in the MEFs using a specific siRNA (). In direct contrast to the overexpression studies, depletion of endogenous C1qbp enhanced staurosporine-induced apoptotic protein processing () and TUNEL staining (). Together, these data strongly indicate that C1qbp acts as an endogenous inhibitor of mitochondrial-dependent apoptosis.
C1qbp stimulates cell proliferation and migration.
We noticed when preparing lysates for protein gel blotting that there was significantly more protein obtained from the C1qbp-infected MEFs than the β-gal-infected cells, suggesting that overexpression of C1qbp may be stimulating proliferation. To test this hypothesis we infected MEFs with the β-gal or C1qbp-encoding adenoviruses and measured proliferation by immunostaining for phosphorylated histone H3. A greater number of cells exhibited phospho-histone H3 staining when infected with C1qbp (), indicative of increased proliferation. Importantly, this increased proliferative response to C1qbp was not simply secondary to increased ATP production, as ATP levels actually decreased in the C1qbp-overexpressing MEFs (). Antithetically, siRNA knockdown of endogenous C1qbp reduced phosphohistone H3 staining () and caused a small but significant increase in cellular ATP ().
We also tested whether C1qbp could influence the migratory capacity of the fibroblasts using a standard wound-healing assay. This was performed in low serum to inhibit proliferation, and indeed, no significant proliferation in any of the infected/transfected cells was observed over the 6 h period (data not shown). The starting wound area was no different between the β-gal- and C1qbp-infected MEFs. However, the ability of the cells to repopulate the wound was significantly increased by C1qbp (), with over 60% of the wound re-covered after 6 h by the C1qbp-infected cells, compared with only 40% in the control-infected cells. Again, siRNA-mediated knockdown of C1qbp had the opposite effect and decreased the rate of wound healing compared with controls (). Thus, it would appear that, in addition to inhibiting cell death, C1qbp also promotes cellular proliferation and movement.
C1qbp is upregulated in human breast cancer cell lines and tumors.
The fact that C1qbp was pro-survival, pro-proliferation and pro-migration, lead us to examine whether C1qbp expression was enhanced in cancer cells. Four different human breast cancer epithelial cell lines, MCF7, BT474, T47D and MDA-MB-231, demonstrated significantly increased C1qbp protein compared with the normal breast epithelial line MCF12A (). Previous studies have suggested that in tumor cells a portion of the C1qbp pool is relocated to the plasma membrane.Citation12,Citation19 However, when we performed immunocytochemistry for C1qbp in the various breast cancer cell lines we found that it co-localized completely with the mitochondrial ATP synthase (Fig. S1) and not with the CellLight plasma membrane marker (Fig. S2). This would indicate that, although elevated, the C1qbp protein was still retained in the mitochondria of these cells. Immunohistochemistry further demonstrated greatly elevated C1qbp protein levels in tumors from both invasive ductal breast carcinoma and breast mucous adenocarcinoma when compared with normal breast tissue ().
Depletion of C1qbp reduces proliferation and migration in breast cancer cells.
To better assess whether C1qbp plays a causative role in breast tumorigenesis, we knocked down C1qbp in the MDA-MB-231 triple-negative breast cancer cell line, which exhibited a marked increase in C1qbp protein levels when compared with MCF12A control cells (see ). We generated 4 stable cell lines where the MDA-MB-231 cells were transfected with either a control shRNA or 3 different C1qbp shRNAs (), resulting in a 40–70% reduction in C1qbp protein (). To assess the effects of C1qbp depletion on proliferation we plated each cell line at an equal density and measured the increase in cell numbers after time. Expression of the C1qbp-specific shRNAs resulted in a reduction of cellular proliferation, with the extent of C1qbp knockdown correlating to degree of growth inhibition (). However, as with the MEF studies, the inhibitory effect on proliferation occurred in the face of actual increases in ATP levels ().
We also determined the effects of C1qbp knockdown on migration in the breast cancer cells using the wound-healing assay. MDA-MB-231 cells transfected with the control shRNA were able to repopulate 40% of the initial wound area over a 6 h period (). In contrast, the migratory capacity was greatly impaired in all 3 of the C1qbp shRNA expressing cell lines when compared with the controls (). Again, no significant proliferation of the different cell lines was observed over the 6 h period under the low serum conditions (data not shown).
Depletion of C1qbp also sensitizes breast cancer cells to doxorubicin.
Our results in MEFs indicated that reducing C1qbp levels would sensitize cells to apoptosis (). We therefore wanted to test whether C1qbp depletion could sensitize the MDA-MB-231 cells to the anticancer agent doxorubicin, which induces cell killing through a mitochondrial-dependent mechanism.Citation20 As has been previously reported in references Citation21 and Citation22, the control MDA-MB-231 cells only showed a modest increase in apoptotic death in response to 24 h of exposure to 2 µM doxorubicin exposure, as determined by TUNEL staining (). In contrast, the cells depleted of C1qbp demonstrated a considerable increase in cell death following doxorubicin exposure ().
