Abstract
Deficiency of the circadian clock transcriptional factor BMAL1 results in the development of premature aging in mice. In agreement with the accelerated aging phenotype, we observed an increase in the number of senescent cells in different tissues (lungs, liver and spleen) of Bmal1-/- mice, which suggests the important role of BMAL1 in the control of senescence in vivo. However, no difference in the rate of proliferation and senescence between primary fibroblasts isolated from wild-type and Bmal1-/- mice has been detected, suggesting that BMAL1 does not play a significant role in replicative senescence in vitro. BMAL1 deficient fibroblasts had an increased sensitivity to hydrogen peroxide treatment, and reduced sensitivity to DNA damaging anticancer drugs etoposide and daunorubicin. Increased sensitivity of Bmal1-/- cells to oxidative stress was p53 independent and correlated with the disrupted regulation of reactive oxygen species (ROS) homeostasis in BMAL1 deficient cells: indeed, circadian oscillations of ROS level can be induced in wild-type but not in Bmal1-/- cells. We propose that BMAL1 is important for the regulation of oxidative stress and DNA damage responses, while deregulation of these processes upon BMAL1 deficiency leads to development of stress induced senescence in vivo.
Introduction
The circadian clock is an internal time-keeping system generating rhythms in behavior, physiology and gene expression with 24 h periodicity.Citation1 The circadian clock regulates many physiological processes, such as metabolism, hormone secretion and sleep.Citation2–Citation6 Disruption of circadian clock function has been linked with an increased risk of cancer, metabolic syndromes and cardiovascular diseases.Citation7–Citation10 Recently the circadian clock was implicated in the regulation of cell cycle and cell proliferation;Citation4,Citation11 it was suggested that disruption of the circadian clock-dependent regulation of cell proliferation may contribute to the development of cancerCitation12 and, probably, aging.Citation13 In agreement with that, animal models of circadian clock deficiency demonstrate changes in the process of aging.Citation14–Citation18
BMAL1 is a transcriptional factor and a major component of the circadian clock.Citation19–Citation21 In complex with transcriptional factors CLOCK or NPAS2, BMAL1 regulates the expression of other circadian clock genes; BMAL1 activity is necessary for generating circadian rhythms in gene expression and behavior. BMAL1 also regulates the expression of a growing number of so-called circadian clock-controlled genes, which are responsible for the circadian output in physiology. Recently, we have demonstrated that BMAL1 deficiency in mice results in a significantly reduced lifespan and premature aging phenotype, thus linking the circadian clock and aging.Citation17 The mechanism of accelerated aging upon BMAL1 deficiency is unknown; regulation of ROS homeostasis was proposed as one potential mechanism.Citation17,Citation22
Aging is associated with accumulation of senescent cells in vivo.Citation23 Senescence is a specific metabolic state of cells characterized by morphologic changes (enlargement),Citation24 reduced ability to proliferateCitation25 and changes in the expression or activity of proteins known as biomarkers of senescence, such as senescence-associated β-galactosidase and others.Citation26,Citation27 Senescent cells are metabolically active and secrete different factors that can affect tissue physiology; thus, accumulation of senescent cells in tissues of old animals can contribute to the development of age-associated pathologies and probably aging itself.Citation28,Citation29 In agreement with a potential role of senescent cells in aging, an increased rate of cellular senescence in vivo and in vitro has been demonstrated in premature aging models.Citation30
Here, we investigate the role of the circadian clock protein BMAL1 in cellular senescence in order to understand the cellular mechanisms of accelerated aging of Bmal1-/- mice. Using tissues from wild-type and Bmal1-/- mice, we found that BMAL1 deficiency leads to increased accumulation of senescent cells in vivo; based on cell culture experiments addressing replicative and stress-induced senescence, we propose the BMAL1-dependent regulation of oxidative stress response as a potential mechanism.
Results
Deficiency of BMAL1 results in an increased amount of senescent cells in vivo.
To study if premature aging of Bmal1-/- is accompanied by accumulation of senescent cells in vivo, we collected different tissues of Bmal1-/- and wild-type mice at the age when Bmal1-/- animals demonstrate multiple features of premature aging (6–10 mo old). Tissues were stained for senescent associated β-galactosidase (SA-β-gal) activity.Citation26 As can be judged by the intensity of developed blue color staining, SA-β-gal activity was significantly increased in the lungs, liver and spleen isolated from Bmal1-/- mice compared with the level of SA-β-gal activity in the same tissues of the age-matched wild-type control (see ). The difference was stronger in the liver and lungs, while in the spleen, the difference between wild type and Bmal1-/- was less obvious; thus, accumulation of senescent cells was tissue-specific. The number of SA-β-gal-positive cells was about 3-fold higher in the lungs (average number of SA-β-gal-positive cells for wild type is 8.8 ± 2.4 and 24.6 ± 6.6 for Bmal1-/-) and in liver (average number of SA-β-gal-positive cells for wild type is 18.6 ± 7.8 and 62.6 ± 4.7 for Bmal1-/-) of Bmal1-/- mice (). We also stained the lungs isolated from 1 mo old wild type and Bmal1-/-, and found that even at this early age, when no signs of premature aging can be observed in Bmal1-/- mice, Bmal1-/- lungs had stronger staining for SA-β-gal (data not shown) than wild-type control. Thus, BMAL1 deficiency results in the increased accumulation of senescent cells in different tissues, which correlates with development of premature aging in Bmal1-/- mice; interestingly, senescent cells can be detected earlier than any morphological signs of premature aging, suggesting that early cellular senescence induced by BMAL1 deficiency may contribute to the development of premature aging of Bmal1-/- mice.
