Abstract
Comment on: Carr SM, et al. EMBO J. 2011; 30:317-27.
The retinoblastoma protein (pRb) is regarded as the archetypal tumor suppressor and has a vital role in regulating progression through the early stages of the cell cycle.Citation1,Citation2 It does this primarily by binding to and controlling key effectors of cell cycle progression, such as the E2F family of transcription factors.Citation1,Citation2 The pRb-E2F interaction is highly significant since the ability of pRb to bind E2F coincides with growth inhibition and cell cycle delay.Citation2,Citation3 This inhibition is mediated by the reduced transcription of E2F target genes and, reflecting the physiological context in which it occurs, might involve several different mechanisms. For example, pRb downregulates E2F-dependent transcription by its direct association with the transcriptional activation domain of E2F-1.Citation3 pRb can also mediate active repression by recruiting chromatin modifying enzymes such as histone deacetylases and methyltransferases. This enables pRb to transcriptionally silence E2F target genes by influencing the chromatin environment.Citation4
Growth control by pRb is influenced by its post-translational modification, the most widely described to date being phosphorylation by the cyclin-dependent kinases (Cdks). As cells progress through G1, a series of phosphorylation events occur on pRb, and the temporal regulation of pRb phosphorylation reflects the cyclical appearance of cyclin/Cdk complexes.Citation5 Phosphorylation of pRb leads to the inactivation of its tumor suppressor activity, resulting in the release of bound E2F transcription factors and the expression of genes required for cell cycle progression and DNA replication. Phosphorylation, however, is not the only post-translational modification to impinge on pRb activity. For example, pRb is also acetylated under conditions of cell cycle exit, such as differentiation and DNA damage.Citation6,Citation7 Further, pRb can be methylated on a C-terminal lysine residue, and this contributes to transcriptional repression via the recruitment of HP1.Citation8 Methylation of lysine residues is known to control protein activity, and this is particularly clear for transcription factors like p53,Citation9 and in the context of chromatin biology.Citation10 The type of methylation event (mono-, di- or tri-), together with its interplay with other post-translational modifications, can provide a complex level of cross-talk, which can result in the finetuning of a biological response.
Indeed, recently, we described a new type of regulation that influences Cdk recognition and phosphorylation of substrate proteins.Citation11 In pRb, a lysine residue located within a region phosphorylated by Cdk kinase was found to be a target for monomethylation by Set7/9, both in vitro and in cells. The lysine residue in question, K810, represents the conserved and essential basic residue found in Cdk substrate proteins (Cdk kinases recognise the concensus sequence S/T-P-X-K/R, where S/T represents the target serine or threonine residue). We demonstrated that methylation at K810 was linked to reduced levels of Cdk phosphorylation at the neighboring serine residues. Interestingly, this interplay between methylation and phosphorylation appeared to be uni-directional, since pRb methylation does not appear to be affected by a prior Cdk phosphorylation event. This suggests that methylation is enforced on the hypophosphorylated, functionally active form of pRb. Indeed, under conditions when pRb is expected to function as a tumor suppressor, such as DNA damage, elevated levels of K810 methylation and a coincident reduction in phosphorylation were apparent. One possible hypothesis for this observed antagonism is that methylation disrupts the interaction between pRb and cyclin/Cdk complexes. This is supported by structural modeling, which suggests that the lysyl side chain of K810 would make a number of important interactions at a cleft formed by the cyclin/Cdk interface, and that these interactions are essential for kinase activity towards the substrate. It is likely that K810 methylation would result in steric clashes with this cyclin/Cdk interface, dislodging the K810 side chain out of its binding pocket. This would reduce the binding efficiency between pRb and cyclin/Cdk and consequently, lead to reduced pRb phosphorylation at the serine sites neighboring K810 (S807/S811).