C1qbp is also upregulated in human lung and colon cancer cell lines and tumors.
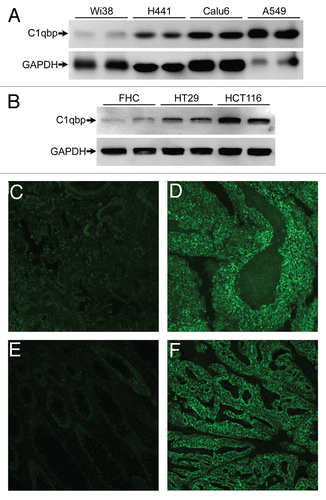
To ascertain whether upregulation of C1qbp expression was restricted to breast tumor cells and tissues, or whether this was general to all carcinomas, we examined C1qbp expression in both cells and tumors derived from human lung and colon cancers. We found gross upregulation of C1qbp in the human lung cancer cell lines H441, Calu-6 and A549 when compared with the normal control Wi38 cell line (). HT-29 and HCT-116 colon cancer cells also exhibited elevated C1qbp in relation to the FHC normal colon cell line (). Increased C1qbp levels in tumors from lung (small cell carcinoma) and colon (well differentiated invasive adenocarcinoma) cancer patients were also observed (). Together these data suggest that C1qbp plays a critical role in the pathogenesis of human tumor formation.
Discussion
Upregulation of C1qbp mRNA and protein levels have been reported for a variety of tumor types, including breast, lung, thyroid, prostate and ovary.Citation11–Citation14 Indeed, in the present study we found that C1qbp expression was profoundly enhanced in both cell lines and tumor sections from breast, lung and colon cancers. However, the role of C1qbp in cancer pathogenesis is controversial, with studies proposing either an oncolyticCitation15–Citation17,Citation23 or an oncogenicCitation8,Citation12–Citation14 function for C1qbp. The results of our study demonstrate that C1qbp is capable of promoting all 3 of the critical contributors to cancerous transformation: specifically increased proliferation, increased cellular mobility and resistance to cell death. Most importantly, we have found that depletion of C1qbp reduces the oncogenic phenotype of the highly aggressive triple-negative MDA-MB-231 breast cancer cell line. Together, our results unequivocally establish a pro-tumor, rather than antitumor, role for C1qbp, indicating that C1qbp could serve as a molecular target for cancer therapeutics.
Mitochondrial C1qbp has been postulated to be a pro-death, and by inference, a tumor suppressive, protein with Hrk, p19ARF and cisplatin-induced apoptosis being reported to be C1qbp-dependent.Citation15,Citation16,Citation23 In direct contrast, we have recently shown, using both gain- and loss-of-function approaches, that C1qbp can in fact suppress oxidative stress-induced mitochondrial dysfunction and necrosis in MEFs.Citation4 In the present investigation, we have extended these findings to show that C1qbp can also suppress staurosporine-induced cell death in MEFs. Interestingly, treatment with staurosporine increased the expression of endogenous C1qbp (). This is similar to what has been observed during c-Myc and cisplatin-induced apoptosis.Citation16,Citation23 Intriguingly, the levels of the exogenous C1qbp also went up, suggesting a post-transcriptional mechanism. It is not entirely clear why this is the case, but it may represent a compensatory response by the cell to try and protect itself against the pro-death effects of the drug. Given that the cell death elicited by staurosporine is apoptotic in nature it would thus appear that C1qbp is capable of inhibiting both mitochondrially driven necrosis and apoptosis. This combination would exert an extremely powerful cytoprotective effect and thus provide an additional mechanism by which cancer cells (1) exhibit reduced cell death and (2) are profoundly resistant to cytotoxic signals and agents. We confirmed this by stably knocking down C1qbp in malignant, drug-resistant MDA-MB-231 breast cancer cells and challenging them with doxorubicin. As would be expected very little cell death was observed in the “normal” MDA-MB-231 cells, either at baseline or in response to the drug. However, by lowering C1qbp levels we greatly increased the degree of doxorubicin-induced mortality. Thus, reductions in C1qbp protein appear to be sufficient to re-sensitize cancer cells to an established anticancer agent. These data are also consistent with the recent study by Fogal et al.Citation8 who demonstrated that knockdown of C1qbp could exacerbate glucose-deprivation evoked death in MDA-MB-435 and MDA-MB-231 cancer cells.