BMAL1 deficiency does not affect cellular replicative senescence in vitro.
The primary mammalian fibroblasts undergo several rounds of proliferation in culture and then reach the arrested stage known as replicative senescence.Citation25 Early replicative senescence was reported for some models of premature aging. Therefore, we decided to investigate if BMAL1 deficiency affects fibroblast replicative senescence in culture. Because the strongest difference in the accumulation of senescent cells was observed in vivo in the lungs, we have chosen primary lung fibroblasts as a model system. Cells were isolated from 6 mo old Bmal1-/- and wild-type mice. As shown in , we did not observe any significant difference in proliferation curves and the onset of replicative senescence between wild-type and Bmal1-/- primary lung fibroblasts. Thus, cell replicative senescence in vitro is not significantly affected by the BMAL1 deficiency and, most likely, cannot be the cause of the increased cellular senescence observed in vivo ().
It is known that mouse primary fibroblasts undergo spontaneous immortalization in culture. We also observed that after several rounds of cell passaging, primary lung fibroblasts became immortalized. Unexpectedly, Bmal1-/- fibroblasts demonstrated a tendency to immortalize earlier when compared with wild-type fibroblasts (average number passages before immortalization for wild type is 14.5 ± 1.5 and 11.4 ± 1.5 for Bmal1-/-). Thus, in contrast to some other models of accelerated aging, BMAL1 deficiency does not result in premature replicative senescence of primary fibroblasts in culture, and different mechanisms are responsible for the accumulation of senescent cells in the tissues of Bmal1-/- mice.
Bmal1-/- cells have increased sensitivity to oxidative stress.
It is unclear if the replicative senescence plays any significant role in the accumulation of senescent cells in vivo;Citation31 the major contributor is probably stress-induced senescence.Citation23 Challenging of cells with different stimuli, such as DNA damage, oncogene activation or aberrant mitogenic signals, can induce premature senescence.Citation23,Citation32,Citation33 Oxidative stress was suggested as one of the major contributors to stress-induced senescence.Citation34 Previously, we have demonstrated that BMAL1 may be involved in reactive oxygen species (ROS) homeostasis;Citation17 treatment with antioxidant N-acetyl cysteine significantly extended the lifespan of Bmal1-/- mice,Citation17,Citation22 suggesting that oxidative stress contributes to the development of premature aging in these animals. Therefore, we decided to investigate the response of Bmal1-/- cells to oxidative stress. Immortalized mouse lung fibroblasts were exposed to different concentrations of hydrogen peroxide for 24, 48 and 72 h. At all tested times, Bmal1-/- cells were significantly more sensitive to hydrogen peroxide than wild-type cells: 24 h WT IC50 = 450 µM, Bmal1-/- IC50 = 385 µM (); 48 h WT IC50 = 200 µM, Bmal1-/- IC50 = 140 µM (); 72 h WT IC50 = 150 µM, Bmal1-/- IC50 = 87.5 µM (). Therefore, BMAL1 deficiency leads to increased cell sensitivity to oxidative stress.
Bmal1-/- cells are more sensitive to oxidative stress-induced growth arrest.
The observed difference in the experiments on sensitivity to hydrogen peroxide prompted us to further investigate the effect of oxidative stress on BMAL1-deficient cells. Oxidative stress induces cell death or growth arrest. First, we investigated if the increased sensitivity to oxidative stress of Bmal1-/- cells was due to the increased level of cell death. Cells were treated with different concentrations of hydrogen peroxide, and the rate of cell death was measured with the Cell Death kit (Sigma). No difference between wild-type and Bmal1-/- cells was detected ().
To compare the effect of oxidative stress on proliferation, an equal number of cells were plated; cells were treated with hydrogen peroxide, and the formed colonies were counted. There was no difference in the number of colonies for the untreated cells. Treatment with hydrogen peroxide resulted in a reduced number of colonies for wild-type and Bmal1-/- cells, and the inhibitory effect was stronger for Bmal1-/- cells (difference between genotypes was statistically significant). At the same time, the relative reduction in the number of colonies after hydrogen peroxide treatment was not as dramatic as the effect of the treatment on colony sizes () or the inhibitory effect observed in the previous set of experiments (). These results suggest that oxidative stress-induced growth arrest was stronger in Bmal1-/- cells. In agreement with that, when cells were treated with hydrogen peroxide only for 24 h and then allowed to grow for additional 120 h in regular growth media, no difference between genotypes was observed (), which indicates that cells arrested by the 24 h hydrogen peroxide treatment resumed proliferation.
Thus, the difference in the sensitivity to oxidative stress between wild-type and Bmal1-/- cells is due to the difference in hydrogen peroxide-induced proliferation arrest rather than the difference in the induced cell death.
Differential effect of BMAL1 deficiency on cell sensitivity to anticancer drugs.
