Abstract
It is now widely recognized that the tumor microenvironment promotes cancer cell growth and metastasis via changes in cytokine secretion and extracellular matrix remodeling. However, the role of tumor stromal cells in providing energy for epithelial cancer cell growth is a newly emerging paradigm. For example, we and others have recently proposed that tumor growth and metastasis is related to an energy imbalance. Host cells produce energy-rich nutrients via catabolism (through autophagy, mitophagy, and aerobic glycolysis), which are then transferred to cancer cells to fuel anabolic tumor growth. Stromal cell-derived L-lactate is taken up by cancer cells and is used for mitochondrial oxidative phosphorylation (OXPHOS) to produce ATP efficiently. However, “parasitic” energy transfer may be a more generalized mechanism in cancer biology than previously appreciated. Two recent papers in Science and Nature Medicine now show that lipolysis in host tissues also fuels tumor growth. These studies demonstrate that free fatty acids produced by host cell lipolysis are re-used via beta-oxidation (beta-OX) in cancer cell mitochondria. Thus, stromal catabolites (such as lactate, ketones, glutamine and free fatty acids) promote tumor growth by acting as high-energy onco-metabolites. As such, host catabolism, via autophagy, mitophagy and lipolysis, may explain the pathogenesis of cancer-associated cachexia and provides exciting new druggable targets for novel therapeutic interventions. Taken together, these findings also suggest that tumor cells promote their own growth and survival by behaving as a “parasitic organism.” Hence, we propose the term “Parasitic Cancer Metabolism” to describe this type of metabolic coupling in tumors. Targeting tumor cell mitochondria (OXPHOS and beta-OX) would effectively uncouple tumor cells from their hosts, leading to their acute starvation. In this context, we discuss new evidence that high-energy onco-metabolites (produced by the stroma) can confer drug resistance. Importantly, this metabolic chemo-resistance is reversed by blocking OXPHOS in cancer cell mitochondria with drugs like Metformin, a mitochondrial “poison.” In summary, parasitic cancer metabolism is achieved architecturally by dividing tumor tissue into at least two well-defined opposing “metabolic compartments:” catabolic and anabolic.
Introduction
The concept that tumor-host interactions are crucial in tumor progression is now well-accepted.Citation1,Citation2 In fact, Stephen PagetCitation3 first proposed the “seed and soil” hypothesis in 1889, which states that cancer cells (“the seeds”) metastasize systemically and grow best in the most tumor-promoting host organs or the most “fertile soil.”Citation4–Citation6 In this context, tumor cells corrupt or transform their microenvironment in order to generate new blood vesselsCitation7,Citation8 to support their oxygen requirements.Citation9,Citation10 Closely linked to this idea, we and others have begun to view cancer as a “parasitic disease” that “steals” energy-rich metabolites from the host microenvironmentCitation11–Citation13 ().
The tumor stroma, which is composed of fibroblasts, adipocytes, endothelial cells and macrophages, lies in extremely close proximity to cancer cells, and can directly promote tumor growth.Citation15–Citation20 Most research on the mechanism(s) by which the stroma promotes tumor growth has focused on changes in the extracellular matrix and the increased secretion of tumor promoting cytokines, such as IL-6, IL-8, SDF1, VEGF and TGFβ.Citation20,Citation21 However, little is known regarding the metabolic properties of the tumor stroma. Several independent studies have recently highlighted the importance of metabolic coupling between cancer cells and other host cells to drive a form of “parasitic cancer metabolism.” This metabolic-coupling promotes epithelial tumor cell growth and metastasis.
Cancer Cells Behave as Metabolic Parasites: Glycolysis in Fibroblasts Promotes Mitochondrial Metabolism in Cancer Cells, Fueling Tumor Growth
Malaria infection is due to an intracellular parasite (Plasmodium) that induces oxidative stress in host cells,Citation22 resulting in the onset of host cell autophagy,Citation23 which supplies the parasites with high-energy metabolites and chemical building blocks. T. cruzi (Chagas disease), another intracellular parasite, also uses the same mechanism(s) involving oxidative stressCitation24,Citation25 and host cell autophagy.Citation26–Citation28 In addition, T. cruzi induces lipolysis of triglycerides in adipocytes, to generate free fatty acids that it can use as a fuel supply.Citation29
Similarly, our group's recent results show that epithelial cancer cells are extracellular parasites that induce oxidative stress in adjacent stromal fibroblasts, by secreting hydrogen peroxide.Citation11,Citation30 This oxidative stress causes stromal fibroblast activation, with the upregulation of HIF1-α activity driving autophagy, mitophagy and aerobic glycolysis in the tumor stroma.Citation31–Citation33 In parallel, oxidative stress also activates NFκB, further accelerating autophagy and the local secretion of inflammatory cytokines from activated fibroblasts. Thus, oxidative stress, autophagy, aerobic glycolysis and inflammation are inextricably linked in the tumor stroma.Citation30,Citation31,Citation34–Citation37 As such, the stroma provides catabolized nutrients to “fuel” the anabolic growth of tumor cells by enhancing their mitochondrial activity.Citation30 L-lactate derived from glycolytic fibroblasts is transferred to cancer cells and is used to generate energy via oxidative mitochondrial metabolism (OXPHOS). Similarly, ketone bodies and glutamine derived from host cell catabolism can also fuel the mitochondrial activity of adjacent epithelial cancer cellsCitation11,Citation38,Citation39 (). We have termed this new form of parasitic cancer metabolism the “reverse Warburg effect” (since increased glycolysis occurs in fibroblasts rather than tumor cells) or the “auotphagic tumor stroma model of cancer” (since tumor cells induce autophagy and mitophagy in adjacent fibroblasts).Citation40–Citation43 (For recent reviews on the conventional “Warburg effect,” please see refs. Citation44–Citation49).
