Abstract
Comment on: Latini P, et al. Cell Cycle 2011; 10:3719-30.
Cockayne syndrome (CS) is a rare autosomal recessive genetic disorder characterized by developmental and progressive defects. Premature aging, cachectic dwarfism and neurological abnormalities are hallmark symptoms of CS,Citation1 and this disease is used as a model system in aging research. Mutations within two genes form the genetic basis: CSA (ERCC8) and CSB (ERCC6), comprising ∼20 and ∼80% of patients, respectively. CS proteins are not only involved in excision repair [transcription coupled-nucleotide excision repair (TC-NER) specifically], but also function in base excision repair (BER), transcription and in mitochondria.
Prevailing theories for CS.
Three prevailing theories for the underlying cellular and molecular causes of CS have been proposed. The first suggests that the principal roles of CS proteins are in TC-NER.Citation2 it is apparent that this alone does not sufficiently account for the multitude of CS-associated disorders. In fact, unlike patients from nuclear DNA repair deficient diseases, CS patients do not display increased cancer incidence and global genome-NER still functions to remove bulky DNA lesions. The second proposes that the CS proteins operate to promote the efficient repair of endogenous DNA damage. CS cells are hypersensitive to agents that increase formation of endogenous-type damage, a lack of CSB leads to increased steady-state levels of oxidative DNA lesions in both nuclear and mitochondrial DNA, and the CSB protein has functional interactions with proteins involved in BER and single-strand break repair.Citation3 The third prevailing theory is that the CS proteins predominantly function in transcription. Both CSA and CSB are components of RNAPII containing complexes, interact with TFIIH and CSB directly stimulates transcriptional elongation by RNAPI and II.Citation4 CSB has also been implicated in regulating genome wide transcriptional programs, as significant transcriptome alterations are seen CSB-deficient cells.Citation5,Citation6 In addition, CSB has been found to be intimately linked to the key transcription factor and tumor suppressor p53.
CS proteins and p53.
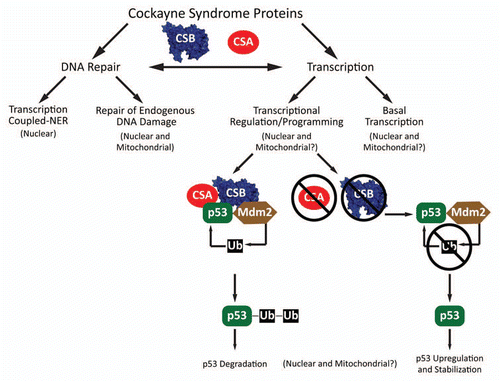
An intriguing connection between CSB and p53 was identified when the two proteins were found to physically interact.Citation7 The fact that p53 responds to stress stimuli and exerts effects on cell survival/death decisions raised questions as to how the CS proteins and p53 functionally interact and influence CS pathologies. The first clues to the CS-p53 connection came when CSB-deficient cells were found to have increased persistent steady-state levels of p53, upregulation and stabilization of p53 after UV irradiation and increased UV-induced apoptotic potential.Citation8 Latini et al.Citation9 show that p53 upregulation and stabilization is common to both CS groups. This increased p53 stability in CS cells was found to be caused by a defect in p53 ubiquitination and subsequent degradation. The cause of the ubiquitination defect was likely due to decreased ubiquitin ligase activity of Mdm2 (the major p53 E3 ubiquitin ligase) in the absence of the CS proteins. CSA and CSB, individually and together, were found to stimulate p53 ubiquitination by Mdm2 and both were found as components of p53-Mdm2 complexes. Overexpression of CSA and CSB lead to increased p53 ubiquitination and ensuing degradation; directly linking the CS proteins to regulation of p53 protein levels and the pro-apoptotic cell signals generated by its transcriptional activity. Supporting the CS-p53 link further, was the finding that the CSB gene is under transcriptional control by p53, adding another feedback loop to the p53 transcriptional program.
The molecular mechanisms underpinning the developmental and progressive defects of CS continue to increase in complexity as the molecular roles of the CS proteins continue to expand (). Defining the molecular roles of the CS proteins may be able to link specific pathway defects to defined symptoms. Some features seen in CS (e.g., neurological features and lack of increased cancer incidence) are compatible with mitochondrial dysfunction; which has been suggested to be the driving force in normal human aging, to play roles in cancer development/treatment, and to underlie neurological diseases. All three proteins (CSA, CSB and p53) are localized to mitochondria and their molecular functions within this organelle are as yet poorly defined, thus a compelling area for future. Studies on CS and the mechanisms of CS protein function remain at the crossroads of DNA repair and transcription and how their interplay affects aging and human disease.
Figures and Tables
Figure 1 Molecular roles of CS proteins. CSA and CSB operate in DNA repair as components of the TC-NER pathway which removes RNA polymerase blocking lesions and in repair of endogenous DNA damage in the nucleus and mitochondria by interacting with BER/SSBR proteins and stimulating removal of base damage. CSB acts in basal nuclear transcription by promoting elongation by RNAPI and RNAPII and plays a larger overall role in transcriptome regulation/programming. CSA and CSB both physically interact with p53 and stimulate Mdm2-dependent ubiquitination and subsequent degradation of p53. In the absence of CSA or CSB, Mdm2 ubiquitination of p53 is lower, stabilizing p53 and promoting pro-apoptotic signals through p53 dependent transcriptional programming.
References
- Natale V. Am J Med Genet 2011; 155A:1081 - 1095; PMID: 21480477; http://dx.doi.org/10.1002/ajmg.a.33933
- Hanawalt PC, et al. Nat Rev Mol Cell Biol 2008; 9:958 - 970; PMID: 19023283; http://dx.doi.org/10.1038/nrm2549
- Stevnsner T, et al. Mech Ageing Dev 2008; 129:441 - 448; PMID: 18541289; http://dx.doi.org/10.1016/j.mad.2008.04.009
- Lainé JP, et al. Trends Genet 2006; 22:430 - 436; PMID: 16797777; http://dx.doi.org/10.1016/j.tig.2006.06.006
- Kyng KJ, et al. Oncogene 2003; 22:1135 - 1149; PMID: 12606941; http://dx.doi.org/10.1038/sj.onc.1206187
- Newman JC, et al. Proc Natl Acad Sci USA 2006; 103:9613 - 9618; PMID: 16772382; http://dx.doi.org/10.1073/pnas.0510909103
- Wang XW, et al. Nat Genet 1995; 10:188 - 195; PMID: 7663514; http://dx.doi.org/10.1038/ng0695-188
- Balajee AS, et al. Oncogene 2000; 19:477 - 489; PMID: 10698517; http://dx.doi.org/10.1038/sj.onc.1203372
- Latini P, et al. Cell Cycle 2011; 10:3719 - 3730