That a mitochondrial protein could modulate mitochondrial-dependent apoptosis was perhaps not that surprising. What was surprising was that modulation of C1qbp could also have profound effects on cell proliferation and migration. Overexpression of C1qbp alone was sufficiently able to increase proliferation in fibroblasts. In contrast, depletion of endogenous C1qbp reduced fibroblast proliferation. Importantly, we were able to recapitulate this finding in the MDA-MB-231 cells, with the degree of inhibition being directly correlated with the degree of C1qbp knockdown. The reduction in cell proliferation, however, was not simply due to decreased cell viability, as reduced C1qbp levels did not affect cell death at baseline in either the MEFs or the cancer cells. Consistent with our findings, while our manuscript was in preparation Amamoto et al. published that knockdown of C1qbp could also reduce proliferation in human prostate cancer cells in association with increased and decreased levels of p21CIP1 and cyclinD1, respectively. Moreover, C1qbp inhibition has been reported to reduce tumor growth in vivo.Citation8
While proliferation rates control the growth of the primary tumor, the metastatic potential of a tumor is more related to the migratory capacity of its constitutive cancer cells. In this regard, C1qbp expression correlated strongly with the appearance of metastases in patients with breast cancer.Citation13 Given this information, we examined the effects of C1qbp modulation on the ability of both MEFs and breast cancer cells to migrate. In the fibroblasts, forced expression of C1qbp significantly enhanced the ability of the cells to repopulate the denuded area in a wound healing assay, whereas reduced C1qbp expression had the opposite effect. Similarly, diminished C1qbp levels profoundly reduced the migration of MDA-MB-231 cells following wounding. Unlike the proliferation results, though, we did not see a gene-dosage effect such that all 3 shRNAs equally attenuated the migratory response in the cancer cells. However, we only measured the cells over 6 h, and it's possible that longer time periods may have revealed a difference between the cell lines.
To our knowledge, this is the first time such a function for C1qbp has been reported and is in contrast to a report that identified C1qbp as a migration-suppressing factor.Citation24 However, that study only used one siRNA and thus off-target effects of this single sequence could not be ruled out. In contrast, in the MDA-MB-231 cells we utilized three shRNAs that targeted different sites on the C1qbp sequence. Hence, we are confident that the effects we observed are indeed specifically due to C1qbp silencing.
The question that arises is how can a protein localized to the mitochondrial matrix affect these various processes, especially proliferation and migration? RNAi-depletion of C1qbp in MDA-MB-435 breast cancer cells reduced expression of key components of the OXPHOS chain, reduced ATP levels and shifted their metabolic profile to a glycolytic program.Citation8 Similarly, Saccharomyces cerevisiae deficient for mam33p, the yeast homolog of C1qbp, exhibit defective oxidative phosphorylation and reduced ATP synthesis.Citation6 This metabolic shift and/or the reduction in ATP could be responsible for reduced proliferation rates observed in our C1qbp-depleted MEFs and MDA-MB-231 cells. However, in contrast to these previous studies, we actually observed a significant increase in ATP levels that was commensurate with the degree of C1qbp reduction. Consistent with this, overexpression of C1qbp, which increased proliferation, concomitantly decreased ATP levels. Thus, changes in ATP levels per se are not likely to be involved.
Another potential mechanism that may contribute to C1qbp's is modulation of mitochondrially derived reactive oxygen species (ROS). C1qbp can affect assembly of complexes I and III, the 2 main sites of mitochondrial ROS production,Citation8 and stable overexpression has been reported to increase mitochondrial ROS in fibroblasts.Citation17 Indeed, enhanced (but non-lethal) mitochondrial ROS have been implicated in the alterations in cell proliferation, migration and death sensitivity required for tumorigenesis. For example, oncogenic Ras-induced cell proliferation and anchorage-independent growth is dependent on increased mitochondrial ROS production,Citation25,Citation26 which in turn activates ERK.Citation25 Mitochondrial ROS-induced activation of the transcription factors HIF1α, AP1 and Ets-1 has also been shown to play an essential role in the enhanced proliferative and migratory capacities of various cancer cells.Citation27–Citation29 Finally, increased mitochondrial oxidants have been demonstrated to induce resistance to chemotherapeutics through the upregulation/activation of cytoprotective factors, such as amphiregulinCitation30 and ERK.Citation31 Thus, it is tempting to speculate that knockdown of C1qbp reduces mitochondrial ROS production in the breast cancer cells and thereby removes one of the major stimuli of cell growth, migration and drug-resistance. Certainly a reduction in ROS would be expected to accelerate oxidative phosphorylation, which in turn would explain the increases in ATP we see with C1qbp knockdown. The effects of C1qbp on mitochondrial ROS is something we are currently pursuing.