Thus, stromal metabolites such as L-lactate, ketones and glutamine promote tumor growth by acting as high-energy onco-metabolites ().Citation50 As such, intracellular parasites and cancer cells use similar metabolic mechanism(s) for survival. These mechanistic insights have important implications for the design of novel therapeutic interventions for cancer treatment and prevention. For example, chloroquine is an effective anti-malarial drug (and also has anticancer activity), because it inhibits autophagy and cuts off “the fuel supply,” by preventing energy transfer from host to parasite.Citation11,Citation42
Although this model of “parasitic metabolism” by tumors has only recently been proposed, energy transfer between cells to fuel growth is in fact not a new invention, but instead reflects the co-optation of a normal physiological process by tumor cells. Metabolic-coupling already exists physiologically in many different organ systems, such as in skeletal muscle, the brain and the ovary (). In skeletal muscle, fast-twitch fibers are glycolytic, and slow-twitch fibers are oxidative. Fast-twitch fibers produce L-lactate, which is transferred to slow-twitch fibers to fuel oxidative mitochondrial metabolism.Citation51 This process is known as the “lactate shuttle.” In the brain, a similar lactate shuttle exists. In this context, astrocytes are glycolytic and neurons are oxidative; this energy-sharing symbiosis has been termed “neuron-glia metabolic coupling.”Citation52,Citation53 Finally, in the ovary, the granulosa cells are glycolytic and the oocyte is oxidative, and this metabolic coupling has been exploited for in vitro fertilization (IVF), to maintain viable oocytes.Citation54,Citation55 Metabolic-coupling also exist between organ systems, such as in the muscle-kidney or muscle-liver glutamine shuttles.Citation56 Therefore, metabolic coupling is a widely used normal physiologic process that has been adopted by epithelial tumor cells, to promote their own growth and survival of the tumor as a “parasitic organism.”
In further support of the idea that a “lactate shuttle” also exists in human tumors, we have shown that the distribution of lactate transporters is highly compartmentalized in human breast cancers (). MCT4, which functions in the extrusion of L-lactate and is a marker of oxidative stress and aerobic glycolysis, is largely confined to cancer-associated fibroblasts in the tumor stroma.Citation14,Citation57 In contrast, MCT1, which drives L-lactate uptake is specifically localized to epithelial cancer cells.Citation57 Importantly, MCT transporters have a broad specificity and can also function in the shuttling of ketone bodies from the tumor stroma to epithelial cancer cells.
Two of our most recent papers published in the journal Cell Cycle directly demonstrate that human breast cancer cells use OXPHOS, mitochondria and parasitic metabolic-coupling.Citation14,Citation58
Since we believe that both skeletal muscle and human tumors are constructed on the same metabolic principles (with adjacent glycolytic and oxidative compartments), we subjected tumors to a specific histochemical stain that has been used for over 50 years to visualize these two compartments in muscle.Citation14 This histochemical stain allows detection of the functional activity of mitochondrial complex IV [cytochrome C oxidase (COX)], a key component of the respiratory chain and OXPHOS.Citation14 In skeletal muscle, fast-twitch fibers are glycolytic and are COX-negative. In contrast, slow-twitch fibers are oxidative and are COX-positive. (left part) shows the distribution of these two metabolic fiber types.
Similar results were obtained in human breast cancers.Citation14 The tumor stroma is largely COX-negative and hence more glycolytic. Conversely, breast cancer epithelial cell “nests” are COX-positive and are therefore more oxidative.Citation14 A representative image is shown in (right part). These findings provide striking functional data to support the idea that tumors use metabolic-coupling and shuttle L-lactate, resulting in a net energy transfer from the tumor stroma to epithelial cancer cells.
Furthermore, using a bioinformatics approach, we demonstrated that epithelial tumor cells laser captured from primary human breast cancers had upregulated mitochondrial genes not seen in tumor fibroblasts. This cancer cell mitochondrial gene signature consisted largely of OXPHOS-related gene transcripts that were upregulated (> 4-fold) in epithelial cancer cells, compared with adjacent stromal tissue. Using this OXPHOS-signature, we showed that this type of oxidative mitochondrial metabolism is significantly upregulated in most human breast cancers (> 2,000 patients examined; p < 10−20), in both ER(+) and ER(−) tumor subtypes.Citation14 In ER(−) patients, this MITO/OXPHOS signature was specifically associated with tumor metastasis.Citation14
In addition, we have used an shRNA approach to genetically create glycolytic fibroblasts, by knocking-down the major mitochondrial transcription factor, namely, TFAM.Citation58 As predicted, these TFAM-deficient fibroblasts underwent metabolic re-programming toward a more glycolytic state, with loss of respiratory chain components as well as increased hydrogen peroxide and L-lactate production.Citation58 These glycolytic fibroblasts also significantly promoted tumor growth using a human tumor xenograft model, employing MDA-MB-231 breast cancer cells.Citation58
Adipocytes Provide Fatty Acids to Ovarian Cancer Cells to Fuel Mitochondrial β-Oxidation and Promote Tumor Growth
A new study by Nieman et al. published in Nature Medicine has now evaluated the growth-promoting properties of the omentum in metastatic ovarian cancer from a metabolic perspective.Citation59 They show that the proximity of adipocytes to ovarian cancer cells leads to lipolysis in adipocytes, with the release of free fatty acids.Citation59 Ovarian cancer cells adjacent to these transformed or activated adipocytes show increased fatty acid uptake and utilization via β-oxidation in mitochondria,Citation59 demonstrating that metabolic-coupling also occurs between these two cellular compartments (summarized in and ).
The lipolytic enzyme, hormone-sensitive lipase (HSL) and the fatty acid transport protein, FABP4, were found to be upregulated in adipocytes in omental metastases as compared with primary ovarian cancers.Citation59 FABP4(−/−) mice injected intraperitoneally with ovarian cancer cells, had a significant reduction in tumor burden as compared with wild-type mice.Citation59 This highlights, for the first time, the critical pro-tumorigenic importance of metabolic-coupling between adipocytes and ovarian cancer cells.