The observed reduced resistance to the hydrogen peroxide treatment of the Bmal1-/- fibroblasts may be a consequence of general changes in genotoxic stress resistance upon BMAL1 deficiency. Alternatively, Bmal1-/- cells can be sensitive to oxidative stress specifically. To investigate if BMAL1 deficiency affects general sensitivity to genotoxic stress, we compared the resistance of wild-type and Bmal1-/- fibroblasts to the toxic action of different anticancer drugs acting through different pathways and affecting cell proliferation/death through different mechanisms. Treatment of cells with the microtubule stabilizing agent Paclitaxel (taxol) did not reveal any difference between wild-type and Bmal1-/- cells (). We also did not detect any significant difference in resistance to antimetabolite 5-Fluorouracil (). At the same time, Bmal1-/- cells were more resistant to the topoisomerase II inhibitor etoposide ( and WT IC50 = 56.33 ng/ml, Bmal1-/- IC50 = 110.9 ng/ml) and the DNA intercalating drug daunorubicin ( and WT IC50 = 45.35 nM, Bmal1-/- IC50 = 95.5 nM).
Based on the results presented in and , we concluded that BMAL1 deficiency specifically affects cell sensitivity to different genotoxic agents: it does not affect sensitivity to some drugs (Paclitaxel, 5-fluorouracil), makes cells more resistant to other agents (etoposide, daunorubicin) and more sensitive to hydrogen peroxide.
Increased sensitivity of Bmal1-/- cells to oxidative stress is p53-independent.
Recently, it was reported that the circadian clock and, specifically, BMAL1 interferes with p53-regulated pathways, and p53-dependent DNA damage response is impaired upon downregulation of BMAL1 expression.Citation35 Interestingly, we found that Bmal1-/- cells are more resistant to some DNA damaging agents, such as etoposide, and more sensitive to hydrogen peroxide; a similar difference in cell sensitivity to DNA damage and oxidative stress was reported for p53-negative cells. Thus, one of the possible mechanisms of the changed sensitivity of Bmal1-/- cells is the effect on the p53 pathway. To study if p53 is involved in the BMAL1-dependent response to oxidative stress, we downregulated p53 activity using GSE56 (a dominant-negative fragment of p53 that can efficiently suppress p53 activity in different cell types).Citation36 We overexpressed GSE56 in wild-type and Bmal1-/- MEFs. As expected, the expression of GSE56 resulted in immortalization of MEFs of both genotypes, which suggests that BMAL1 does not play a significant role in immortalization induced by p53 deficiency. The generated wild-type and Bmal1-/- cells were treated with hydrogen peroxide, similar to the protocol for the fibroblast isolated from the lungs. Wild-type MEFs were still more resistant to the toxic effect of hydrogen peroxide, thus suppression of p53 activity did not affect the difference in sensitivity to hydrogen peroxide between Bmal1-/- and wild-type cells (). Based on these results, we conclude that the increased sensitivity of Bmal1-/- cells to oxidative stress does not depend on p53 function.
BMAL1 deficiency impairs the circadian oscillation of ROS in culture.
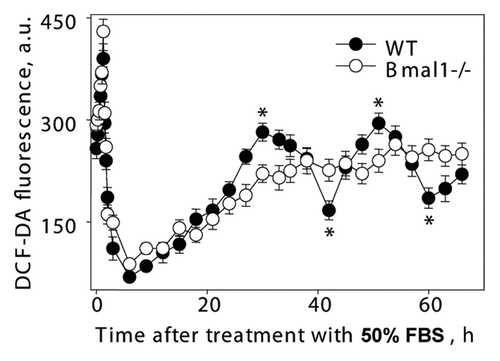
Previously, it was reported that ROS levels demonstrate circadian oscillations in tissues of wild-type mice. Circadian rhythms in vivo (rhythms in gene expression, electrical activity and metabolism) can be modeled in cell culture.Citation37 We used established methods for induction of circadian oscillation in cell cultureCitation37 to investigate if BMAL1 directly regulates ROS homeostasis in cells. Cells kept in monolayer for 72 hrs were treated with 50% FBS for 2 h and transferred to serumfree media. Treatment with mitogenic stimuli induces transient induction of ROS level (80–83); in agreement with that, the treatment with 50% FBS resulted in sharp changes in ROS levels, which were similar in both wild-type and Bmal1-/- cells (see , time points between 0 and 3 h), suggesting that the normal response of ROS levels to serum stimulation was not compromised in Bmal1-/- cells. After a sharp peak of induction, ROS levels decreased in wild-type and Bmal1-/- cells with similar kinetics and reached a minimum in 6 h. After that, the ROS level slowly increased and reached another peak 30 h after the serum treatment in wild-type cells. One more peak was detected 51 h after the induction. Thus, serum treatment induced circadian oscillation of ROS levels in wild-type MEFs (two peaks with period about 21 h were detected). In contrast, the ROS levels in Bmal1-/- cells initially increased with a similar kinetics for 21 hrs after treatment but then reached plateau and did not demonstrate any rhythmic oscillations. Thus, circadian rhythms in oscillation of intracellular ROS level detected in cell culture depend on the expression of BMAL1. These results suggest that BMAL1 is directly involved in the regulation of ROS homeostasis in cells, and that BMAL1 activity may contribute to the observed difference in the response of the BMAL1-deficient cells to oxidative stress.
Discussion
Aging is a complex biological process; our understanding of the mechanisms of aging is in its emerging state. Recent studies demonstrated that the rate of aging is regulated: for example, activation of the TOR or IGF pathways accelerates aging, while suppression of these pathways was shown to increase longevity.Citation38,Citation39 At the same time, the existence of anti-aging or gerosuppressor systems has also been shown:Citation40 downregulation of pathways controlling genome stability resulted in premature aging and upregulation increased longevity. Like other physiological systems, the activity of the circadian clock is affected by agingCitation41 and, conversely, this may contribute to the control of aging. Several recent reports connect the circadian clock and aging in different organismsCitation14–Citation18,Citation42 including humans, suggesting an evolutionarily conserved connection between these two phenomena, although the molecular and cellular mechanisms remain mostly unknown.