In addition to mitochondria, C1qbp has been proposed to localize to multiple subcellular compartments, including the golgi,Citation32 endoplasmic reticulumCitation33 and nucleus.Citation34 Moreover, unlike normal cells, tumor cells have been reported to express C1qbp on their cell surface.Citation12,Citation20 Thus, it is possible that an extramitochondrial pool of C1qbp is responsible for regulating cell death, cell proliferation and movement in the MEFs and breast cancer cells. However, in the present study we found that C1qbp expression in the various breast cancer cell lines is still restricted to the mitochondrion and does not co-localize with the plasma membrane. We have also confirmed these results in the various lung and colon cancer cell lines (data not shown). Moreover, we have previously demonstrated that the adenovirally encoded C1qbp we employed in the MEF culture system is only targeted to the mitochondria.Citation4 Yet this exogenously expressed C1qbp was still capable of inhibiting cell death, stimulating proliferation and enhancing cell migration. These results would thus argue against a role for non-mitochondrial C1qbp in our findings.
In summary, we have demonstrated that the mitochondrial matrix protein C1qbp significantly contributes to the hyperproliferative, hypermigratory and cytoprotected phenotype of a cancer cell, in this case one derived from breast tumor. Given that C1qbp is upregulated in a variety of cancers, including lung and colon, it is likely that it plays a similar role in these tumor types. Further studies investigating the precise mechanisms by which this mitochondrial protein (dys)regulates these cellular processes are clearly necessary. However, it would appear that C1qbp is a genuine target for the development of novel therapies for the treatment of a wide spectrum of cancers.
Materials and Methods
Reagents.
Lipofectamine RNAiMAX and the CellLight™ Plasma Membrane-CFP marker were from Invitrogen; DMEM medium, DMEM/F-12 medium, Hanks's buffered saline solution (HBSS), horse serum and fetal bovine serum were from Hyclone; epidermal growth factor was from Calbiochem; Arrest-In transfection reagent was obtained from Thermo Open Biosystems; the In Situ Cell Death Detection (TUNEL) kit was from Roche; human control and tumor tissue slides were purchased from Millipore and Abcam; all other chemicals/reagents were from Sigma-Aldrich.
Cell culture.
All experiments involving the harvesting of mouse tissues and embryos were approved by the University of Missouri-Columbia Animal Care and Use Committee and conformed to the NIH guidelines for the use and care of animals. Primary cultured mouse embryonic fibroblasts (MEFs) were obtained from E13.5–15.5 C57/B6 mouse embryos by trypsin digestion as previously described in reference Citation35. The MEFs were then maintained in DMEM medium supplemented with 10% v/v FBS. Human breast normal and cancer cells (MCF12A, BT-474, MCF-7, T47D, MDA-MB-231), lung normal and cancer cells (Wi38, H441, Calu-6, A549) and colon normal and cancer cells (FHC, HT-29, HCT-116) were obtained from the ATCC and cultured as recommended by the ATCC. All of the cell lines were passaged for less than 6 mo.
Adenovirus and siRNAs.
Replication-deficient adenoviruses for β-galactosidase and mouse C1qbp (with a c-terminal Myc tag) have been previously described in reference Citation4. MEFs were typically infected with adenovirus at a multiplicity of infection (MOI) of 30–120 plaque-forming units for 2 h at 37°C. The MEFs were then cultured for another 48 h in virus-free media before analysis. The siRNA against mouse C1qbp (5′-GGA GGG AUA CAA ACU AUA CAC UCA A-3′), as well as a non-specific control siRNA, were obtained from Invitrogen. MEFs were transfected with 100 nM siRNA using Lipofectamine RNAiMAX and then cultured for 48 h prior to the various experiments. For the stable transfection experiments, the aggressive, metastatic MDA-MB-231 breast cancer cells were transfected with pGIPZ-based plasmids (Thermo Open Biosystems) expressing either a non-specific shRNA or 3 different human-specific C1qbp shRNAs (#1: 5′-GAU ACU AAU UAU ACA CUC A-3′; #2: 5′-CUA GAC AUG UGC UUU GAA A-3′ and #3: 5′-CCG ACG GAG ACA AAG CUU U-3′) using Arrestin-In transfection reagent. The cells were then passaged for 3 d before switching to media containing 1 mg/mL puromycin to select for the shRNA-expressing cells. The cells were then maintained in the puromycin-containing media.
Fluorescence microscopy.
Apoptosis was assessed in cells using a commercially available TUNEL staining kit according to the manufacturer's instructions, and fluorescence images were collected using an inverted fluorescent microscope (Olympus IX51) connected to a digital camera. For immunocytochemistry, cells were fixed in 4% paraformaldehyde. After permeabilization the slides were incubated overnight with anti-C1qbp (Sigma), anti-ATP Synthase (Mitosciences), or anti-phospho-histone H3 (Millipore). The cells were then incubated with the appropriate fluorophore-conjugated secondary antibody (Alexa, Invitrogen) and visualized. In some cases, the cells were transfected with the CellLight™ Plasma Membrane-CFP marker 24 h prior to fixation. This construct is the myristoylation/palmitoylation targeting sequence of Lck fused to CFP. For the proliferation studies, MDA-MB-231 cells were incubated with the DNA stain bis-benzamide to determine the total number of nuclei.
Protein gel blotting.