The investigators also demonstrated in vivo and in vitro that homing, migration and invasion of ovarian cancer cells is induced by omental adipocytes via IL-8, IL-6, MCP-1 and TIMP-1, since neutralizing antibodies led to decreased homing.Citation59 This was confirmed in vivo for IL-8 by using an IL-8 receptor inhibitor. Inflammatory cytokines such as IL-6 and IL-8 are involved in promoting catabolic processes, including lipolysis and glycolysis.Citation60–Citation63 In addition, tumor cells can induce the production of inflammatory cytokines, including IL-6 and IL-8, and transform the stroma to a highly catabolic state with increased autophagy.Citation35 These studies once again stress the link between inflammation and autophagy-based catabolism in the stroma and tumor progression.
In summary, metabolic coupling between adipocytes and cancer cells favors tumor growth, and this can be reversed by inhibiting lipid transport from adipocytes to carcinoma cells.Citation59
Adipocyte Lipolysis Promotes Cancer-Associated Cachexia and Leads to a Generalized Catabolic State, Driving Tumor Growth
Cancer-associated cachexia (CAC) is a well-known complication of cancer that frequently is the cause of death in cancer patients. CAC leads to abnormal whole body catabolic metabolism, similar to starvation.Citation64 The mechanism(s) underlying tumor-associated cachexia are poorly understood, and no widely effective strategies, including increased calorie intake, exist to manage the condition.Citation65
The study by Das et al. recently published in the journal Science presents important new data regarding CAC. By studying the effects of adipose tissue metabolism on muscle and fat mass,Citation66 the investigators discovered that inhibition of lipolysis halts muscle and fat-related cachexia, as observed in lipase-knockout mice. Triglycerides are metabolized to fatty acids and glycerol by adipose triglyceride lipase (ATGL) and hormone-sensitive lipase (HSL). Inhibition of ATGL and, to a lesser degree, of HSL, prevented cachexia and weight loss.Citation66 In fact, ATGL-knockout mice had similar weights as control mice that were not injected with cancer cells.Citation66 Also, by studying white adipose tissue (WAT) in cancer patients, they found that ATGL activity was highest and WAT mass was the lowest in patients who died with CAC, as compared with other cancer patients.Citation66 These data directly show that metabolic-coupling exists between different organ systems in cancer patients. It is well-known that chemokines can alter the metabolism of different organs, but this study goes on to show that the metabolites themselves act as chemokines, inducing changes in the metabolism of other organs.Citation66
Clinical Implications of Metabolic Coupling for Cancer Treatment
The discovery of metabolic-coupling between tumor cells and their hosts opens many new avenues for drug discovery involving novel therapeutic targets. Thus, like diabetes, cancer should be viewed as a systemic metabolic disease of energy imbalance. In fact, similar metabolic profiles (with insulin resistance and increased lipolysis) are found in patients with diabetes mellitus and those with advanced cancers and cachexia.Citation67–Citation69 Anti-diabetic drugs such as metformin, which inhibits both lipolysis and mitochondrial oxidative phosphorylation,Citation70,Citation71 need to be further investigated for the effective management of cancer patients with “parasitic metabolism” or “metabolic coupling,” between cancer cells and the host ().
Metformin is a known “mitochondrial poison” that mechanistically functions as a complex I inhibitor that blocks mitochondrial oxidative phosphorylation. As a consequence, Metformin also activates AMP kinaseCitation72 and increases insulin-sensitivity, thus decreasing glucose blood levels,Citation73 which would be conceptually beneficial in treating cancers. Epidemiologic studies support the use of metformin in cancer treatment and suggest a protective effect against cancer developmentCitation74 and cancer progression in diabetics receiving metformin.Citation75 Thus, the anticancer activity of metformin may stem from its antimitochondrial activity, thereby preventing cancer cells from using the energy-rich onco-metabolites (L-lacate, ketones, glutamine and fatty acids) derived from the tumor stroma.
Recently, we used a mitochondrial complex I activity stain to functionally visualize the anti-mitochondrial activity of Metformin in human breast cancer tumor samples.Citation14 As with complex IV staining, complex I mitochondrial activity was mainly localized to the epithelial cancer cell compartment.Citation14 Strikingly, tumor tissue frozen sections treated with Metformin revealed a near complete absence of Complex I activity,Citation14 consistent with the idea that Metformin does indeed function as a Complex I inhibitor in vivo ().
Reversing Drug Resistance: Targeting Cancer Cell Mitochondria for Destruction
Importantly, stromal-epithelial metabolic-coupling also induces chemo-resistance.Citation76,Citation77 Thus, resistance to standard anticancer therapies may be both a metabolic and stromal phenomenon, related to mitochondrial “health” or “well-being” in cancer cells.Citation76,Citation77
Mitochondria fulfill a dual role in cell metabolism. During anabolic cell growth, they are the “powerhouse” of the cell and fuel proliferation. However, during periods of stress and cell death (apoptosis), they act as “sensors” to decide whether a given cell will undergo suicide or programmed cell death. In this context, the mitochondria are also the cell's executioners. Most chemotherapeutic agents act via the induction of apoptosis in cancer cells, placing mitochondria at the center stage of both cancer cell metabolism and drug resistance ().
In this context, we have shown that energy-rich metabolites derived from stromal cells maintain mitochondrial “well-being” in cancer cells, thereby conferring drug resistance.Citation76,Citation77 More specifically, simple metabolites (such as L-lactate, ketones and glutamine) promote mitochondrial “health” in cancer cells, effectively shutting off their apoptotic machinery, resulting in protection against cell death, even when challenged with toxic drugs.Citation76,Citation77 Similarly, we can overcome metabolite-induced chemo-resistance in cancer cells by using mitochondrial poisons (such as Metformin or Arsenic) or by using drug combinations that function as mitochondrial poisons (Tamoxifen + Dastatinib).Citation76,Citation77 Conversely, we can genetically pheno-copy the protective effects of these onco-metabolites by overexpressing recombinant proteins that maintain mitochondrial “health,” such as TIGAR,Citation78,Citation79 directly conferring chemoresistance in cancer cells.Citation76,Citation77
Thus, metabolic-coupling promotes tumor growth and makes epithelial cancer cells more resistant to conventional therapies. As such, drugs that can target mitochondrial function in cancer cells or drugs that halt glycolysis, lipolysis or catabolism in the surrounding tumor stroma may be beneficial in preventing tumor progression and metastasis.