Accumulation of senescent cells in aged tissues contributes to the pathology of aging.Citation23 Cell senescence can be induced through different mechanisms.Citation23 While the replicative senescence was the first type of experimentally induced senescence, its role in the age-associated senescence in vivo is unclear.Citation31 In contrast to that, stress-induced senescence significantly contributes to the age-associated senescence and was suggested as one anticancer mechanisms in vivo.Citation23 We investigated the effect of BMAL1 deficiency on senescence in vivo and in vitro. In situ staining revealed a significantly increased number of cells positive for SA-β-gal in the liver, lungs and spleen of Bmal1-/- mice. The difference in the number of senescent cells between wild type and Bmal1-/- can be detected already in one month old mice, before the premature aging phenotype of Bmal1-/- mice develops. Most likely, the increased number of senescent cells in the tissues of Bmal1-/- mice is a consequence of stress-induced senescence. Indeed, we did not detect any significant difference in replicative senescence, but observed an increased sensitivity of Bmal1-/- cells to hydrogen peroxide. Oxidative stress is a powerful activator of the stressinduced cellular senescence and is probably one of the major causes of the stress-induced senescence in vivo.Citation23,Citation34
Previously, we and others demonstrated that the circadian clock is involved in the regulation of ROS levels in vivo and in vitro.Citation17,Citation43 These results further support the idea that circadian clock-dependent control of ROS homeostasis and oxidative stress response plays an essential role in the regulation of aging. Indeed, accumulation of senescent cells in BMAL1-deficient animals has been observed in the tissues that normally are subjects for increased levels of oxidative damage. For example, the lung cells are exposed to a high level of oxygen; the liver cells are exposed to different organic toxins originating from food; the spleen has a large number of immune cells, many of which produce high levels of ROS.
Previously it was reported that BMAL1 regulates organism and cell sensitivity to genotoxic stressCitation44 and is involved in the control of cell cycleCitation45,Citation46 and DNA damage-induced cell cycle arrest.Citation35 While BMAL1 deficiency reduces proliferation of primary hepatocytesCitation46 and hair follicle epithelium,Citation45 we did not observe a reduced rate of proliferation in BMAL1-deficient fibroblasts. Downregulation of BMAL1 expression with shRNAs in tumor cells made cells more resistant to DNA damage-induced arrest.Citation35 We also observed that Bmal1-/- cells are more resistant to some DNA damaging agents, such as etoposide or daunorubicin; at the same time, no difference was observed for another DNA damaging agent, such as 5-fluorouracil. It was suggested that BMAL1 regulates cell cycle arrest through the interference with the p53 pathway and regulation of p21/Inkk1 expression;Citation35 however, we observed a p53-independent effect of BMAL1. Several recent reports addressed the role of the circadian clock and different circadian clock proteins in cell death and proliferation; however, the results of different studies often contradict each other.Citation14,Citation35,Citation44–Citation47 We suggest that different circadian proteins play different roles in the control of cell death and proliferation; additionally, even for the same protein, the effect is cell type- and treatment-specific.
A connection between the circadian clock and aging was recently suggested.Citation48 Oxidative stress and DNA damage responses are among pathways implicated in the control of aging. The role demonstrated here of the circadian clock transcriptional factor BMAL1 in the regulation of oxidative stress response provides a mechanistic link between aging and the circadian clock. However, several important questions need to be answered. Does early accumulation of senescent cells in the tissues of Bmal1-/- mice contribute to the development of aging in these mice? Is the control of ROS homeostasis and oxidative stress response the only mechanism through which the circadian clock protein BMAL1 regulates senescence in vivo or can another clock control pathway, such as DNA repair,Citation49,Citation50 contribute? Is accelerated accumulation of senescent cells caused by the disruption of the circadian clock or is it BMAL1 deficiency-specific? Recently an increased level of senescent cells was reported in vasculature of Per2-deficient mice,Citation51 arguing that the circadian clock mechanism may be important.
Finally, more and more data are accumulated on the role of the circadian clock in the genotoxic stress response; the chrono-therapeutic approach was proposed in order to improve the efficiency of anticancer treatments.Citation52 However, a complex relationship between the circadian clock and genotoxic stress response suggests that further research is necessary.Citation10 Based on the presented data, we propose that the circadian clock may act as a gerosuppressor system and rational induction of circadian clock activity can probably delay the development of age-associated pathology and aging.
Materials and Methods
Animals.
Bmal1-/- mice were obtained from Dr. C. Bradfield (University of Wisconsin);Citation19 details of target gene knockout strategies and animal generations can be found in the above cited publications. Mice were backcrossed to C57BL/6J inbred strain (The Jackson Laboratory) for 10 generations. Wild-type and knockout mice were generated by breeding of heterozygotic parents. Animals were maintained on a 12:12 light:dark cycle, with lights on at 7:00 a.m., on a regular diet. All animal studies were conducted in accordance with the regulations of the Committee on Animal Care and Use at Cleveland State University.
Tissue collection, β-gal detection and histology.