Cells were lysed in buffer containing 150 mM NaCl, 10 mM Tris (pH 7.4), 1 mM EDTA and 1% Triton-X100. Proteins were resolved by SDS-PAGE using 10–15% acrylamide, transferred onto PVDF membranes and blotted using the following commercially available antibodies: anti-C1qbp from Sigma; anti-GAPDH from Santa Cruz Biotechnology and anti-cleaved caspase-3, anti-PARP and anti-Myc-tag from Cell Signaling. Membranes were then incubated with the appropriate alkaline phosphatase-linked secondary antibody (Santa Cruz) and visualized by enhanced chemifluorescence (Amersham).
ATP levels.
The ATP levels in total MEF and MDA-MB-231 cell lysates were determined using the CellTiter-GLO luminescence assay from Promega.
Wound healing assay.
MEFs were seeded onto fibronectin-coated 12-well plates and infected with either β-gal or C1qbp adenoviruses or transfected with control or C1qbp siRNAs. Alternatively, stably transfected MDA-MB-231 cells were seeded onto fibronectin-coated 12-well plates. Twenty-four h later, the MEFs or the MDA-MB-231 cells were switched to their respective media containing only 0.5% FBS and incubated overnight. The next day a “wound” was created in each well using a P200 pipette tip. The media was replaced with fresh 0.5% FBS-containing media and the wound area photographed using an inverted phase contrast microscope (Olympus IX51). The wound area was then photographed every hour for 6 h following the initial wound. The wound area for each time point was then quantified using NIH ImageJ freeware.
Immunohistochemistry.
Histological slides of human normal and tumor tissue sections were deparaffinized and boiled in antigen retrieval solution (Abcam). After blocking, the slides were incubated with anti-C1qbp (Sigma) followed by incubation with a FITC-conjugated secondary antibody (Alexa, Invitrogen). Fluorescence images were then collected using an inverted fluorescent microscope (Olympus IX51) connected to a digital camera.
Statistical analyses.
Statistical significance was calculated by the Student t-test. A p value < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 C1qbp suppresses staurosporine-induce apoptosis in fibroblasts. (A) Mouse embryonic fibroblasts (MEFs) were infected with an adenovirus encoding Myc-tagged C1qbp, or β-galactosidase (β-gal, control) and then protein gel blotted for Myc, cleaved caspase-3 and cleaved PARP following exposure to 300 nM staurosporine for 18 h. GAPDH was used to demonstrate equivalent loading. (B) TUNEL staining in the infected MEFs exposed to increasing concentrations of staurosporine for 18 h. (C) MEFs were transfected with 100 nM of control (Con) or C1qbp-specific siRNA and protein gel blotted for C1qbp, cleaved caspase-3, cleaved PARP and GAPDH following exposure to 300 nM staurosporine for 18 h. (D) TUNEL staining in the siRNA-transfected MEFs exposed to increasing concentrations of staurosporine for 18 hrs. The results shown are representative of 3 and 4 independent experiments performed in duplicate. Error bars indicate SEM and *p < 0.05 vs. β-gal or Con siRNA.
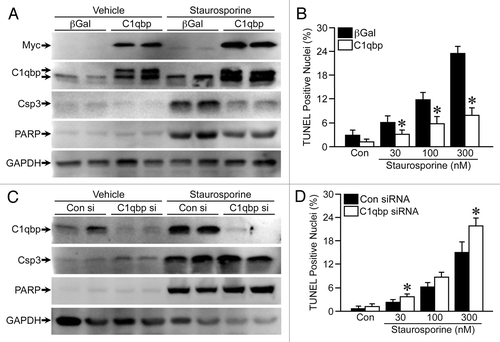
Figure 2 C1qbp promotes proliferation and migration in fibroblasts. (A) Phospho-histone H3 (pH3) staining, a marker of cell proliferation, in MEFs either infected with β-galactosidase (β-gal) or C1qbp adenoviruses or transfected with control (Con) or C1qbp siRNAs. (B) ATP levels in the virus-infected and siRNA-transfected MEFs. (C) Wound-healing assay in MEFs infected with either β-gal or C1qbp adenoviruses. (D) Wound-healing assay in MEFs transfected with either Con or C1qbp siRNAs. The results shown are representative of 3 and 4 independent experiments performed in duplicate. Error bars indicate SEM and *p < 0.05 vs. β-gal or Con siRNA.