Ultimately, this new concept of “parasitic cancer metabolism” () could radically change how we treat cancer patients and stimulate new metabolic strategies for cancer prevention and therapy.Citation13,Citation43,Citation50,Citation81
Note Added In Proof
After this Perspective was accepted for publication in Cell Cycle, another paper appeared in the journal Science, showing that baseline mitochondrial dysfunction in cancer cells is a positive predictor of the therapeutic response to conventional chemotherapy. Thus, as we proposed based on our experiments, mitochondrial dysfunction in cancer cells sensitizes them to chemotherapy, resulting in a good clinical response and increased overall survival in cancer patients. As such, mitochondrial dysfunction (i.e., the conventional Warburg effect) in tumor cells predicts chemo-sensitivity in cancer patients, whereas mitochondrial “health and well-being” in cancer cells (i.e., indicative of “Parasitic Cancer Metabolism”) predicts chemo-resistance. These clinical findings directly support our new paradigm, which is based on (1) mitochondrial function and (2) energy transfer in cancer metabolism.
For further details, please see: Chonghaile TN, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VD, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science 2011; 334:1129–33; PMID:22033517.
Figures and Tables
Figure 1 Energy transfer in cancer metabolism. The tumor stroma generates catabolites that are transferred to cancer cells for anabolic growth. The stroma has high levels of autophagy, mitophagy, glycolysis and lipolysis, while epithelial cancer cells have high mitochondrial mass and activity (oxidative phosphorylation and β oxidation). Reproduced and modified with permission from reference Citation14.
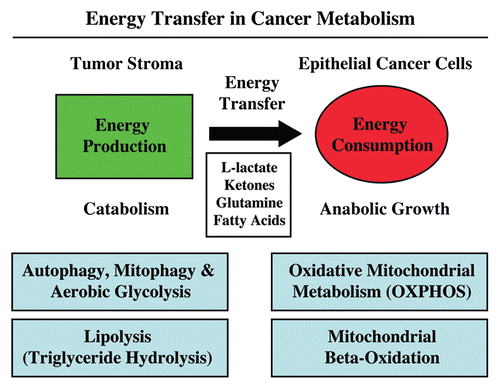
Figure 2 Onco-metabolites derived from the tumor stroma promote anabolic cancer cell growth via the TCA cycle and oxidative mitochondrial metabolism. Note that various stromally derived onco-metabolites (L-lactate, ketones, free fatty acids and glutamine; shown in red) all feed into the TCA/Krebs cycle via either Acetyl-CoA or Alpha-Keto-Glutarate, promoting oxidative mitochondrial metabolism (OXPHOS) in epithelial cancer cells. The end result is highly efficient ATP production in aggressive cancer cells.
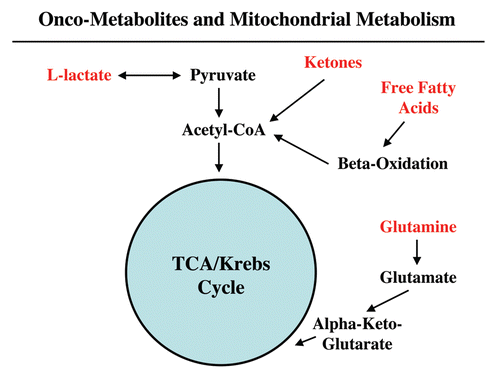
Figure 3 Physiologic energy transfer. Metabolic-coupling is a normal and widespread physiological phenomenon that is required to maintain homeostasis or energy balance. Metabolic-coupling occurs in organ systems throughout the body, including skeletal muscle, the brain and the ovary. In all three tissues, a “lactateshuttle” exists. In this context, glycolytic cells (fast-twitch muscle fibers, astrocytes and cumulus/granulosa cells) are metabolically coupled to oxidative cells (slow-twitch muscle fibers, neurons and oocytes). L-lactate is generated in glycolytic cells from glucose and is transferred to oxidative cells, which is efficiently used to make large amounts of ATP, via oxidative mitchondrial metabolism. Monocarboxylate transporters (MCTs) shuttle lactate from one cell type to another.
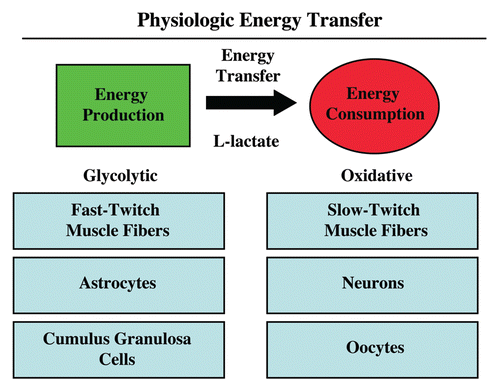
Figure 4 Shuttling the onco-metabolite L-lactate from the tumor stroma to epithelial cancer cells. Previously, we proposed that a “lactate shuttle” also exists in human breast cancers. More specifically, the distribution of lactate transporters is highly compartmentalized in human breast cancers. Note that MCT4 (a marker of aerobic glycolysis and L-lactate secretion) is largely confined to cancer-associated fibroblasts in the tumor stroma. Conversely, MCT1 (a marker of L-lactate uptake) is localized to epithelial cancer cells. Because of their broad specificity, the same MCT transporters can also function in the shuttling of ketone bodies from the tumor stroma to epithelial cancer cells. Arrows indicate the direction of energy transfer. Reproduced and modified with permission from reference Citation57.