For histological analysis, animals were sacrificed and organs were immediately removed and frozen in OST solution. Four µm sections were fixed in 4% paraformaldehyde/PBS solution and stained for β gal activity X-gal/potassium ferricyanide/potassium ferrocyanide protocol. Sections were analyzed under microscopes and photographed. Numbers of blue cells on stained tissue images was counted independently by three persons blind to genotype. Sections were counterstained with hematoxylin/eosin according to standard protocol to confirm normal tissue histology.
Cell culture.
All cells were cultured in Dulbecco's modified Eagle's medium (LRI) supplemented with 10% fetal bovine serum premium (ATLANTA Biologicals) and 10,000 units Of Penicillin G and 10,000 µg/ml streptomycin (LRI). Cells were maintained at 37°C in humid atmosphere with 5% CO2 in air.
To generate primary lung fibroblasts, lungs isolated from WT AND Bmal1 KO mice were cut to small pieces and placed in cell growth media. After 72 h, media were replaced, and all unattached cells were removed.
To generate culture of mouse embryonic fibroblasts (MEFs), mice were bred and dams were sacrificed (euthanized using carbon dioxide inhalation) on day 13–14 of gestation and cell culture was generated similar to lung fibroblast culture. Constructs for expression of dominant-negative p53 fragment GSE56 were previously described.
Replicative senescence experiments were performed according to following protocol: cells were collected on indicated days; at every passage, total number of cells were counted and cells were plated at density 104/cm2.
H2O2 and anticancer drug sensitivity assay.
WT and BMAL1-/- cells were plated in 96-well plates at a density of 1 × 103 cells per well. Twenty-four h after plating, cells were treated with different concentrations of hydrogen peroxide, taxol, daunorubicin, etoposide and 5′FU (Sigma-Aldrich) for 24, 48 and 72 h. Cells survival was estimated using two independent protocols. MTT assay, incorporation and detection of MTT reagent (Sigma) was performed according to the manufacturer protocol. In an independent set of experiments, treated cells were fixed with 0.2% glutaraldehyde (Fischer Scientific) and stained with 0.05% crystal violet solution dye was extracted with 1% SDS solution and amount was estimated as optical density at 570 nM using VictorCitation3 1,420 plate reader. Cell death was estimated using Cyto-tox ONE Homogeneous Membrane Integrity assay Kit according to manufacturer's instructions (Promega).
Colony formation assay.
WT and BMAL1 immortalized lung fibroblasts were plated in 6-well plates at a density 5 × 102 cells per well. After 24 h of plating, cells were treated with 250 nM H2O2 for the duration of the experiment. In some experiments, cells were treated with H2O2 for 24 h and then transferred to regular growth media. After 7 d, cells were fixed with 0.2% glutaraldehyde and stained with 0.05% crystal violet solution. Number of colonies were counted independently by three persons blind to genotype.
Measurement of ROS levels.
ROS levels were determined in tissue extracts using ROS-sensitive fluorescent dye as described.Citation53 Briefly, cells were mixed with dichlorodihydrofluorescein (DCF) in PBS and incubated in the dark at 370°C for 30 min. Fluorescence of dye loaded cells was detected with FACScalibur cell sorter and analyzed with appropriate software.
Statistical analysis.
In all cell survival experiments, at least three replicas were used All experiments were repeated at least three times. Unpaired t-test was used for statistical analysis. p < 0.05 was considered as statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Figures and Tables
Figure 1 BMAL1 deficiency leads to accumulation of senescent cells in vivo. Representative histological images of the lungs (left column), liver (middle column) and spleen (right column) isolated from 6 mo old wildtype (upper row) or Bmal1-/- (lower row) mice were stained for senescent associated β-galactosidase.
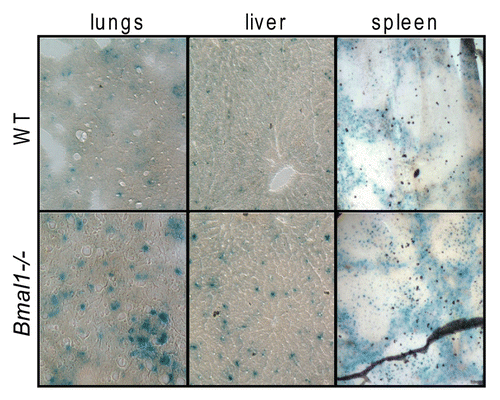
Figure 2 BMAL1 deficiency does not affect fibroblast replicative senescence in vitro. Growth curves of primary lung fibroblasts isolated from wild type (closed circles) and Bmal1-/- (open circles). On indicated days cells were counted and plated with equal density.
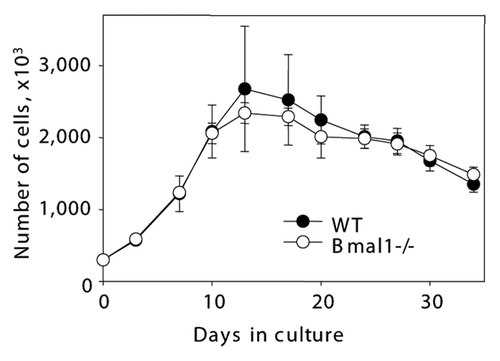
Figure 3 BMAL1-deficient cells have increased sensitivity to hydrogen peroxide. Relative survival of wild-type (black circles) and Bmal1-/- (open circles) fibroblasts treated with indicated concentrations of hydrogen peroxide for 24 h (A), 48 h (B) and 72 h (C). Survival of untreated cells was set as 100%. *p < 0.01.