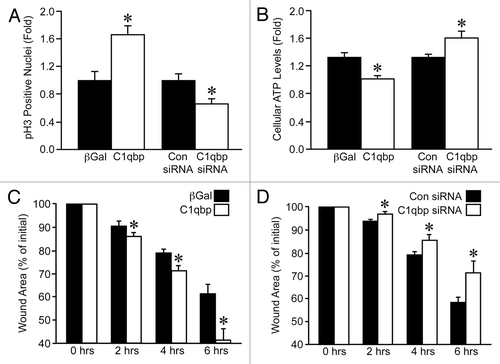
Figure 3 Mitochondrial C1qbp is upregulated in human breast cancer cells and tumors. (A) Protein gel blotting for C1qbp in the normal breast epithelial line MCF12A and in the MCF7, BT474, T47D and MDA-MB-231 breast cancer cell lines. GAPDH was used as a loading control. The results shown are representative of 3 and 4 independent experiments performed in duplicate. (B–G) Fluorescent immunohistochemical staining for C1qbp in sections from: (B and C) normal human breast tissue; (D and E) breast invasive ductal carcinoma and (F and G) breast mucous adenocarcinoma. Results are representative of staining from 4 different patient samples in each group.
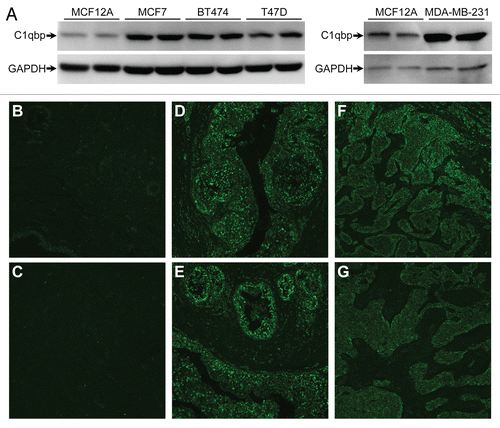
Figure 4 Knockdown of C1qbp reduces proliferation in MDA-MB-231 breast cancer cells. (A) MDA-MB-231 cells were stably transfected with either a control shRNA (Con) or one of 3 different C1qbp-specific shRNAs and then protein gel blotted for C1qbp. GAPDH was used as a loading control. (B) Quantification of the efficiency of C1qbp knockdown in the MDA-MB-231 shRNA cell lines. (C) Each of the MDA-MB-231 shRNA cell lines was plated at an equal density, and the increase in cell numbers after 48 hrs measured by bis-benzamide staining of the nuclei. (D) ATP levels in the control and C1qbp shRNA MDA-MB-231 cell lines. The results shown are representative of 3 and 4 independent experiments performed in duplicate. Error bars indicate SEM and *p < 0.05 vs. Con sh.
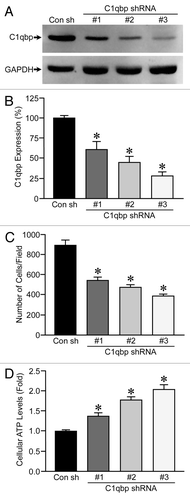
Figure 5 Knockdown of C1qbp reduces migration and resistance to doxorubicin in MDA-MB-231 breast cancer cells. (A) Wound-healing assay in MDA-MB-231 cells stably transfected with either a control shRNA (Con) or one of 3 different C1qbp-specific shRNAs. (B) TUNEL staining in the different MDA-MB-231 shRNA cell lines at baseline and following exposure to 2 µM doxorubicin for 24 h. The results shown are representative of 3 and 4 independent experiments performed in duplicate. Error bars indicate SEM and *p < 0.05 vs. Con shRNA.
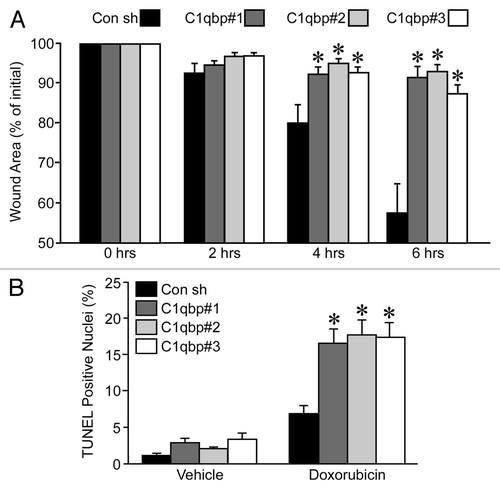
Figure 6 C1qbp is upregulated in human lung and colon cancer cell lines and tumors. (A) Protein gel blotting for C1qbp in the normal lung Wi38 cell line and in the H441, Calu-6 and A549 lung cancer cell lines. GAPDH was used as a loading control. (B) Protein gel blotting for C1qbp and GAPDH in the normal colon FHC cell line and in the HT-29 and HCT-116 colon cancer cell lines. (C–F) Fluorescent immunostaining for C1qbp in sections from: (C) normal human lung tissue; (D) small cell lung carcinoma; (E) normal human colon tissue and (F) colon invasive adenocarcinoma (well differentiated, mucinous). The results shown are representative of 3 and 4 independent experiments performed in duplicate (cell lines) or 4 different patient samples (tumors).
Additional material
Download Zip (2.1 MB)Acknowledgments
This work was supported by National Institutes of Health grants HL092327 and HL094404 (to Christopher P. Baines).