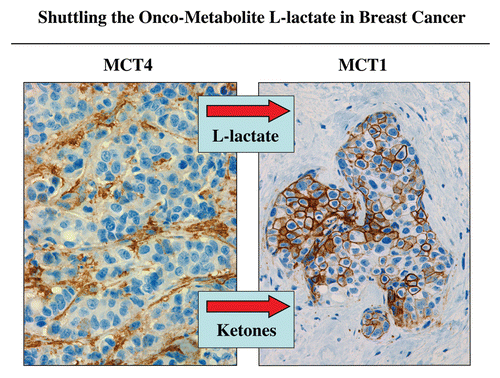
Figure 5 Metabolic compartmentalization of mitochondrial activity in skeletal muscle and human breast cancer. Frozen sections from skeletal muscle tissue or human breast cancers were subjected to a routine histochemical stain that detects the functional activity of mitochondrial complex IV [cytochrome C oxidase (COX)]. This allows the visualization of oxidative mitochondrial metabolism in tissue sections. COX-positive cells are positively stained brown (see red arrows). In skeletal muscle, note that fast-twitch fibers are glycolytic (Gly) and are COX-negative, while slow-twitch fibers are oxidative (Ox) and are COX-positive. In breast cancers, the tumor stroma is glycolytic (Gly) and is COX-negative, while epithelial tumor cell nests are oxidative (Ox) and are COX-positive. These results support the idea that tumors show metabolic compartmentalization and specialization, as occurs in skeletal muscle tissue. Reproduced and modified with permission from reference Citation14.
![Figure 5 Metabolic compartmentalization of mitochondrial activity in skeletal muscle and human breast cancer. Frozen sections from skeletal muscle tissue or human breast cancers were subjected to a routine histochemical stain that detects the functional activity of mitochondrial complex IV [cytochrome C oxidase (COX)]. This allows the visualization of oxidative mitochondrial metabolism in tissue sections. COX-positive cells are positively stained brown (see red arrows). In skeletal muscle, note that fast-twitch fibers are glycolytic (Gly) and are COX-negative, while slow-twitch fibers are oxidative (Ox) and are COX-positive. In breast cancers, the tumor stroma is glycolytic (Gly) and is COX-negative, while epithelial tumor cell nests are oxidative (Ox) and are COX-positive. These results support the idea that tumors show metabolic compartmentalization and specialization, as occurs in skeletal muscle tissue. Reproduced and modified with permission from reference Citation14.](/cms/asset/8a9bc08c-ad6c-43cb-a946-f1e54724863f/kccy_a_10918487_f0005.gif)
Figure 6 Uncoupling parasitic cancer metabolism. Drugs such as chloroquine (which inhibits autophagy) and metformin (which inhibits lipolysis), will prevent energy transfer to cancer cells and tumor growth. In this scenario, mitochondrial poisons (such as metformin, arsenic and others) could also be used to uncouple tumor cells from the energy-producing host stroma.
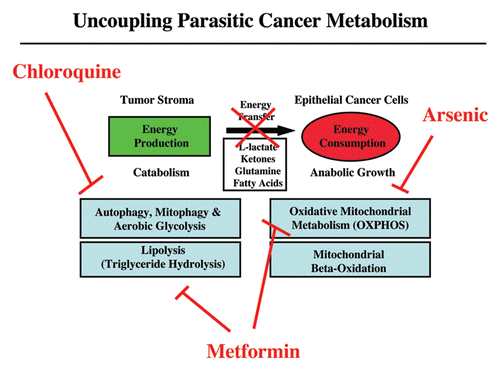
Figure 7 Visualizing the anti-mitochondrial effects of Metformin in human breast cancer tumor tissue. Frozen sections from human breast cancers were subjected to mitochondrial Complex I (NADH) activity staining. NADH-positive cells are positively stained blue (see red arrows). In breast cancers, note that the tumor stroma is glycolytic and is NADH-negative, while epithelial tumor cell nests are oxidative and are NADH-positive. Note that treatment with Metformin, a known complex I inhibitor, prevented the NADH-staining of epithelial cancer cell nests. Positive controls with skeletal muscle were performed in parallel to ensure the specificity of the staining procedure and allowed us to detect complex I-positive muscle fibers (blue), which represent oxidative slow-twitch fibers. Reproduced and modified with permission from reference Citation14.
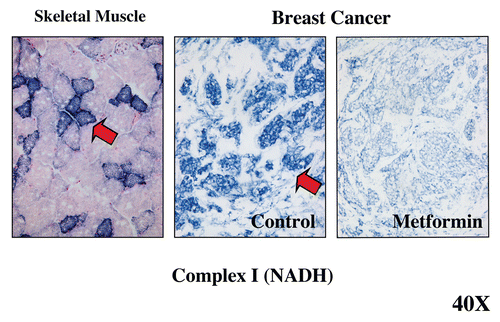
Figure 8 Stromal onco-metabolites confer drug resistance by promoting mitochondrial health or well-being, providing an escape from stress-induced apoptosis. Simple stromally derived metabolites (such as L-lactate, ketones and glutamine) promote mitochondrial “health” in cancer cells, effectively shutting off their apoptotic machinery, resulting in protection against cell death, even when challenged with anticancer drugs. In contrast, we can overcome metabolite-induced drug resistance in cancer cells by using mitochondrial poisons (such as Metformin).
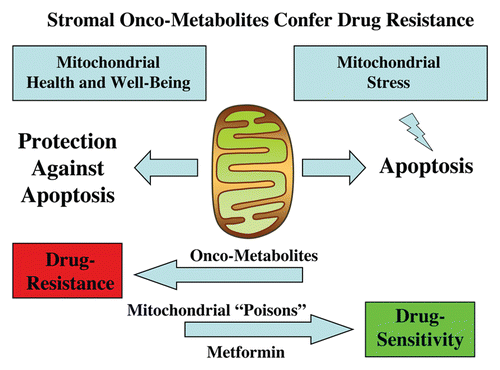
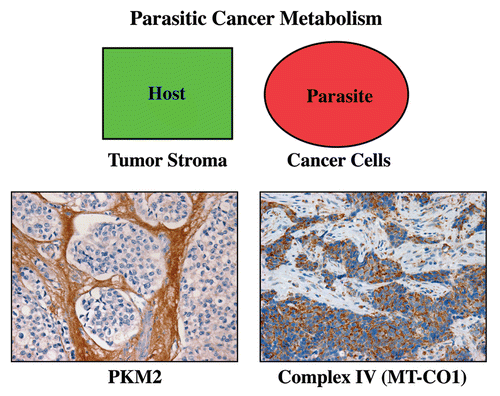
Figure 9 Metabolic compartments in parasitic cancer metabolism. In summary, we believe that cancer cells act as metabolic parasites and extract nutrients from host cells by inducing catabolic processes (autophagy, mitophagy, aerobic glycolysis and lipolysis). As a consequence, the tumor stroma shows a shift toward aerobic glycolysis, and epithelial cancer cells show functional hyper-activation of oxidative mitochondrial metabolism (OXPHOS). In support of this model, cancer-associated fibroblasts and the tumor stroma overexpress PKM2 (a rate-limiting glycolytic enzyme, left panel). Conversely, breast cancer epithelial cells upregulate MT-CO1 (a key component of mitochondrial complex IV, right panel). The metabolic compartmentalization of PKM2 and MT-CO1 were visualized by immunostaining with specific antibody probes (brown reaction product). Reproduced and modified with permission from references Citation14 and Citation80.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660), and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council. F.S.M. was supported by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) [# 576200/2008.5, #473670/2008-9], and the Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG) [#14916]. H.B.T. was funded by NIH grant AI-76248.