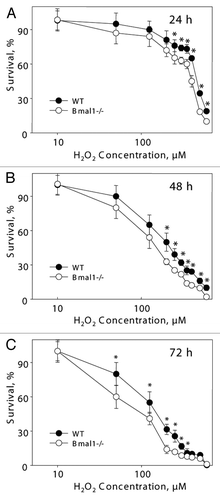
Figure 4 Increased sensitivity of BMAL1-deficient cells to hydrogen peroxide is due to increased growth inhibition. (A) Relative cell death of wildtype (black circles) and Bmal1-/- (open circles) fibroblasts treated with indicated concentrations of hydrogen peroxide for 4 h were calculated based on fluorescent signal using kit. Signal of untreated cells was set as 0 and signal of cells lysed with SDS was set as 1.0. (B) Numbers of colonies formed by wild-type (black bars) and Bmal1-/- (gray bars) fibroblasts. Equal amount of cells were plated, cells grew in regular growth media for 96 h (U/T), or in the presence of 250 mM of hydrogen peroxide for 96 h (250 mM H2O2), or treated with 250 mM of hydrogen peroxide for 24 h and then in regular growth media for additional 72 h (250 mM H2O2/media). (C) Representative images of colony formation experiment. *p < 0.05.
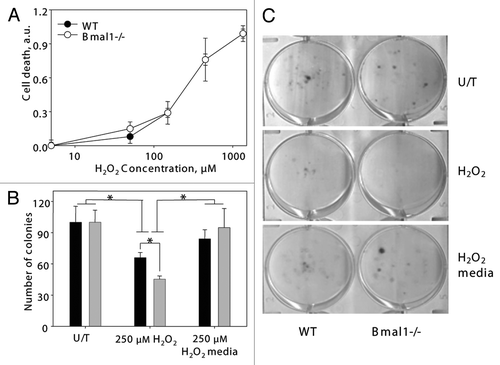
Figure 5 BMAL1 deficiency cells have differential sensitivity to anticancer drugs. Relative survival of wild-type (black circles) and Bmal1-/- (open circles) fibroblasts treated with indicated concentrations of paclitaxel (A), 5-fluorouracil (B), daunorubicin (C) and etoposide (D). Survival of untreated cells was set as 100%. *p < 0.01.
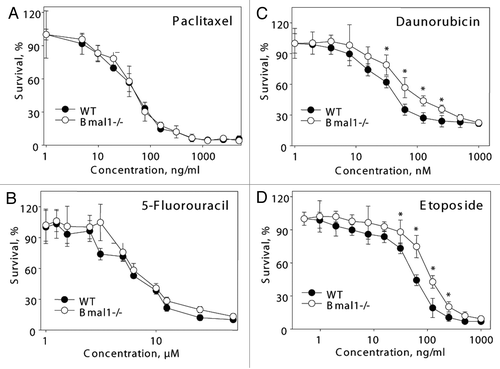
Figure 6 Increased sensitivity of BMAL1-deficient cells to hydrogen peroxide is p53-independent. Relative survival of GSE 56 (dominant negative fragment of p53) wild-type (black circles) and Bmal1-/- (open circles) fibroblasts treated with indicated concentrations of hydrogen peroxide for 72 h. Survival of untreated cells was set as 100%. *p < 0.01.
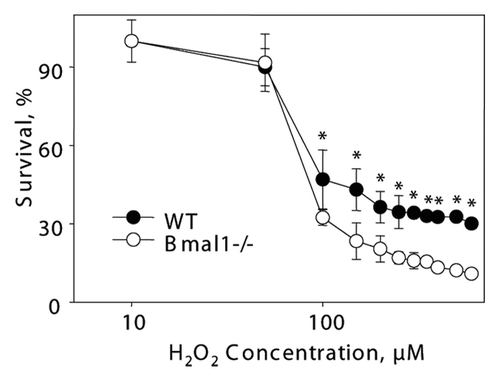
Figure 7 Circadian rhythms of intracellular ROS level can be induced in cell culture. Confluent wild-type (black circles) and Bmal1-/- (open circles) fibroblasts were maintained in DMEM with 5% FBS for 7 d, at time 0 h media were replaced with media with 50% FBS (serum shock); cells were incubated for 2 h and then 50% FBS media was replaced with serum free media; cells were collected every 10 min during serum shock and every 3 h after serum replacement and ROS level was measured as described in Materials and Methods. *Statistically significant (p < 0.001) difference between peak and throw for wild-type cells.
Acknowledgments
We thank William Samsa for editorial help. This work was supported by AHA grant 0835155N to and NIH grants 1R03AG033881 R.V.K.