References
- Storrs SB, Kolb WP, Pinckard RN, Olson MS. Characterization of the binding of purified human C1q to heart mitochondrial membranes. J Biol Chem 1981; 256:10924 - 10929; PMID: 6974732
- Ghebrehiwet B, Lim BL, Kumar R, Feng X, Peerschke EI. gC1q-R/p33, a member of a new class of multifunctional and multicompartmental cellular proteins, is involved in inflammation and infection. Immunol Rev 2001; 180:65 - 77; PMID: 11414365; http://dx.doi.org/10.1034/j.1600-065X.2001.1800106.x
- Peerschke EI, Petrovan RJ, Ghebrehiwet B, Ruf W. Tissue factor pathway inhibitor-2 (TFPI-2) recognizes the complement and kininogen binding protein gC1qR/p33 (gC1qR): implications for vascular inflammation. Thromb Haemost 2004; 92:811 - 819; PMID: 15467913
- McGee AM, Baines CP. Complement 1q-binding protein inhibits the mitochondrial permeability transition pore and protects against oxidative stress-induced death. Biochem J 2011; 433:119 - 125; PMID: 20950273; http://dx.doi.org/10.1042/BJ20101431
- Dedio J, Jahnen-Dechent W, Bachmann M, Müller-Esterl W. The multiligand-binding protein gC1qR, putative C1q receptor, is a mitochondrial protein. J Immunol 1998; 160:3534 - 3542; PMID: 9531316
- Muta T, Kang D, Kitajima S, Fujiwara T, Hamasaki N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J Biol Chem 1997; 272:24363 - 24370; PMID: 9305894; http://dx.doi.org/10.1074/jbc.272.39.24363
- Seytter T, Lottspeich F, Neupert W, Schwarz E. Mam33p, an oligomeric, acidic protein in the mitochondrial matrix of Saccharomyces cerevisiae is related to the human complement receptor gC1q-R. Yeast 1998; 14:303 - 310; PMID: 9559539; http://dx.doi.org/10.1002/(SICI)1097-0061(19980315)14:4<303::AID-YEA217>3.0.CO;2-N
- Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol 2010; 30:1303 - 1318; PMID: 20100866; http://dx.doi.org/10.1128/MCB.01101-09
- Solaini G, Sgarbi G, Baracca A. Oxidative phosphorylation in cancer cells. Biochim Biophys Acta 2011; 1807:534 - 542; PMID: 20849810; http://dx.doi.org/10.1016/j.bbabio.2010.09.003
- Kaipparettu BA, Ma Y, Wong LJ. Functional effects of cancer mitochondria on energy metabolism and tumorigenesis: utility of transmitochondrial cybrids. Ann NY Acad Sci 2010; 1201:137 - 146; PMID: 20649550; http://dx.doi.org/10.1111/j.1749-6632.2010.05621.x
- Rubinstein DB, Stortchevoi A, Boosalis M, Ashfaq R, Ghebrehiwet B, Peerschke EI, et al. Receptor for the globular heads of C1q (gC1q-R, p33, hyaluronan-binding protein) is preferentially expressed by adenocarcinoma cells. Int J Cancer 2004; 110:741 - 750; PMID: 15146564; http://dx.doi.org/10.1002/ijc.20105
- Fogal V, Zhang L, Krajewski S, Ruoslahti E. Mitochondrial/cell-surface protein p32/gC1qR as a molecular target in tumor cells and tumor stroma. Cancer Res 2008; 68:7210 - 7218; PMID: 18757437; http://dx.doi.org/10.1158/0008-5472.CAN-07-6752
- Chen YB, Jiang CT, Zhang GQ, Wang JS, Pang D. Increased expression of hyaluronic acid binding protein 1 is correlated with poor prognosis in patients with breast cancer. J Surg Oncol 2009; 100:382 - 386; PMID: 19565630; http://dx.doi.org/10.1002/jso.21329
- Amamoto R, Yagi M, Song Y, Oda Y, Tsuneyoshi M, Naito S, et al. Mitochondrial p32/C1QBP is highly expressed in prostate cancer and is associated with shorter prostate-specific antigen relapse time after radical prostatectomy. Cancer Sci 2011; 102:639 - 647; PMID: 21205079; http://dx.doi.org/10.1111/j.1349-7006.2010.01828.x
- Sunayama J, Ando Y, Itoh N, Tomiyama A, Sakurada K, Sugiyama A, et al. Physical and functional interaction between BH3-only protein Hrk and mitochondrial pore-forming protein p32. Cell Death Differ 2004; 11:771 - 781; PMID: 15031724; http://dx.doi.org/10.1038/sj.cdd.4401418
- Kamal A, Datta K. Upregulation of hyaluronan binding protein 1 (HABP1/p32/gC1qR) is associated with Cisplatin induced apoptosis. Apoptosis 2006; 11:861 - 874; PMID: 16544101; http://dx.doi.org/10.1007/s10495-006-5396-4