References
- Rønnov-Jessen L, Bissell MJ. Breast cancer by proxy: can the microenvironment be both the cause and consequence?. Trends Mol Med 2009; 15:5 - 13; PMID: 19091631; http://dx.doi.org/10.1016/j.molmed.2008.11.001
- Kenny PA, Lee GY, Bissell MJ. Targeting the tumor microenvironment. Front Biosci 2007; 12:3468 - 3474; PMID: 17485314; http://dx.doi.org/10.2741/2327
- Paget S. The distribution of secondary growths in cancer of the breast. Lancet 1889; 133:571 - 573; http://dx.doi.org/10.1016/S0140-6736(00)49915-0
- Hart IR. ‘Seed and soil’ revisited: mechanisms of site-specific metastasis. Cancer Metastasis Rev 1982; 1:5 - 16; PMID: 6764375; http://dx.doi.org/10.1007/BF00049477
- Hart IR, Fidler IJ. Role of organ selectivity in the determination of metastatic patterns of B16 melanoma. Cancer Res 1980; 40:2281 - 2287; PMID: 7388794
- Hart IR, Talmadge JE, Fidler IJ. Metastatic behavior of a murine reticulum cell sarcoma exhibiting organ-specific growth. Cancer Res 1981; 41:1281 - 1287; PMID: 7011533
- Harris AL. Antiangiogenesis for cancer therapy. Lancet 1997; 349:13 - 15; PMID: 9164441; http://dx.doi.org/10.1016/S0140-6736(97)90014-3
- Horak ER, Leek R, Klenk N, LeJeune S, Smith K, Stuart N, et al. Angiogenesis, assessed by platelet/endothelial cell adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet 1992; 340:1120 - 1124; PMID: 1279332; http://dx.doi.org/10.1016/0140-6736(92)93150-L
- Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1971; 133:275 - 288; PMID: 4332371; http://dx.doi.org/10.1084/jem.133.2.275
- Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Ann Surg 1972; 175:409 - 416; PMID: 5077799; http://dx.doi.org/10.1097/00000658-197203000-00014
- Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol 2011; 43:1045 - 1051; PMID: 21300172; http://dx.doi.org/10.1016/j.biocel.2011.01.023
- Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol 2011; 178:1949 - 1952; PMID: 21514412; http://dx.doi.org/10.1016/j.ajpath.2011.03.002
- Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res 2011; 13:213; PMID: 21867571; http://dx.doi.org/10.1186/bcr2892
- Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of Oxidative Mitochondrial Metabolism in Epithelial Cancer Cells In Situ: Visualizing the Therapeutic Effects of Metformin in Tumor Tissue. Cell Cycle 2011; 10; In press PMID: 21558814
- Steidl C, Lee T, Shah SP, Farinha P, Han G, Nayar T, et al. Tumor-associated macrophages and survival in classic Hodgkin's lymphoma. N Engl J Med 2010; 362:875 - 885; PMID: 20220182; http://dx.doi.org/10.1056/NEJMoa0905680
- Jacobs TW, Byrne C, Colditz G, Connolly JL, Schnitt SJ. Radial scars in benign breast-biopsy specimens and the risk of breast cancer. N Engl J Med 1999; 340:430 - 436; PMID: 9971867; http://dx.doi.org/10.1056/NEJM199902113400604
- Maeshima AM, Niki T, Maeshima A, Yamada T, Kondo H, Matsuno Y. Modified scar grade: a prognostic indicator in small peripheral lung adenocarcinoma. Cancer 2002; 95:2546 - 2554; PMID: 12467069; http://dx.doi.org/10.1002/cncr.11006
- Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res 1999; 59:5002 - 5011; PMID: 10519415
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392 - 401; PMID: 16572188; http://dx.doi.org/10.1038/nrc1877
- McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol 2010; 28:4022 - 4028; PMID: 20644094; http://dx.doi.org/10.1200/JCO.2010.28.4257
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005; 121:335 - 348; PMID: 15882617; http://dx.doi.org/10.1016/j.cell.2005.02.034
- Narsaria N, Mohanty C, Das BK, Mishra SP, Prasad R. Oxidative Stress in Children with Severe Malaria. J Trop Pediatr 2011; In press PMID: 21602230; http://dx.doi.org/10.1093/tropej/fmr043
- Dey S, Guha M, Alam A, Goyal M, Bindu S, Pal C, et al. Malarial infection develops mitochondrial pathology and mitochondrial oxidative stress to promote hepatocyte apoptosis. Free Radic Biol Med 2009; 46:271 - 281; PMID: 19015023; http://dx.doi.org/10.1016/j.freeradbiomed.2008.10.032
- Ba X, Gupta S, Davidson M, Garg NJ. Trypanosoma cruzi induces the reactive oxygen species-PARP-1-RelA pathway for upregulation of cytokine expression in cardiomyocytes. J Biol Chem 2010; 285:11596 - 11606; PMID: 20145242; http://dx.doi.org/10.1074/jbc.M109.076984
- Wen JJ, Gupta S, Guan Z, Dhiman M, Condon D, Lui C, et al. Phenyl-alpha-tert-butyl-nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic Rats. J Am Coll Cardiol 2010; 55:2499 - 2508; PMID: 20510218; http://dx.doi.org/10.1016/j.jacc.2010.02.030
- Gupta S, Dhiman M, Wen JJ, Garg NJ. ROS signalling of inflammatory cytokines during Trypanosoma cruzi infection. Adv Parasitol 2011; 76:153 - 170; PMID: 21884891; http://dx.doi.org/10.1016/B9780-12-385895-5.00007-4
- Gupta S, Wen JJ, Garg NJ. Oxidative Stress in Chagas Disease. Interdiscip Perspect Infect Dis 2009; 2009:190354; PMID: 19547716
- Romano PS, Arboit MA, Vazquez CL, Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 2009; 5:6 - 18; PMID: 19115481; http://dx.doi.org/10.4161/auto.5.1.7160