References
- Ko CH, Takahashi JS. Molecular components of the mammalian circadian clock. Hum Mol Genet 2006; 15:271 - 277; PMID: 16987893; http://dx.doi.org/10.1093/hmg/ddl207
- Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell 2008; 134:728 - 742; PMID: 18775307; http://dx.doi.org/10.1016/j.cell.2008.08.022
- Reddy AB, O'Neill JS. Healthy clocks, healthy body, healthy mind. Trends Cell Biol 2010; 20:36 - 44; PMID: 19926479; http://dx.doi.org/10.1016/j.tcb.2009.10.005
- Borgs L, Beukelaers P, Vandenbosch R, Belachew S, Nguyen L, Malgrange B. Cell “circadian” cycle: new role for mammalian core clock genes. Cell Cycle 2009; 8:832 - 837; PMID: 19221497; http://dx.doi.org/10.4161/cc.8.6.7869
- Zhdanova IV. Sleep and its regulation in zebrafish. Rev Neurosci 2011; 22:27 - 36; PMID: 21615259; http://dx.doi.org/10.1515/RNS.2011.005
- Gachon F, Bonnefont X. Circadian clock-coordinated hepatic lipid metabolism: only transcriptional regulation?. Aging (Albany NY) 2010; 2:101 - 106; PMID: 20354271
- Dardente H, Cermakian N. Molecular circadian rhythms in central and peripheral clocks in mammals. Chronobiol Int 2007; 24:195 - 213; PMID: 17453843; http://dx.doi.org/10.1080/07420520701283693
- Yu EA, Weaver DR. Disrupting the circadian clock: gene-specific effects on aging, cancer, and other phenotypes. Aging (Albany NY) 2011; 3:479 - 493; PMID: 21566258
- Young ME, Bray MS. Potential role for peripheral circadian clock dyssynchrony in the pathogenesis of cardiovascular dysfunction. Sleep Med 2007; 8:656 - 667; PMID: 17387040; http://dx.doi.org/10.1016/j.sleep.2006.12.010
- Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science 2010; 330:1349 - 1354; PMID: 21127246; http://dx.doi.org/10.1126/science.1195027
- Geyfman M, Andersen B. Clock genes, hair growth and aging. Aging (Albany NY) 2010; 2:122 - 128; PMID: 20375466
- Gery S, Koeffler HP. Circadian rhythms and cancer. Cell Cycle 2010; 9:1097 - 1103; PMID: 20237421; http://dx.doi.org/10.4161/cc.9.6.11046
- Khapre RV, Samsa WE, Kondratov RV. Circadian regulation of cell cycle: Molecular connections between aging and the circadian clock. Ann Med 2010; 42:404 - 415; PMID: 20568980; http://dx.doi.org/10.3109/07853890.2010.499134
- Antoch MP, Gorbacheva VY, Vykhovanets O, Toshkov IA, Kondratov RV, Kondratova AA, et al. Disruption of the circadian clock due to the Clock mutation has discrete effects on aging and carcinogenesis. Cell Cycle 2008; 7:1197 - 1204; PMID: 18418054; http://dx.doi.org/10.4161/cc.7.9.5886
- Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002; 111:41 - 50; PMID: 12372299; http://dx.doi.org/10.1016/S0092-8674(02)00961-3
- Dubrovsky YV, Samsa WE, Kondratov RV. Deficiency of circadian protein CLOCK reduces lifespan and increases age-related cataract development in mice. Aging (Albany NY) 2010; 2:936 - 944; PMID: 21149897
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev 2006; 20:1868 - 1873; PMID: 16847346; http://dx.doi.org/10.1101/gad.1432206
- Krishnan N, Kretzschmar D, Rakshit K, Chow E, Giebultowicz JM. The circadian clock gene period extends healthspan in aging Drosophila melanogaster. Aging (Albany NY) 2009; 1:937 - 948; PMID: 20157575
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000; 103:1009 - 1017; PMID: 11163178; http://dx.doi.org/10.1016/S0092-8674(00)00205-1
- Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science 1998; 280:1564 - 1569; PMID: 9616112; http://dx.doi.org/10.1126/science.280.5369.1564
- Hogenesch JB, Gu YZ, Jain S, Bradfield CA. The basic-helix-loop-helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA 1998; 95:5474 - 5479; PMID: 9576906; http://dx.doi.org/10.1073/pnas.95.10.5474
- Kondratov RV, Vykhovanets O, Kondratova AA, Antoch MP. Antioxidant N-acetyl-L-cysteine ameliorates symptoms of premature aging associated with the deficiency of the circadian protein BMAL1. Aging (Albany NY) 2009; 1:979 - 987; PMID: 20157581
- Campisi J. Senescent cells, tumor suppression and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513 - 522; PMID: 15734683; http://dx.doi.org/10.1016/j.cell.2005.02.003
- Goldstein S. Replicative senescence: the human fibroblast comes of age. Science 1990; 249:1129 - 1133; PMID: 2204114; http://dx.doi.org/10.1126/science.2204114
- Hayflick L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp Cell Res 1965; 37:614 - 636; PMID: 14315085; http://dx.doi.org/10.1016/0014-4827(65)90211-9
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92:9363 - 9367; PMID: 7568133; http://dx.doi.org/10.1073/pnas.92.20.9363
- Shelton DN, Chang E, Whittier PS, Choi D, Funk WD. Microarray analysis of replicative senescence. Curr Biol 1999; 9:939 - 945; PMID: 10508581; http://dx.doi.org/10.1016/S0960-9822(99)80420-5
- Coppé JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008; 6:2853 - 2868; PMID: 19053174; http://dx.doi.org/10.1371/journal.pbio.0060301