- Chowdhury AR, Ghosh I, Datta K. Excessive reactive oxygen species induces apoptosis in fibroblasts: role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR). Exp Cell Res 2008; 314:651 - 667; PMID: 18166172; http://dx.doi.org/10.1016/j.yexcr.2007.10.033
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292:727 - 730; PMID: 11326099; http://dx.doi.org/10.1126/science.1059108
- Sánchez-Martín D, Cuesta AM, Fogal V, Ruoslahti E, Alvarez-Vallina L. The multicompartmental p32/gClqR as a new target for antibody-based tumor targeting strategies. J Biol Chem 2011; 286:5197 - 5203; PMID: 21156793; http://dx.doi.org/10.1074/jbc.M110.161927
- Jung K, Reszka R. Mitochondria as subcellular targets for clinically useful anthracyclines. Adv Drug Deliv Rev 2001; 49:87 - 105; PMID: 11377805; http://dx.doi.org/10.1016/S0169-409X(01)00128-4
- Muñoz-Gámez JA, Martín-Oliva D, Aguilar-Quesada R, Cañuelo A, Nuñez MI, Valenzuela MT, et al. PARP inhibition sensitizes p53-deficient breast cancer cells to doxorubicin-induced apoptosis. Biochem J 2005; 386:119 - 125; PMID: 15456408; http://dx.doi.org/10.1042/BJ20040776
- Gariboldi MB, Ravizza R, Molteni R, Osella D, Gabano E, Monti E. Inhibition of Stat3 increases doxorubicin sensitivity in a human metastatic breast cancer cell line. Cancer Lett 2007; 258:181 - 188; PMID: 17920763; http://dx.doi.org/10.1016/j.canlet.2007.08.019
- Itahana K, Zhang Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell 2008; 13:542 - 553; PMID: 18538737; http://dx.doi.org/10.1016/j.ccr.2008.04.002
- Seo M, Lee WH, Suk K. Identification of novel cell migration-promoting genes by a functional genetic screen. FASEB J 2010; 24:464 - 478; PMID: 19812375; http://dx.doi.org/10.1096/fj.09-137562
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA 2010; 107:8788 - 8793; PMID: 20421486; http://dx.doi.org/10.1073/pnas.1003428107
- Hole PS, Pearn L, Tonks AJ, James PE, Burnett AK, Darley RL, et al. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34+ hematopoietic progenitor cells. Blood 2010; 115:1238 - 1246; PMID: 20007804; http://dx.doi.org/10.1182/blood-2009-06-222869
- Comito G, Calvani M, Giannoni E, Bianchini F, Calorini L, Torre E, et al. HIF-1α stabilization by mitochondrial ROS promotes Met-dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic Biol Med 2011; 51:893 - 904; PMID: 21703345; http://dx.doi.org/10.1016/j.freeradbiomed.2011.05.042
- Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, et al. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci USA 2010; 107:4675 - 4680; PMID: 20176937; http://dx.doi.org/10.1073/pnas.0907705107
- Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res 2009; 69:2375 - 2383; PMID: 19276362; http://dx.doi.org/10.1158/0008-5472.CAN-08-3359
- Chang CJ, Yin PH, Yang DM, Wang CH, Hung WY, Chi CW, et al. Mitochondrial dysfunction-induced amphiregulin upregulation mediates chemo-resistance and cell migration in HepG2 cells. Cell Mol Life Sci 2009; 66:1755 - 1765; PMID: 19337692; http://dx.doi.org/10.1007/s00018-009-8767-5
- Chan DW, Liu VW, Tsao GS, Yao KM, Furukawa T, Chan KK, et al. Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 2008; 29:1742 - 1750; PMID: 18632752; http://dx.doi.org/10.1093/carcin/bgn167
- Sengupta A, Banerjee B, Tyagi RK, Datta K. Golgi localization and dynamics of hyaluronan binding protein 1 (HABP1/p32/C1QBP) during the cell cycle. Cell Res 2005; 15:183 - 186; PMID: 15780180; http://dx.doi.org/10.1038/sj.cr.7290284
- van Leeuwen HC, O'Hare P. Retargeting of the mitochondrial protein p32/gC1Qr to a cytoplasmic compartment and the cell surface. J Cell Sci 2001; 114:2115 - 2123; PMID: 11493647
- Chattopadhyay C, Hawke D, Kobayashi R, Maity SN. Human p32 interacts with B subunit of the CCAAT-binding factor, CBF/NF-Y, and inhibits CBF-mediated transcription activation in vitro. Nucleic Acids Res 2004; 32:3632 - 3641; PMID: 15243141; http://dx.doi.org/10.1093/nar/gkh692
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434:658 - 662; PMID: 15800627; http://dx.doi.org/10.1038/nature03434