- Nagajyothi F, Desruisseaux MS, Machado FS, Upadhya R, Zhao D, Schwartz GJ, et al. Response of adipose tissue to early infection with Trypanosoma cruzi. J Infect Dis 2012; In press
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer-associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276; PMID: 20814239; http://dx.doi.org/10.4161/cc.9.16.12553
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer-associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515 - 3533; PMID: 20855962; http://dx.doi.org/10.4161/cc.9.17.12928
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer-associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle 2010; 9:3534 - 3551; PMID: 20864819; http://dx.doi.org/10.4161/cc.9.17.12908
- Migneco G, Whitaker-Menezes D, Chiavarina B, Castello-Cros R, Pavlides S, Pestell RG, et al. Glycolytic cancer-associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010; 9:2412 - 2422; PMID: 20562527; http://dx.doi.org/10.4161/cc.9.12.11989
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle 2010; 9:2201 - 2219; PMID: 20519932; http://dx.doi.org/10.4161/cc.9.11.11848
- Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle 2011; 10:1784 - 1793; PMID: 21566463; http://dx.doi.org/10.4161/cc.10.11.15674
- Korkaya H, Wicha MS. Inflammation and autophagy conspire to promote tumor growth. Cell Cycle 2011; 10:2623 - 2624; PMID: 21829104; http://dx.doi.org/10.4161/cc.10.16.16414
- Witkiewicz AK, Kline J, Queenan M, Brody JR, Tsirigos A, Bilal E, et al. Molecular profiling of a lethal tumor microenvironment, as defined by stromal caveolin-1 status in breast cancers. Cell Cycle 2011; 10:1794 - 1809; PMID: 21521946; http://dx.doi.org/10.4161/cc.10.11.15675
- Bonuccelli G, Tsirigos A, Whitaker-Menezes D, Pavlides S, Pestell RG, Chiavarina B, et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010; 9:3506 - 3514; PMID: 20818174; http://dx.doi.org/10.4161/cc.9.17.12731
- Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011; 10:1271 - 1286; PMID: 21512313; http://dx.doi.org/10.4161/cc.10.8.15330
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer-associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001; PMID: 19923890; http://dx.doi.org/10.4161/cc.8.23.10238
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505; PMID: 20861672; http://dx.doi.org/10.4161/cc.9.17.12721
- Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle 2010; 9:4297 - 4306; PMID: 21051947; http://dx.doi.org/10.4161/cc.9.21.13817
- Pavlides S, Vera I, Gandara R, Sneddon S, Pestell R, Mercier I, et al. Warburg Meets Autophagy: Cancer-associated Fibroblasts Accelerate Tumor Growth and Metastasis Via Oxidative Stress, Mitophagy and Aerobic Glycolysis. Antioxid Redox Signal 2012; In press PMID: 21883043
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029 - 1033; PMID: 19460998; http://dx.doi.org/10.1126/science.1160809
- Kaelin WG Jr, Thompson CB. Q&A: Cancer: clues from cell metabolism. Nature 2010; 465:562 - 564; PMID: 20520704; http://dx.doi.org/10.1038/465562a
- Koppenol WH, Bounds PL, Dang CV. Otto Warburg's contributions to current concepts of cancer metabolism. Nat Rev Cancer 2011; 11:325 - 337; PMID: 21508971; http://dx.doi.org/10.1038/nrc3038
- Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer 2011; 11:85 - 95; PMID: 21258394; http://dx.doi.org/10.1038/nrc2981
- Semenza GL. A return to cancer metabolism. J Mol Med (Berl) 2011; 89:203 - 204; PMID: 21301793
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344 - 1348; PMID: 21127245; http://dx.doi.org/10.1126/science.1193497
- Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and Cancer Metabolism in the Tumor Microenvironment: Markers, Models and Mechanisms. Annual Reviews of Pathology: Mechanisms of Disease 2012; 7 In press
- Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol 2009; 587:5591 - 5600; PMID: 19805739; http://dx.doi.org/10.1113/jphysiol.2009.178350
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA 1994; 91:10625 - 10629; PMID: 7938003; http://dx.doi.org/10.1073/pnas.91.22.10625
- Hashimoto T, Hussien R, Cho HS, Kaufer D, Brooks GA. Evidence for the mitochondrial lactate oxidation complex in rat neurons: demonstration of an essential component of brain lactate shuttles. PLoS ONE 2008; 3:2915; PMID: 18698340; http://dx.doi.org/10.1371/journal.pone.0002915
- Heller DT, Cahill DM, Schultz RM. Biochemical studies of mammalian oogenesis: metabolic cooperativity between granulosa cells and growing mouse oocytes. Dev Biol 1981; 84:455 - 464; PMID: 20737884; http://dx.doi.org/10.1016/0012-1606(81)90415-2
- Johnson MT, Freeman EA, Gardner DK, Hunt PA. Oxidative metabolism of pyruvate is required for meiotic maturation of murine oocytes in vivo. Biol Reprod 2007; 77:2 - 8; PMID: 17314311; http://dx.doi.org/10.1095/biolreprod.106.059899
- Stumvoll M, Perriello G, Meyer C, Gerich J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int 1999; 55:778 - 792; PMID: 10027916; http://dx.doi.org/10.1046/j.1523-755.1999.055003778.x
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle 2011; 10:1772 - 1783; PMID: 21558814; http://dx.doi.org/10.4161/cc.10.11.15659