- Faragher RG, Kipling D. How might replicative senescence contribute to human ageing?. Bioessays 1998; 20:985 - 991; PMID: 10048298; http://dx.doi.org/10.1002/(SICI)1521-1878(199812)20:12<985::AIDBIES4>3.3.CO;2-1
- Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell 2009; 8:311 - 323; PMID: 19627270; http://dx.doi.org/10.1111/j.1474-9726.2009.00481.x
- Smith JR, Pereira-Smith OM. Replicative senescence: implications for in vivo aging and tumor suppression. Science 1996; 273:63 - 67; PMID: 8658197; http://dx.doi.org/10.1126/science.273.5271.63
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593 - 602; PMID: 9054499; http://dx.doi.org/10.1016/S0092-8674(00)81902-9
- Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene 1998; 16:1113 - 1123; PMID: 9528853; http://dx.doi.org/10.1038/sj.onc.1201862
- Chen Q, Fischer A, Reagan JD, Yan LJ, Ames BN. Oxidative DNA damage and senescence of human diploid fibroblast cells. Proc Natl Acad Sci USA 1995; 92:4337 - 4341; PMID: 7753808; http://dx.doi.org/10.1073/pnas.92.10.4337
- Mullenders J, Fabius AW, Madiredjo M, Bernards R, Beijersbergen RL. A large scale shRNA barcode screen identifies the circadian clock component ARNTL as putative regulator of the p53 tumor suppressor pathway. PLoS ONE 2009; 4:4798; PMID: 19277210; http://dx.doi.org/10.1371/journal.pone.0004798
- Ossovskaya VS, Mazo IA, Chernov MV, Chernova OB, Strezoska Z, Kondratov R, et al. Use of genetic suppressor elements to dissect distinct biological effects of separate p53 domains. Proc Natl Acad Sci USA 1996; 93:10309 - 10314; PMID: 8816796; http://dx.doi.org/10.1073/pnas.93.19.10309
- Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998; 93:929 - 937; PMID: 9635423; http://dx.doi.org/10.1016/S0092-8674(00)81199-X
- Brown-Borg HM. Hormonal regulation of longevity in mammals. Ageing Res Rev 2007; 6:28 - 45; PMID: 17360245; http://dx.doi.org/10.1016/j.arr.2007.02.005
- Blagosklonny MV. Aging: ROS or TOR. Cell Cycle 2008; 7:3344 - 3354; PMID: 18971624; http://dx.doi.org/10.4161/cc.7.21.6965
- Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasiprogram. Cell Cycle 2010; 9:3151 - 3156; PMID: 20724817; http://dx.doi.org/10.4161/cc.9.16.13120
- Hofman MA, Swaab DF. Living by the clock: the circadian pacemaker in older people. Ageing Res Rev 2006; 5:33 - 51; PMID: 16126012; http://dx.doi.org/10.1016/j.arr.2005.07.001
- Bauer J, Antosh M, Chang C, Schorl C, Kolli S, Neretti N, et al. Comparative transcriptional profiling identifies takeout as a gene that regulates life span. Aging (Albany NY) 2010; 2:298 - 310; PMID: 20519778
- Hardeland R, Coto-Montes A, Poeggeler B. Circadian rhythms, oxidative stress and antioxidative defense mechanisms. Chronobiol Int 2003; 20:921 - 962; PMID: 14680136; http://dx.doi.org/10.1081/CBI-120025245
- Gorbacheva VY, Kondratov RV, Zhang R, Cherukuri S, Gudkov AV, Takahashi JS, et al. Circadian sensitivity to the chemotherapeutic agent cyclophosphamide depends on the functional status of the CLOCK/BMAL1 transactivation complex. Proc Natl Acad Sci USA 2005; 102:3407 - 3412; PMID: 15689397; http://dx.doi.org/10.1073/pnas.0409897102
- Lin KK, Kumar V, Geyfman M, Chudova D, Ihler AT, Smyth P, et al. Circadian clock genes contribute to the regulation of hair follicle cycling. PLoS Genet 2009; 5:1000573; PMID: 19629164; http://dx.doi.org/10.1371/journal.pgen.1000573
- Gréchez-Cassiau A, Rayet B, Guillaumond F, Teboul M, Delaunay F. The circadian clock component BMAL1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J Biol Chem 2008; 283:4535 - 4542; PMID: 18086663; http://dx.doi.org/10.1074/jbc.M705576200
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, et al. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA 2007; 104:3342 - 3347; PMID: 17360649; http://dx.doi.org/10.1073/pnas.0611724104
- Kondratov RV. A role of the circadian system and circadian proteins in aging. Ageing Res Rev 2007; 6:12 - 27; PMID: 17369106; http://dx.doi.org/10.1016/j.arr.2007.02.003
- Kang TH, Reardon JT, Kemp M, Sancar A. Circadian oscillation of nucleotide excision repair in mammalian brain. Proc Natl Acad Sci USA 2009; 106:2864 - 2867; PMID: 19164551; http://dx.doi.org/10.1073/pnas.0812638106
- Kang TH, Sancar A. Circadian regulation of DNA excision repair: implications for chrono-chemotherapy. Cell Cycle 2009; 8:1665 - 1667; PMID: 19411851; http://dx.doi.org/10.4161/cc.8.11.8707
- Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, et al. Vascular disease in mice with a dysfunctional circadian clock. Circulation 2009; 119:1510 - 1517; PMID: 19273720; http://dx.doi.org/10.1161/CIRCULATIONAHA.108.827477
- Hrushesky WJ. Circadian timing of cancer chemotherapy. Science 1985; 228:73 - 75; PMID: 3883493; http://dx.doi.org/10.1126/science.3883493
- Radak Z, Chung HY, Naito H, Takahashi R, Jung KJ, Kim HJ, et al. Age-associated increase in oxidative stress and nuclear factor kappaB activation are attenuated in rat liver by regular exercise. FASEB J 2004; 18:749 - 750; PMID: 14766800