Intriguingly, methylation of K810 also influenced phosphorylation events throughout pRb, even at distant Cdk target sites. This suggests that the ability of Set7/9 to methylate pRb and thereby hinder phosphorylation at S807/S811, subsequently impacts on phosphorylation at other Cdk sites throughout pRb. Whilst the exact mechanism for this global regulation of phosphorylation has yet to be elucidated, a couple of possibilities exist. Firstly, phosphorylation of S807/S811 could represent a priming event that is required for efficient phosphorylation of subsequent Cdk target sites in pRb, perhaps by regulating the recognition by or recruitment of other cyclin/Cdk complexes. There is evidence to suggest, at least, that S807/S811 phosphorylation by cyclin C/Cdk3 occurs earlier in the cell cycle than other cyclin-directed activities, and that this phosphorylation is important for transition of cells from G0 to G1.Citation12 Secondly, methylation of K810 may lead to the recruitment of proteins containing methyl-recognition motifs, which subsequently impact on or interfere with Cdkdependent phosphorylation of pRb. Either way, it is clear that pRb methylation has an important role in regulating cell cycle progression, since disrupting methylation at K810 resulted in decreased G1 arrest, and this was co-incident with decreased association of pRb with the promoters of E2F-1 responsive genes and aberrant transcriptional repression.
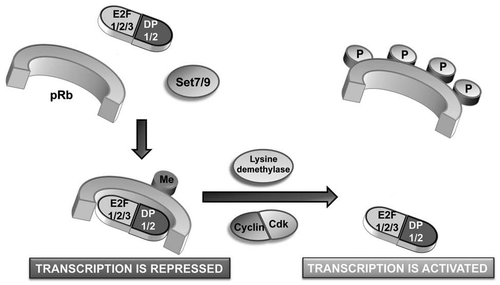
It is intriguing to see that a type of post-translational code can govern Cdk-dependent growth control. This is another striking example of interplay between two different types of post-translational modification, a feature that is becoming more widely accepted as important for the functional regulation of non-histone proteins. Lysine modification of pRb within a Cdk concensus site provides an alternative and direct substrate-based mechanism to regulate Cdk activity (), which likely acts in concert with other levels of Cdk control such as the induction of Cdk inhibitors like p21.Citation13 Together, both mechanisms may act as a fail-safe to limit pRb phosphorylation by Cdks during stress responses. Since cell cycle arrest in response to stress is primarily a protective mechanism, it seems likely that once this stress is relieved, the methyl mark on pRb would be removed by a demethylation event, permitting cells to re-enter the cell cycle (). It will be of interest not only to identify which demethylase enzyme is involved in this process, but also to revisit other known Cdk substrates and ask whether this methylation-phosphorylation switch is utilized often and in which signaling pathways.
Figures and Tables
Figure 1 Model depicting the effect of methylation on Cdk-dependent growth control. Under conditions of DNA damage, pRb is methylated at K810 by Set7/9. This locks pRb in a hypophosphorylated, growth-arresting state, thereby limiting E2F target gene expression. Once the stress has been overcome, demethylase enzymes will remove the methyl mark, permitting pRb phosphorylation by cyclin/Cdk complexes. This will relieve cell cycle arrest by facilitating expression of E2F target genes.
Acknowledgements
Supported by Cancer Research UK, the Medical Research Council, Leukaemia Research Fund, and the Association for International Cancer Research.
Comment on: Carr SM, et al. EMBO J 2011; 30:317 - 327
Related Research Data
References
- Weinberg RA. Cell 1995; 81:323 - 330
- Stevens C, et al. Arch Biochem Biophys 2003; 412:157 - 169
- Dyson N. Genes Dev 1998; 12:2245 - 2262
- Van den Heuval S, et al. Nat Rev Mol Cell Biol 2008; 9:713 - 724
- Mittnacht S. Genes Dev 1998; 8:21 - 27
- Chan HM, et al. Nat Cell Biol 2001; 3:667 - 674
- Markham D, et al. EMBO Rep 2006; 7:192 - 198
- Munro S, et al. Oncogene 2010; 29:2357 - 2367
- Scoumanne A, et al. Histol Histopathol 2008; 23:1143 - 1149
- Martin C, et al. Nat Rev Mol Cell Biol 2005; 6:838 - 849
- Carr SM, et al. EMBO J 2011; 30:317 - 327
- Ren S, et al. Cell 2004; 117:239 - 251
- Vidal A, et al. Gene 2000; 247:1 - 15