- Balliet RM, Capparelli C, Guido C, Pestell TG, Martinez-Outschoorn UE, Lin Z, et al. Mitochondrial Oxidative Stress in Cancer-associated Fibroblasts Drives Lactate Production, Promoting Breast Cancer Tumor Growth: Understanding the Aging & Cancer Connection. Cell Cycle 2011; 10:4065 - 4073
- Nieman KM, Kenny HA, Penicka C, Ladanyi A, Buell-Gutbrod R, Zillhardt M, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med 2011; In press PMID: 22037646; http://dx.doi.org/10.1038/nm.2492
- van Hall G, Steensberg A, Sacchetti M, Fischer C, Keller C, Schjerling P, et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans. J Clin Endocrinol Metab 2003; 88:3005 - 3010; PMID: 12843134; http://dx.doi.org/10.1210/jc.2002-021687
- Staiger H, Woll J, Haas C, Machicao F, Schleicher ED, Fritsche A, et al. Selective association of plasma non-esterified fatty acid species with circulating interleukin-8 concentrations in humans. Exp Clin Endocrinol Diabetes 2009; 117:21 - 27; PMID: 19053024; http://dx.doi.org/10.1055/s-0028-1085459
- Debigaré R, Marquis K, Cote CH, Tremblay RR, Michaud A, LeBlanc P, et al. Catabolic/anabolic balance and muscle wasting in patients with COPD. Chest 2003; 124:83 - 89; PMID: 12853506; http://dx.doi.org/10.1378/chest.124.1.83
- Buck M, Chojkier M. Muscle wasting and dedifferentiation induced by oxidative stress in a murine model of cachexia is prevented by inhibitors of nitric oxide synthesis and antioxidants. EMBO J 1996; 15:1753 - 1765; PMID: 8617220
- Evans WJ, Morley JE, Argiles J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008; 27:793 - 799; PMID: 18718696; http://dx.doi.org/10.1016/j.clnu.2008.06.013
- Kumar NB, Kazi A, Smith T, Crocker T, Yu D, Reich RR, et al. Cancer cachexia: traditional therapies and novel molecular mechanism-based approaches to treatment. Curr Treat Options Oncol 2010; 11:107 - 117; PMID: 21128029; http://dx.doi.org/10.1007/s11864-010-0127-z
- Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 2011; 333:233 - 238; PMID: 21680814; http://dx.doi.org/10.1126/science.1198973
- Tayek JA. A review of cancer cachexia and abnormal glucose metabolism in humans with cancer. J Am Coll Nutr 1992; 11:445 - 456; PMID: 1506607
- Groop LC, Bonadonna RC, DelPrato S, Ratheiser K, Zyck K, Ferrannini E, et al. Glucose and free fatty acid metabolism in non-insulin-dependent diabetes mellitus. Evidence for multiple sites of insulin resistance. Clin Invest 1989; 84:205 - 213; PMID: 2661589; http://dx.doi.org/10.1172/JCI114142
- Nurjhan N, Consoli A, Gerich J. Increased lipolysis and its consequences on gluconeogenesis in non-insulin-dependent diabetes mellitus. J Clin Invest 1992; 89:169 - 175; PMID: 1729269; http://dx.doi.org/10.1172/JCI115558
- Riccio A, Del Prato S, Vigili de Kreutzenberg S, Tiengo A. Glucose and lipid metabolism in non-insulin-dependent diabetes. Effect of metformin. Diabete Metab 1991; 17:180 - 184; PMID: 1936473
- El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem 2000; 275:223 - 228; PMID: 10617608; http://dx.doi.org/10.1074/jbc.275.1.223
- Zakikhani M, Dowling RJ, Sonenberg N, Pollak MN. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila) 2008; 1:369 - 375; PMID: 19138981; http://dx.doi.org/10.1158/1940-6207.CAPR-08-0081
- Phoenix KN, Vumbaca F, Fox MM, Evans R, Claffey KP. Dietary energy availability affects primary and metastatic breast cancer and metformin efficacy. Breast Cancer Res Treat 2010; 123:333 - 344; PMID: 20204498; http://dx.doi.org/10.1007/s10549-009-0647-z
- Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care 2009; 32:1620 - 1625; PMID: 19564453; http://dx.doi.org/10.2337/dc08-2175
- Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, et al. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol 2009; 27:3297 - 3302; PMID: 19487376; http://dx.doi.org/10.1200/JCO.2009.19.6410
- Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: Therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle 2011; 10:2521 - 2528; PMID: 21768775; http://dx.doi.org/10.4161/cc.10.15.16584
- Martinez-Outschoorn UE, Lin Z, Ko YH, Flomenberg N, Wang C, Pavlides S, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther 2011; 12:924 - 938; PMID: 22041887
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009; 28:3015 - 3026; PMID: 19713938; http://dx.doi.org/10.1038/emboj.2009.242
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107 - 120; PMID: 16839880; http://dx.doi.org/10.1016/j.cell.2006.05.036
- Bonuccelli G, Whitaker-Menezes D, Castello-Cros R, Pavlides S, Pestell RG, Fatatis A, et al. The reverse Warburg effect: glycolysis inhibitors prevent the tumor promoting effects of caveolin-1 deficient cancer-associated fibroblasts. Cell Cycle 2010; 9:1960 - 1971; PMID: 20495363; http://dx.doi.org/10.4161/cc.9.10.11601
- Sotgia F, Martinez-Outschoorn UE, Lisanti MP. Mitochondrial oxidative stress drives tumor progression and metastasis: should we use antioxidants as a key component of cancer treatment and prevention?. BMC Med 2011; 9:62; PMID: 21605374; http://dx.doi.org/10.1186/1741-7015-9-62